3.3.1. The H2O/OH Bands
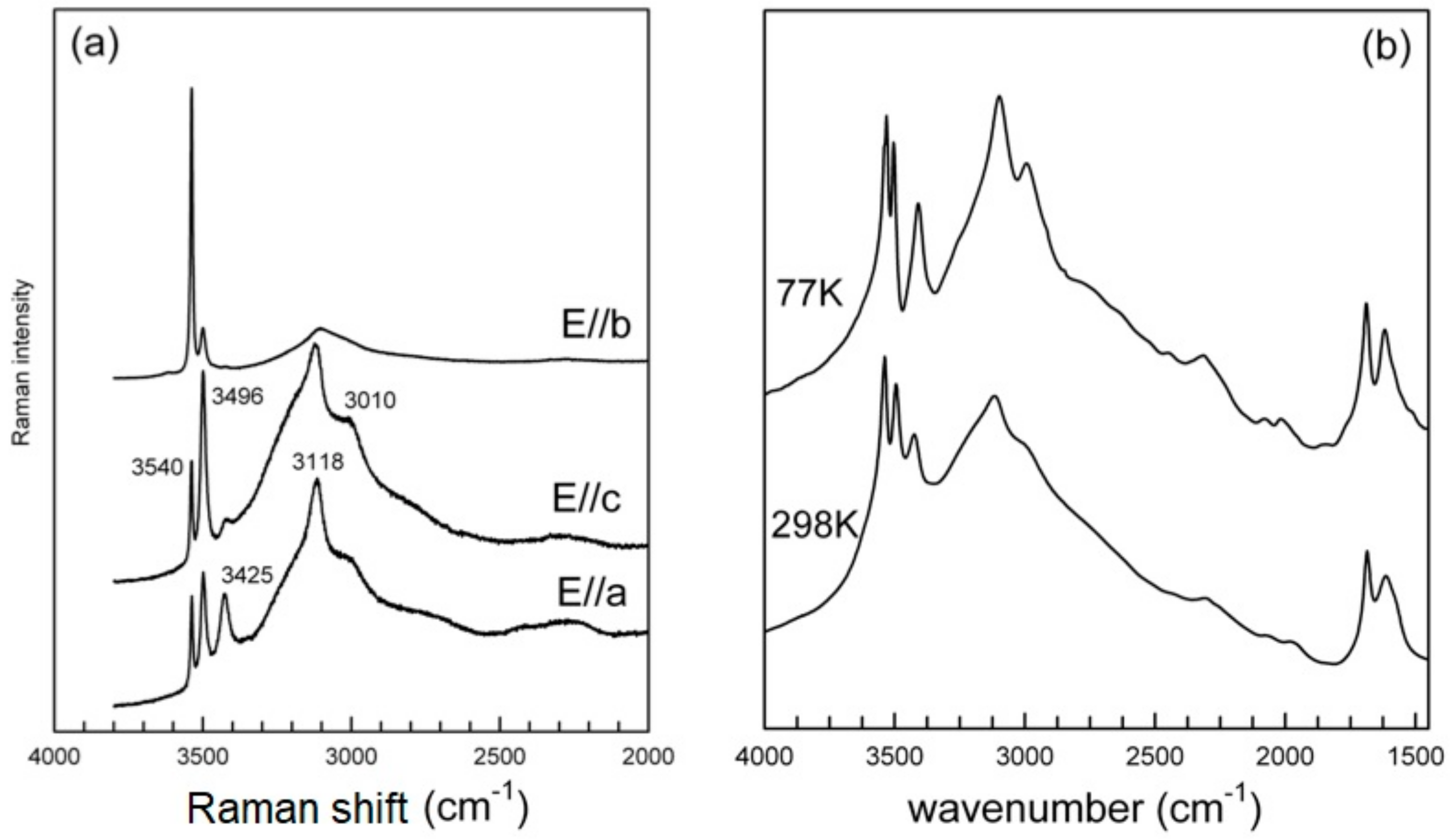
Single-crystal Raman spectra collected in the principal H
2O-stretching region (3800–2000 cm
−1) for different orientation of the laser beam with respect to the crystal orientation are compared in
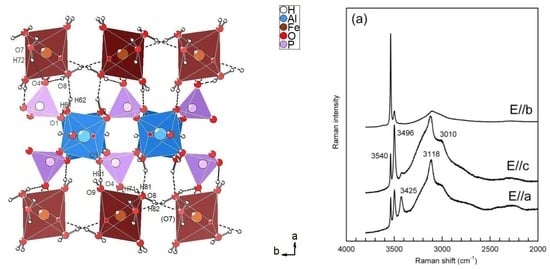
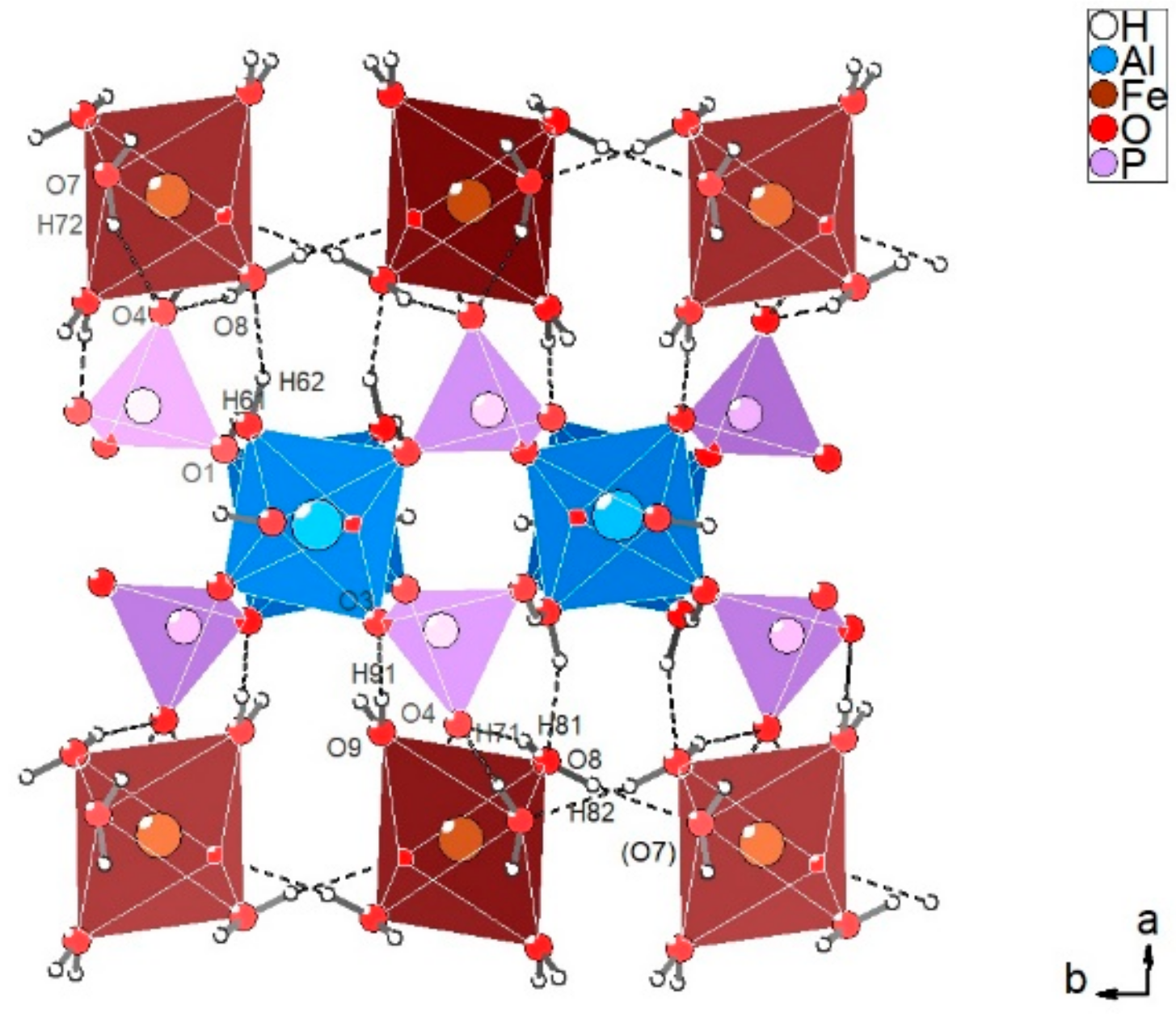
Figure 6a. In particular, the spectra were collected on a platy cleavage fragment, with the laser oriented either parallel or perpendicular to the elongation of the plate (top and middle), or by orienting the plate on one edge (bottom). As described above, the structure of metavauxite is strongly layered (
Figure 3b), and thus shows a perfect cleavage, however never reported in the description of its physical properties (see, for example, the pertinent mindat.org database entry [
37]). Considering the structural arrangement, the observed cleavage is expected to be parallel to the
b–
c plane (
Figure 3). Spectra collected by rotating the plate 90 degrees show a strong pleochroism of the Raman signal in the H
2O-stretching region (
Figure 6a). All spectra consist of three well-defined peaks at 3540, 3496, and 3425 cm
−1 and a broad scattering extending from 3400 to 2000 cm
−1, which is clearly due to the contribution of several overlapping components; some of these are resolved at 3118 and 3010 cm
−1 (
Figure 6a), while an evident shoulder is apparent around 3190 cm
−1. Rotation of the crystal with respect to the laser beam is correlated with a change in intensity of all components, which is particularly evident for the higher-frequency peak at 3540 cm
−1, whose scattering increases by one order of magnitude when the beam is normal to the plate elongation (
Figure 6a, top).
The powder FTIR pattern (
Figure 6b) collected at room-T (298K) is almost identical to the Raman spectrum and shows three well-resolved peaks on the higher-frequency side of the broad absorption, with wavenumbers coincident (within 1–2 cm
−1) to the corresponding Raman peaks (
Figure 6b and
Table 6).
In the H
2O bending (ν
2) region (
Figure 6b) two peaks at 1687 and 1614 cm
−1 are present. At 77K (
Figure 6b), there is a slight shift (
Table 6) and a sharpening of all bands that are, thus, better resolved; see, in particular, the peak around 3010 cm
−1; no new components, notably bands due to ice formation, are observed.
In the metavauxite structure, there are four independent H
2O molecules and one OH group (
Table 2); considering that the H
2O molecules are all involved in hydrogen bonds of different strengths with the surrounding oxygen atoms (
Table 4), up to eight stretching bands originating from the single O–H bonds in the H
2O molecules are ideally expected. In addition, the O–H stretching mode of the hydroxyl groups, and the first overtone of the H
2O bendings (2ν
2) also occur in this range, thus contributing to the complexity of the pattern (
Figure 6). Decomposition of the broad absorption into different bands involves significant constraints to be introduced. However, the observed band wavenumbers, based on the empirical O
donor…O
acceptor distance–frequency correlation of Libowitzky [
38], are all compatible with the bond distances reported in
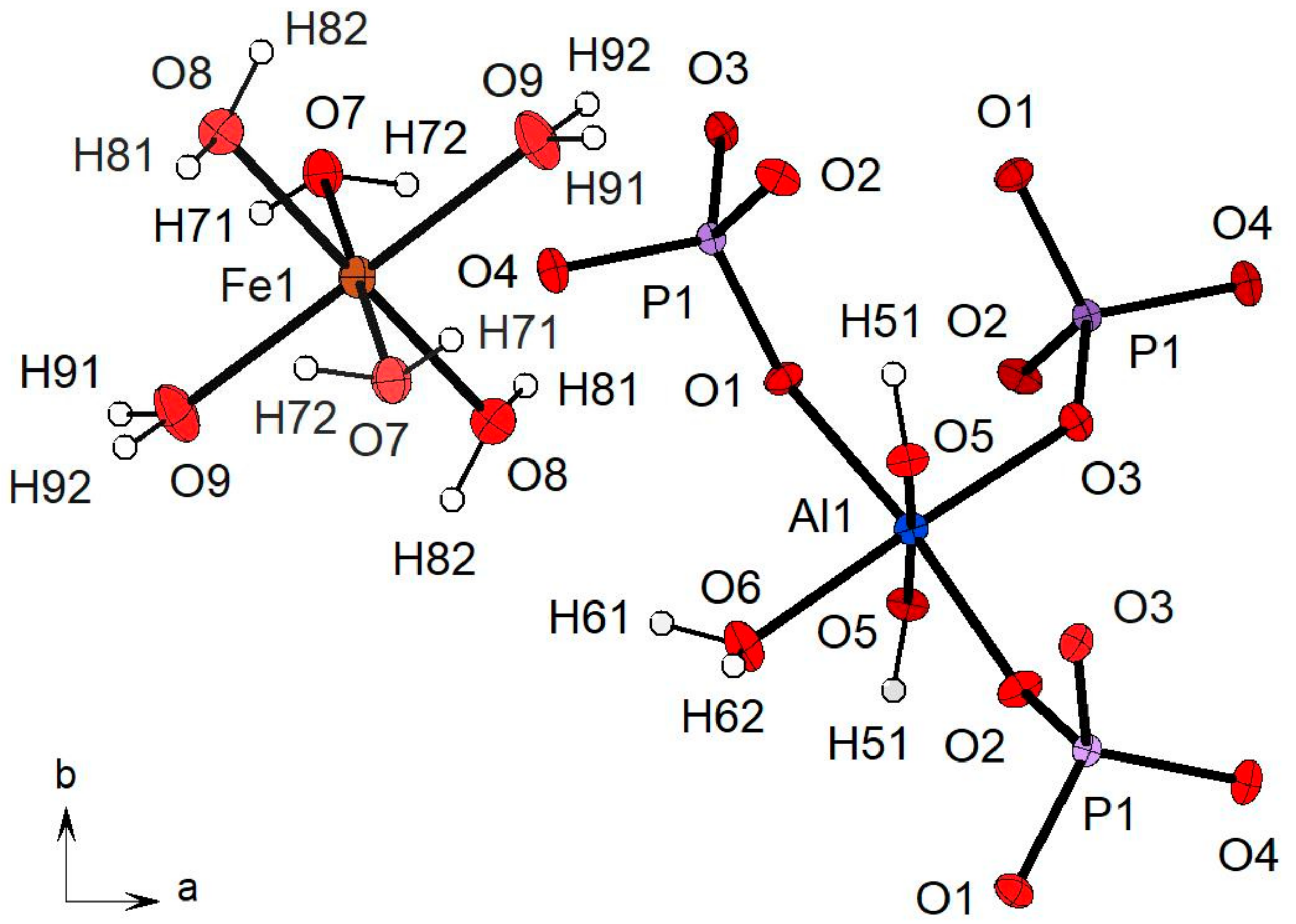
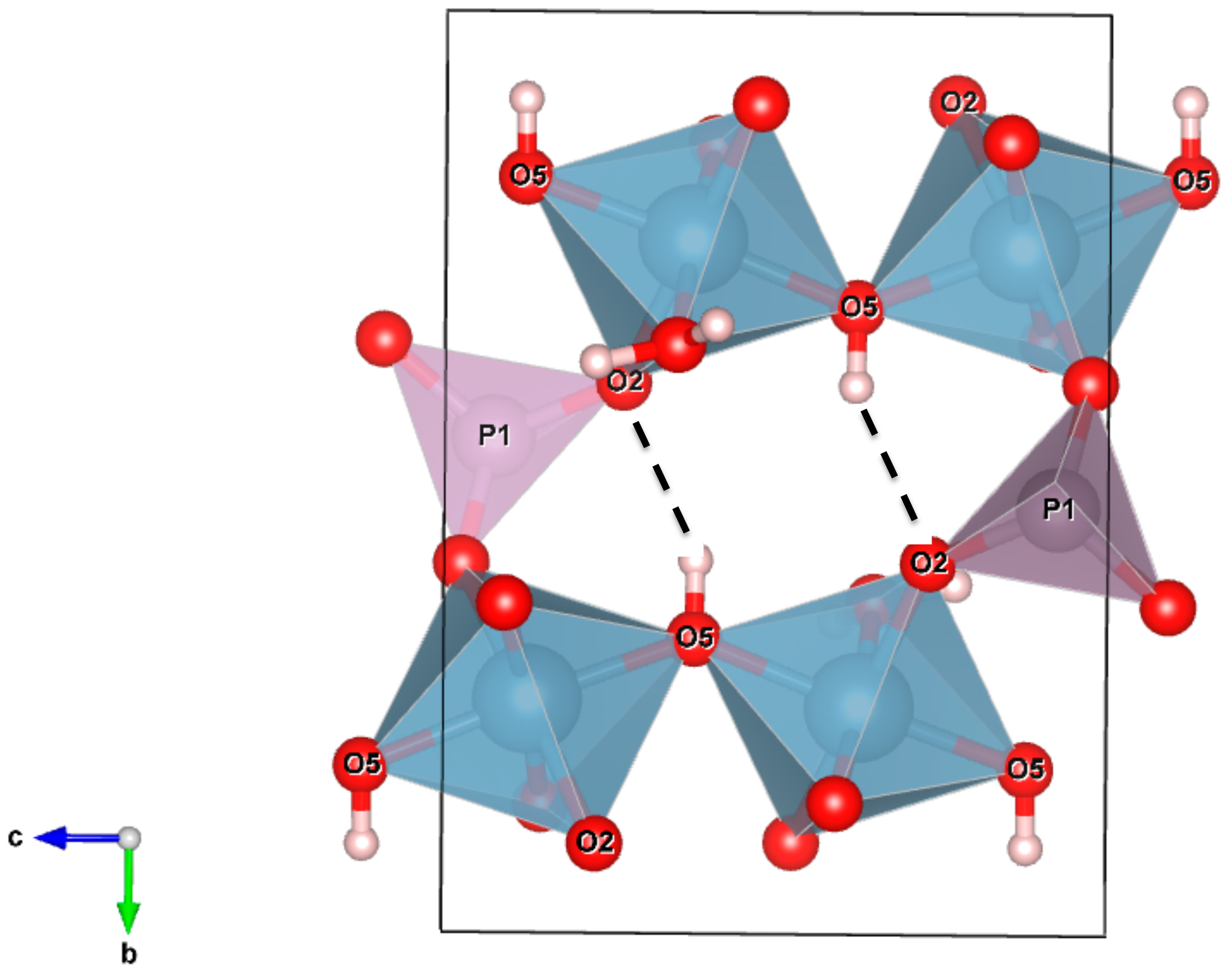
Table 4. In particular, the sharp peak at 3540 cm
−1 can be assigned to the O5–H5 hydroxyl group; the calculated wavenumber is, in fact, exactly coincident with the O
donor–O
acceptor distance (2.978 A) observed via the structure refinement (
Table 4). The local configuration of this O–H group is shown in
Figure 7. The O5 oxygen is directly bonded to two nearest-neighbor Al cations and to the H51 hydrogen; this arrangement can be expressed as Al1Al1–O5…H51. If the H51 hydrogen were free from any interaction with the next-nearest-neighbor surrounding oxygens, the vibrational frequency would be around 3670 cm
−1. This is the case of vauxite, where the OH-stretching mode (ν
OH) is close to 3660 cm
−1 [
5]. In metavauxite, the bond valence sum resulting from the structure refinement (
Table 5) indicates that the O5–H51 hydroxyl group is involved in a weak but significant hydrogen bridge with the next-nearest-neighbor O2 oxygen connecting the Al1 octahedron with the P1 tetrahedron (
Figure 7).
In such a case, the O–H bond strength is lowered, and the corresponding vibrational mode is shifted by~120 cm
−1 toward lower wavenumbers. This assignment is also consistent with the strong pleochroism observed for the Raman spectra in the O–H stretching region. Inspection of
Figure 3a shows that the elongation of the crystal plate must coincide with the
c crystallographic direction, i.e., with the direction of the octahedral chains. This being the case, the spectrum of
Figure 6a (top) has been collected with the E electric vector of the incident beam closely parallel to the O5–H5 bond direction (
Figure 3a, see also
Figure 7), causing the significant enhancement of the intensity of the Raman scattering.
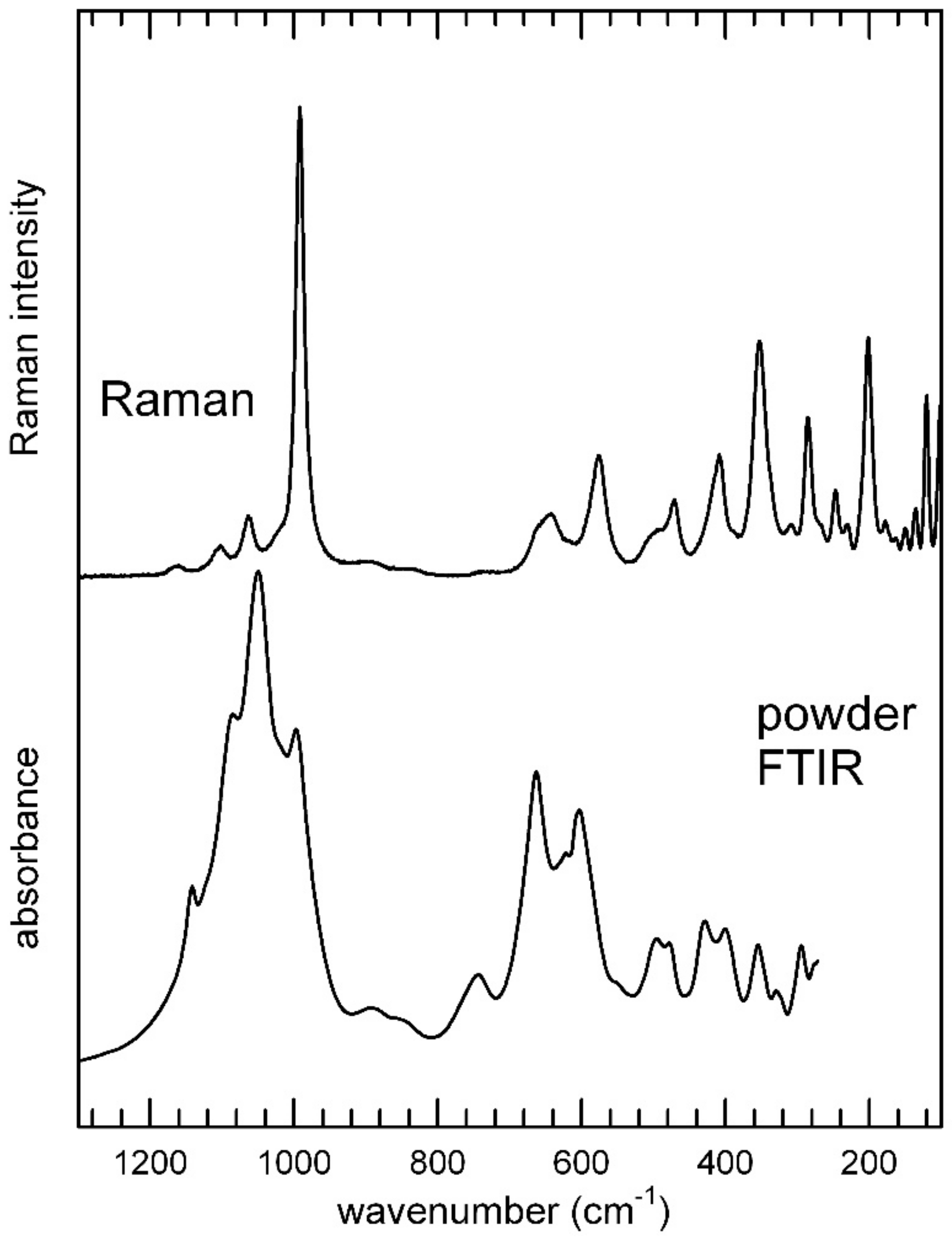
3.3.2. The Low-Frequency Region
The Raman spectrum in the low–frequency 1400–400 cm
−1 region is given in
Figure 8 (top) in comparison with the FTIR powder spectrum (
Figure 8, bottom). Measured band positions (wavenumbers, cm
−1) are listed in
Table 6, where the band assignments are based on literature data for similar materials [
39,
40,
41,
42,
43]. The free phosphate ion, PO
43− with ideal
Td point symmetry has four modes of vibration: The symmetric stretching ν
1(A
1) at 980 cm
−1, the symmetric bending ν
2(E) at 420 cm
−1, the asymmetric stretching ν
3(F
2) at 1082 cm
−1, and the asymmetric bending ν
4(F
2) at 567 cm
−1 (e.g., [
39,
40]). In accordance with selection rules, the triply degenerate asymmetric stretching and bending modes (F
2) are both Raman and infrared active, whereas the non-degenerate symmetric stretching (A
1) and the doubly degenerate symmetric bending (E) are Raman active only. In the IR pattern, these latter modes became apparent only when the symmetry of the PO
43− ion is lowered [
39,
40]. In such a case, shifts of the absorption bands with respect to the ideal values, band splitting, and appearance of ideally non active IR/Raman modes are observed. Both the FTIR and Raman patterns (
Figure 8 and
Table 6) suggest that the point symmetry of the PO
43− group in metavauxite is reduced from
Td to
Cs, in accordance with the structure refinement that provides four slightly different P1–O distances in the unique P tetrahedron (
Table 3). The intense peaks observed in the antisymmetric stretching range at 1142, 1084, and 1049 cm
−1, respectively (FTIR), and the weak peaks at 1159, 1102, and 1063 cm
−1 (Raman) can, thus, be assigned to the ν
3 mode.
One component at 992 (Raman) and 997 (IR) cm
−1 occur as symmetric stretching mode. The antisymmetric bending modes (ν
4,
Table 6) are intense in the FTIR spectrum (in the 600–450 cm
−1 range), while being relatively weak in the Raman pattern.
The ν
2(PO
4)
3− modes, are observed at wavenumbers >400 cm
−1 (e.g., [
2,
41]) in the Raman spectrum as medium intense absorptions (
Figure 7). Finally, Raman peaks that are found below 200 cm
−1 are generally classified as lattice modes (e.g., [
44]).

 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}