Systematic Design of Crystal Structure for Hofmann-Like Spin Crossover Fe(L)2[Ag(CN)2]2 Complexes

 , ,
, ,
Abstract
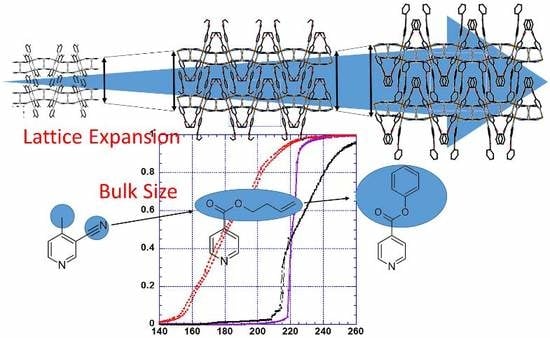
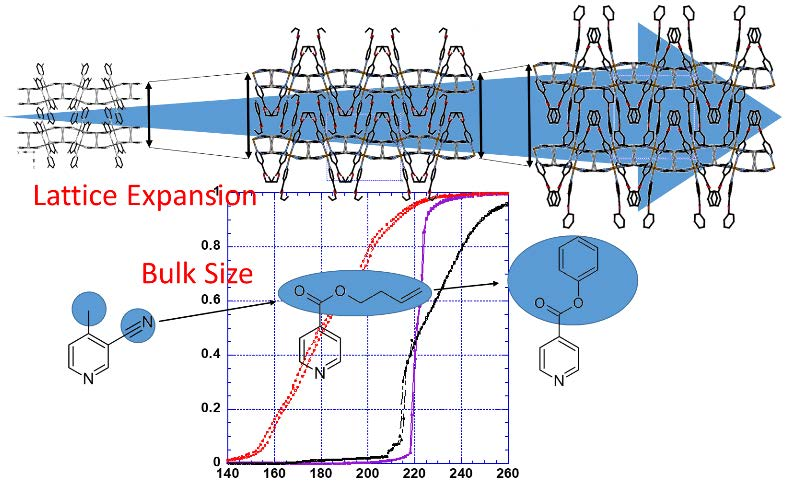
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis
2.2.1. Preparation of Compound 1
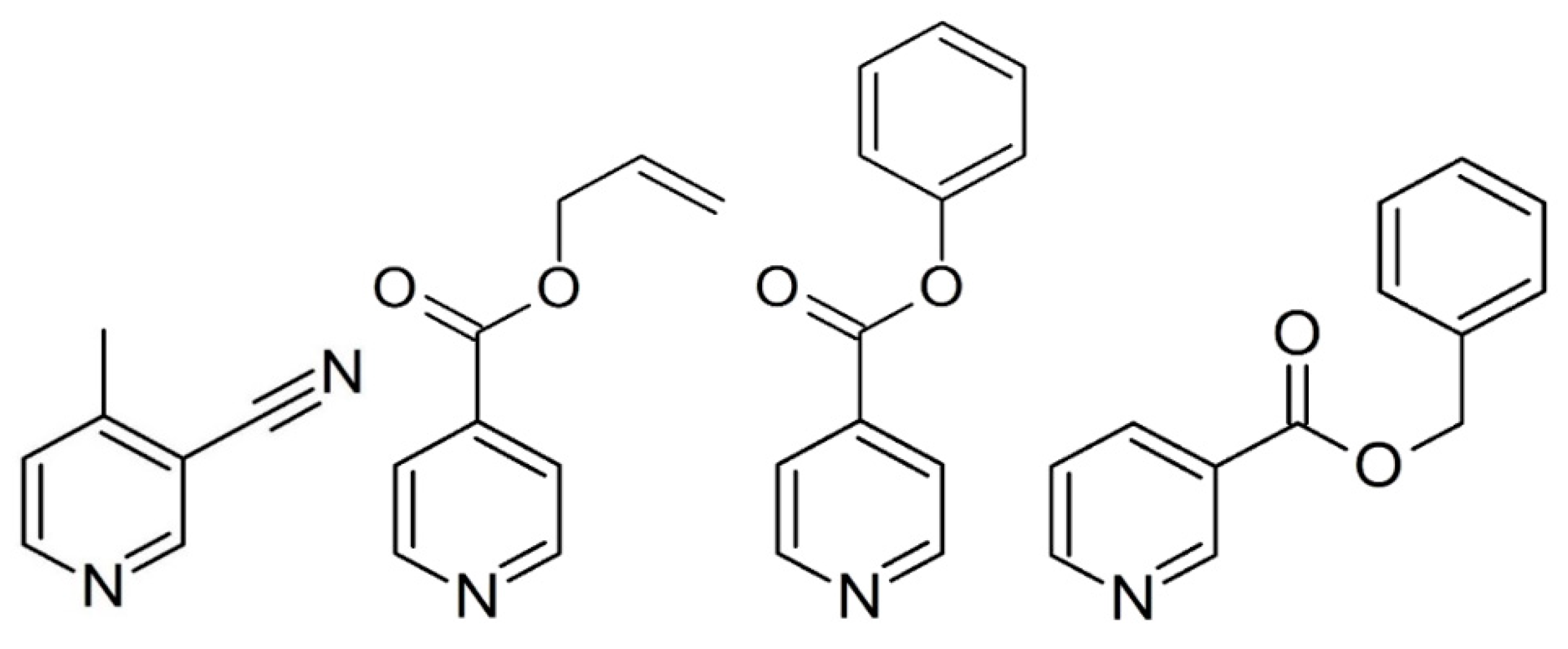
2.2.2. Preparation of Compounds 2–4
2.3. X-Ray Crystallography
2.4. Magnetic Measurements
3. Results
3.1. Crystal Structures
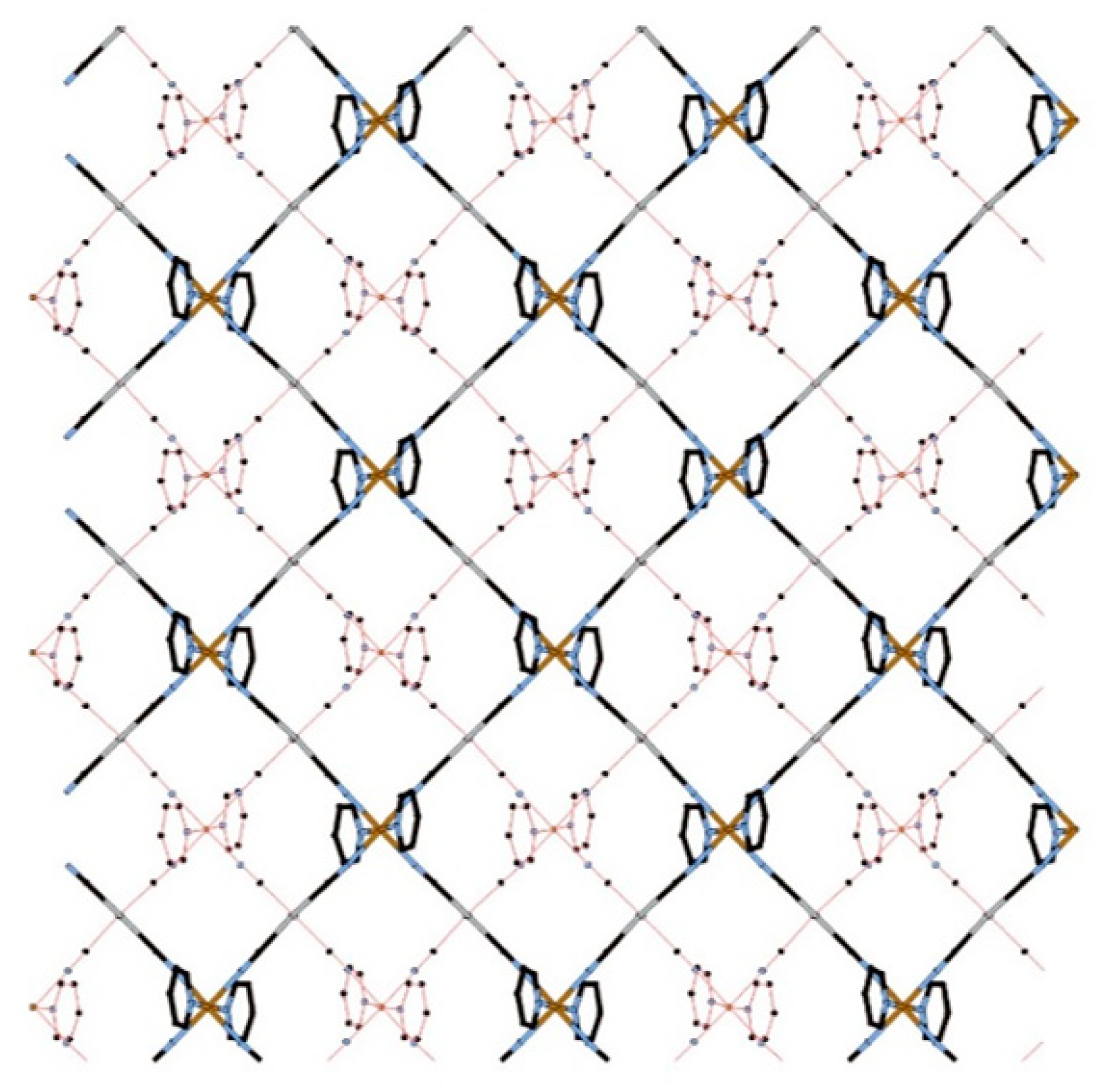
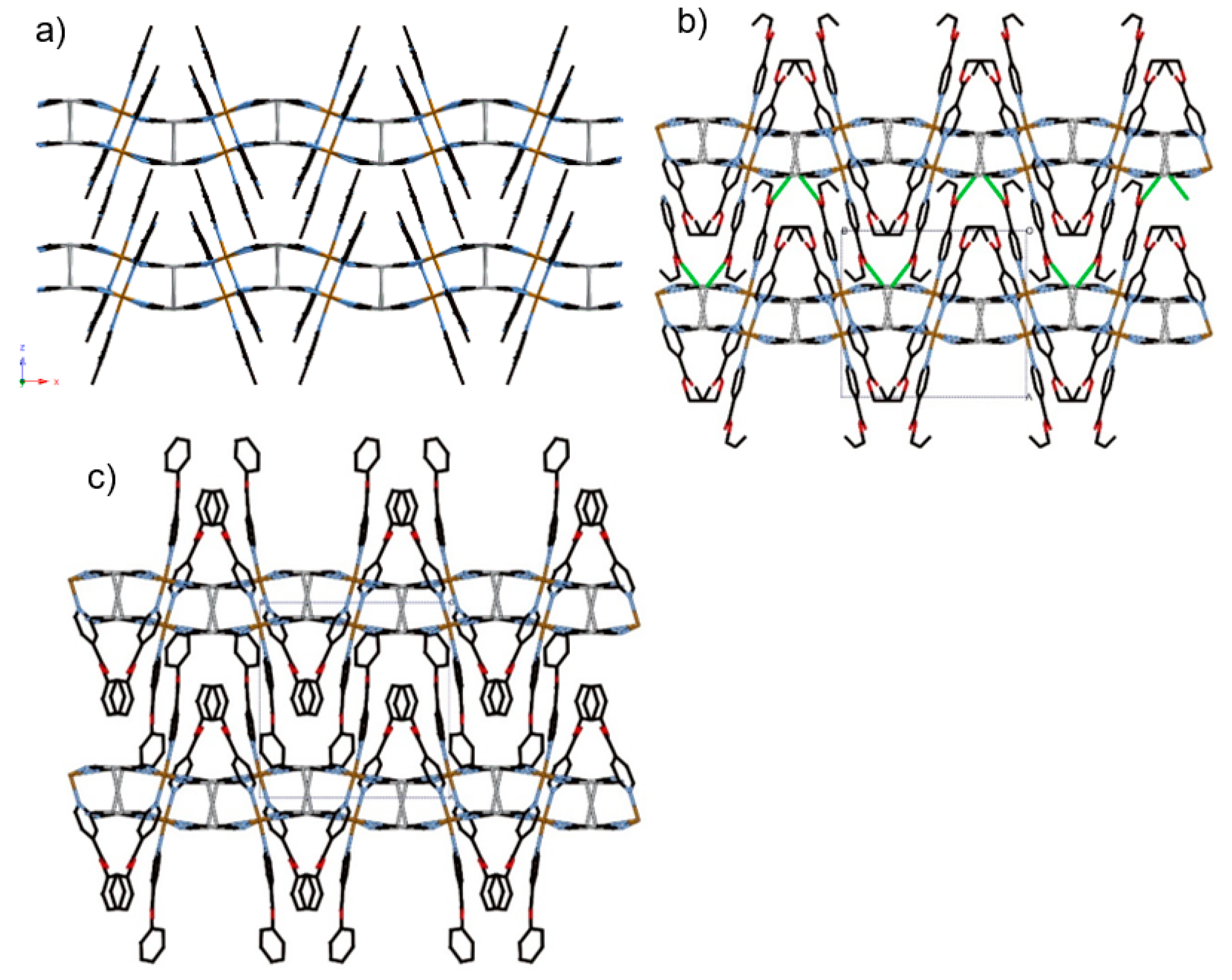
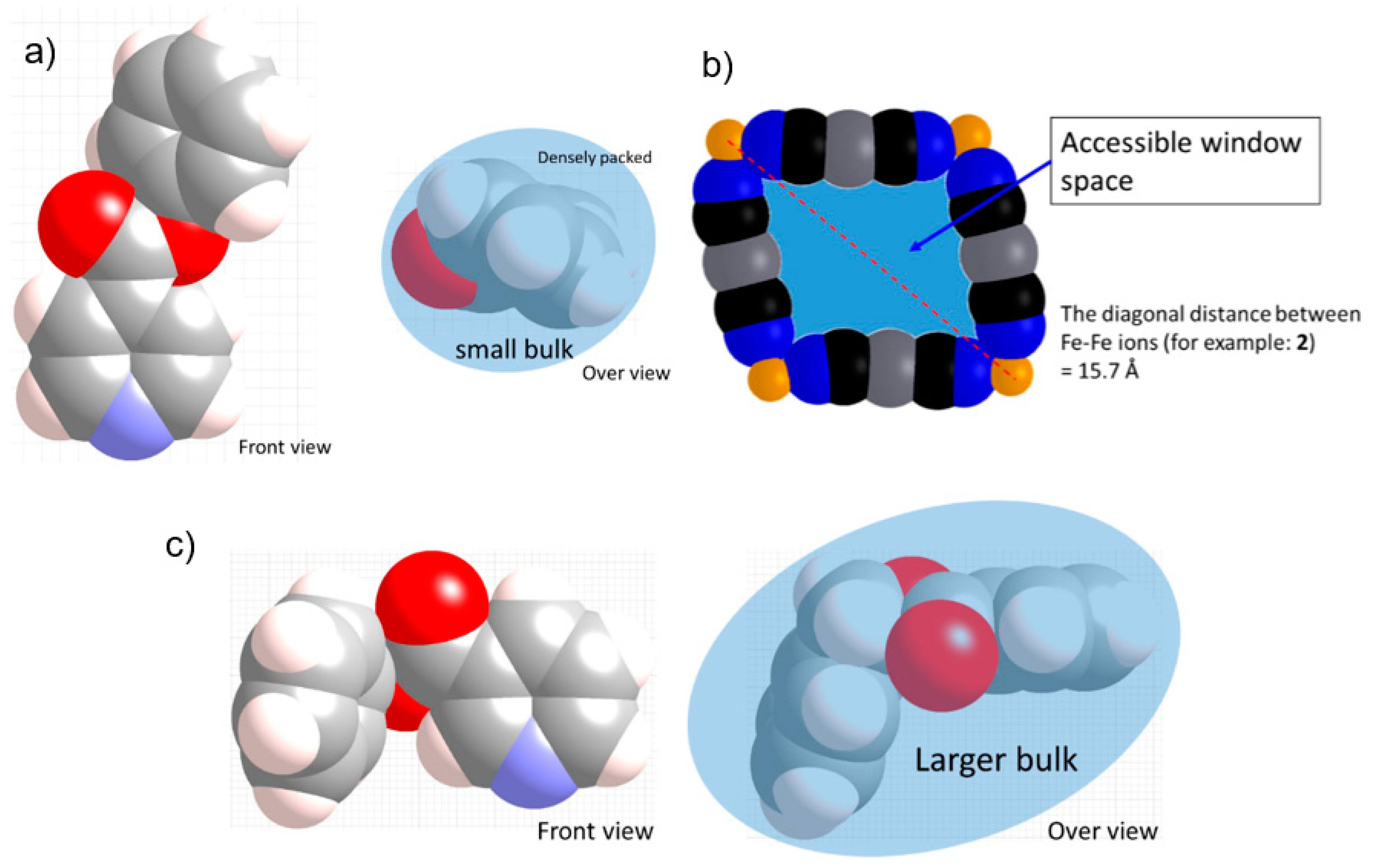
3.1.1. Overview of Bilayer Structure for 1–3
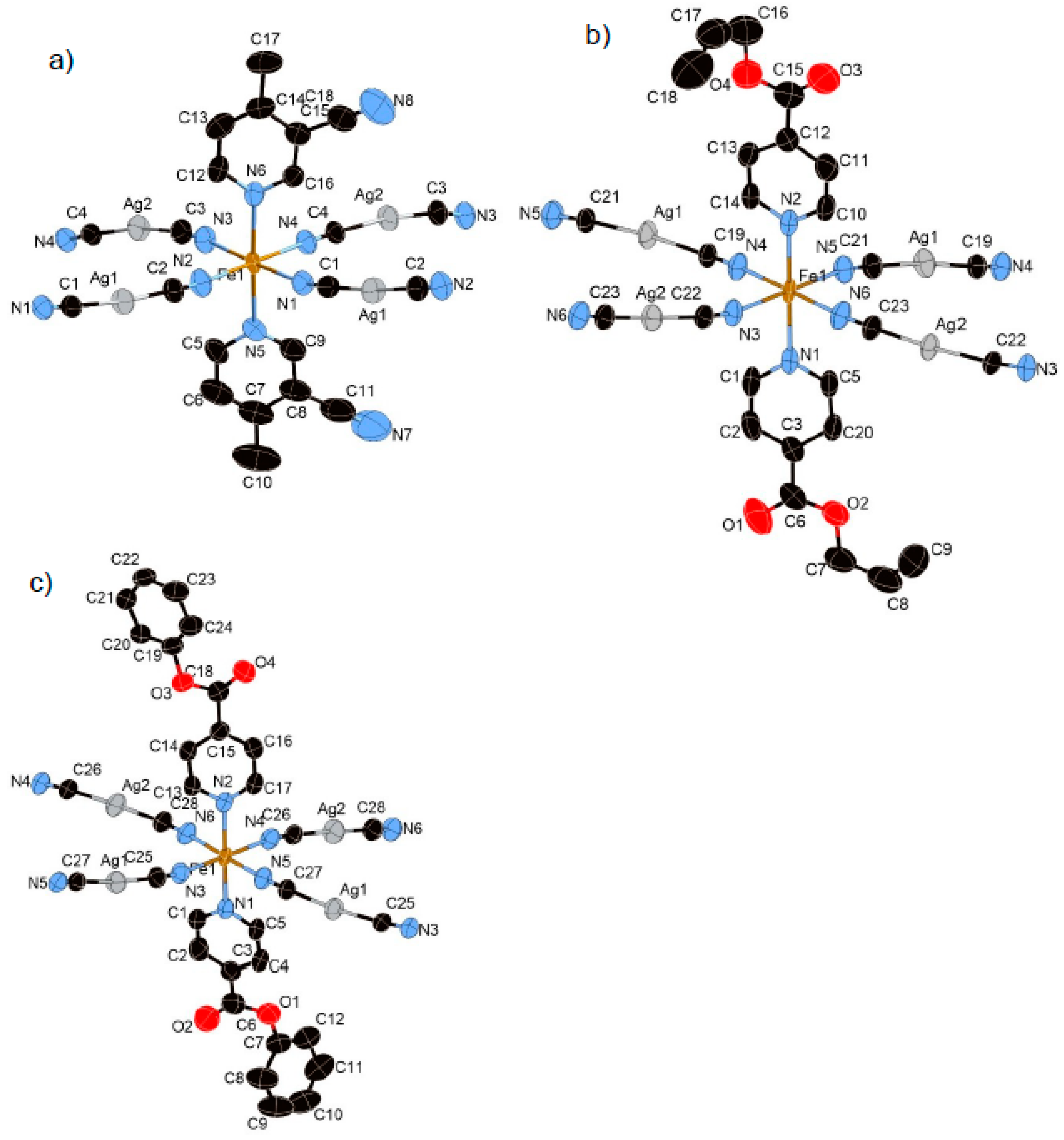
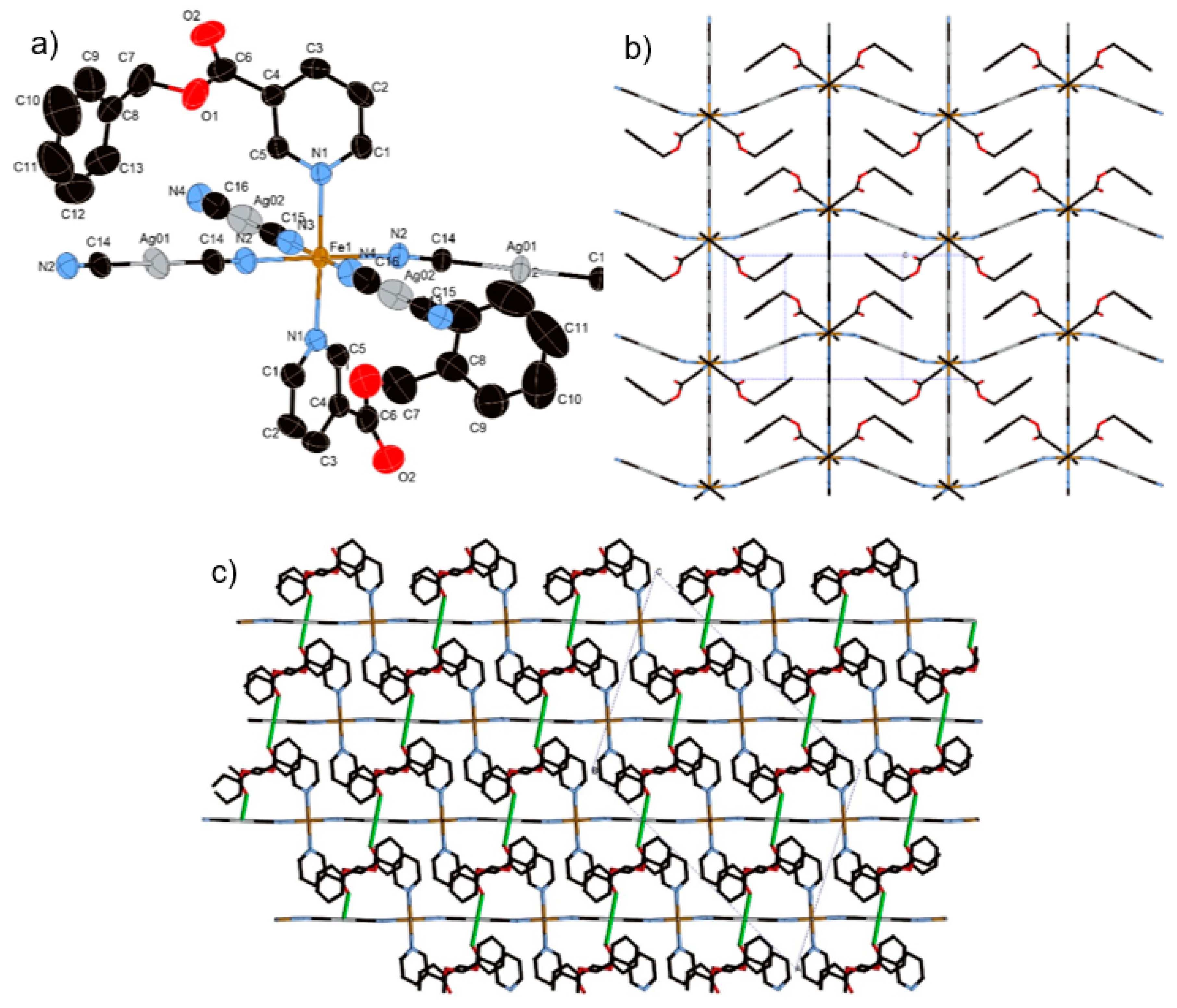
3.1.2. Structure of Compound 1 (T = 296 K and 100 K)
3.1.3. Structure of Compound 2 (T = 250 K and 85 K)
3.1.4. Structure of Compound 3 (T = 250 K and 85 K)
3.1.5. Structure of Compound 4 (T = 275 K and 90 K)
3.2. Magnetic Properties
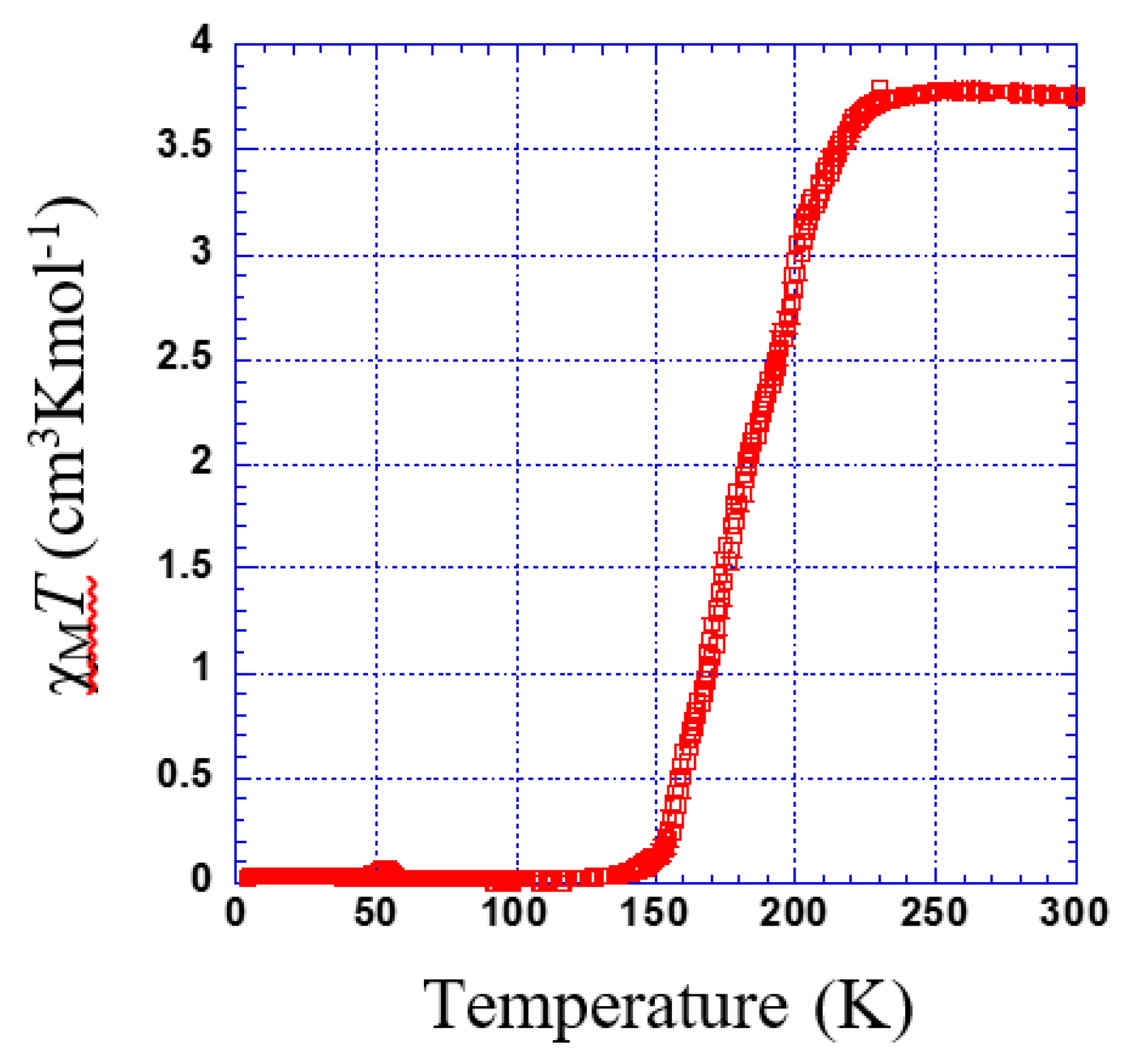
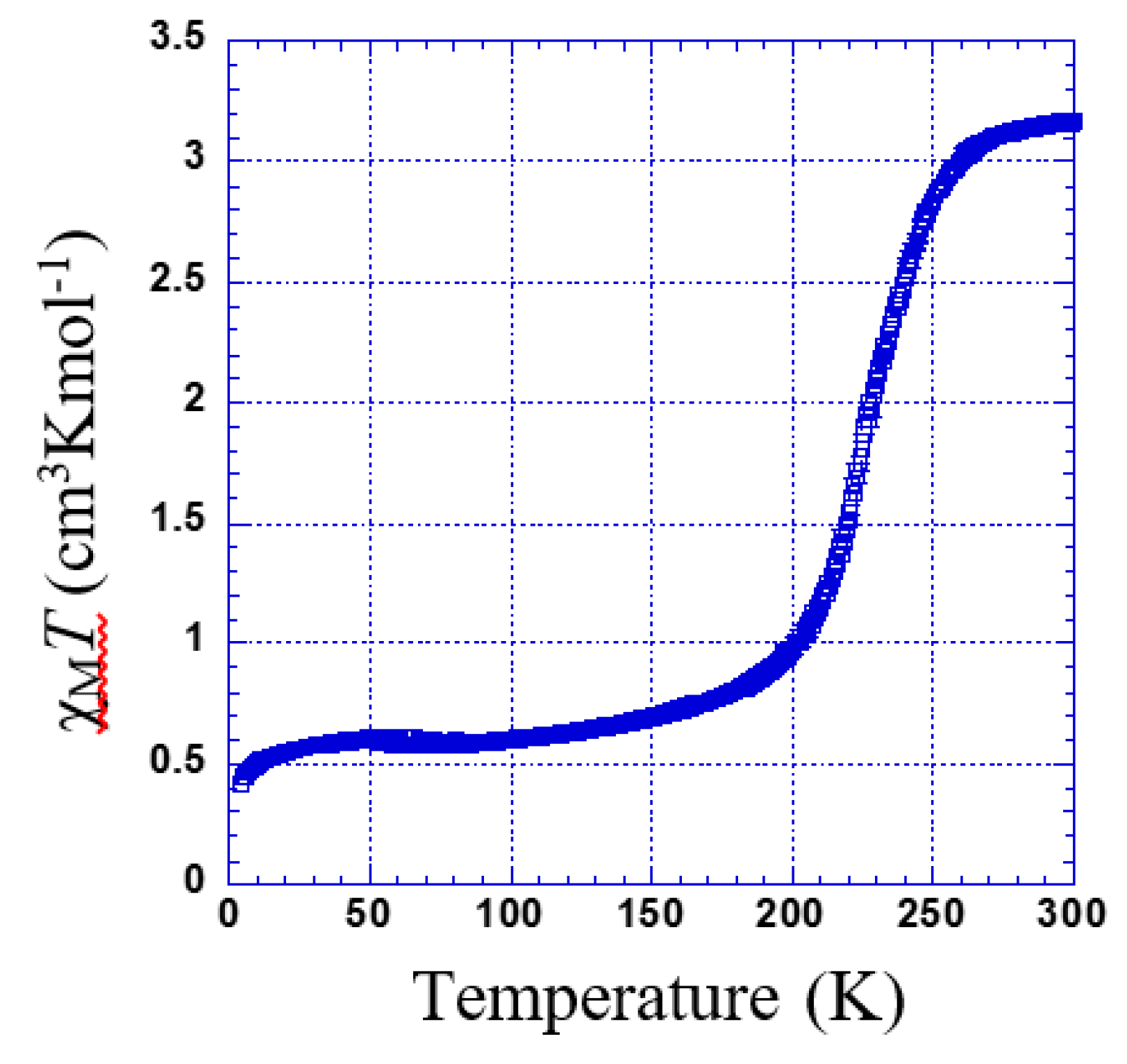
3.2.1. Thermal Dependence Magnetic Behavior of Compound 1
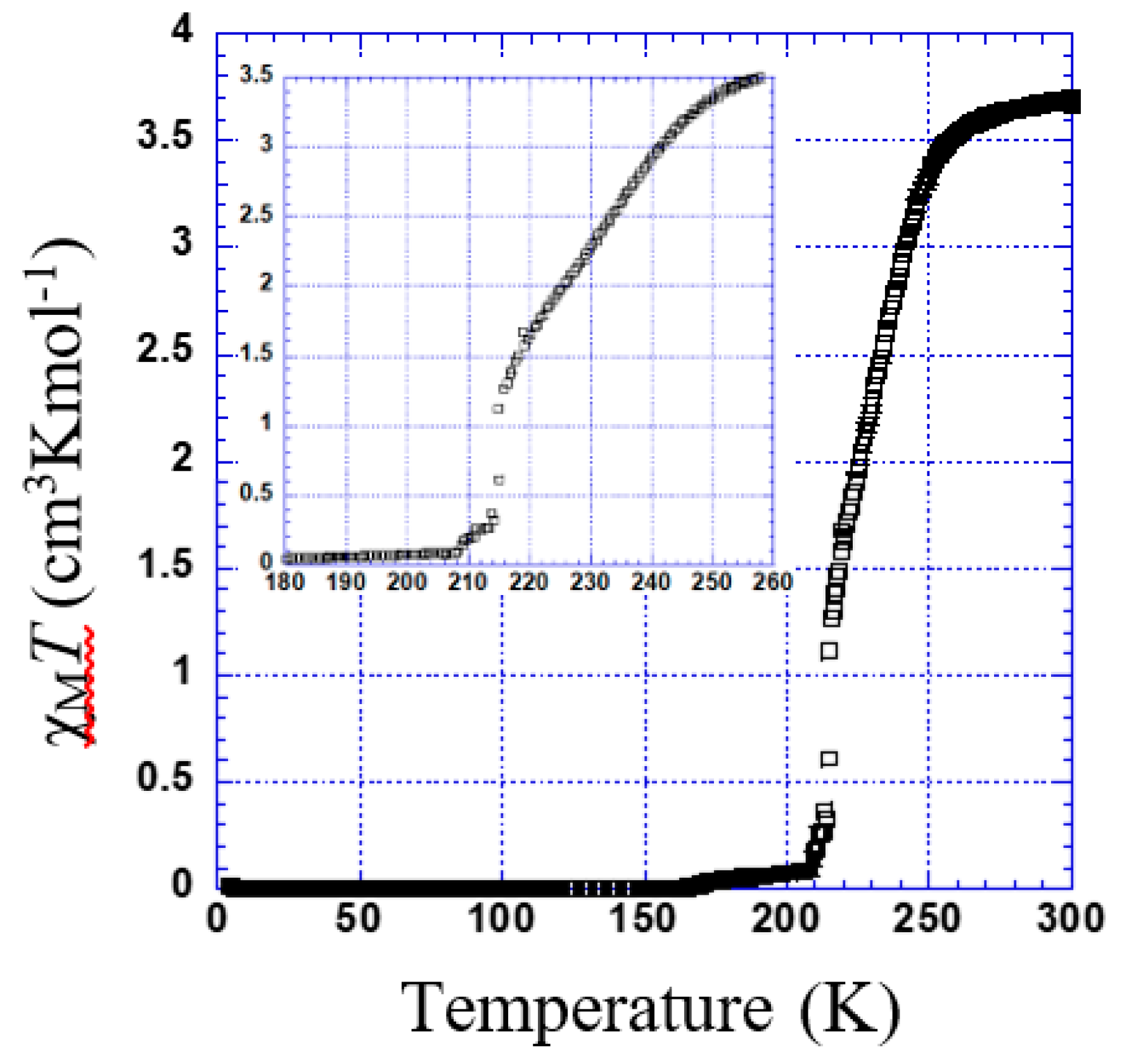
3.2.2. Thermal Dependence Magnetic Behavior of Compound 2
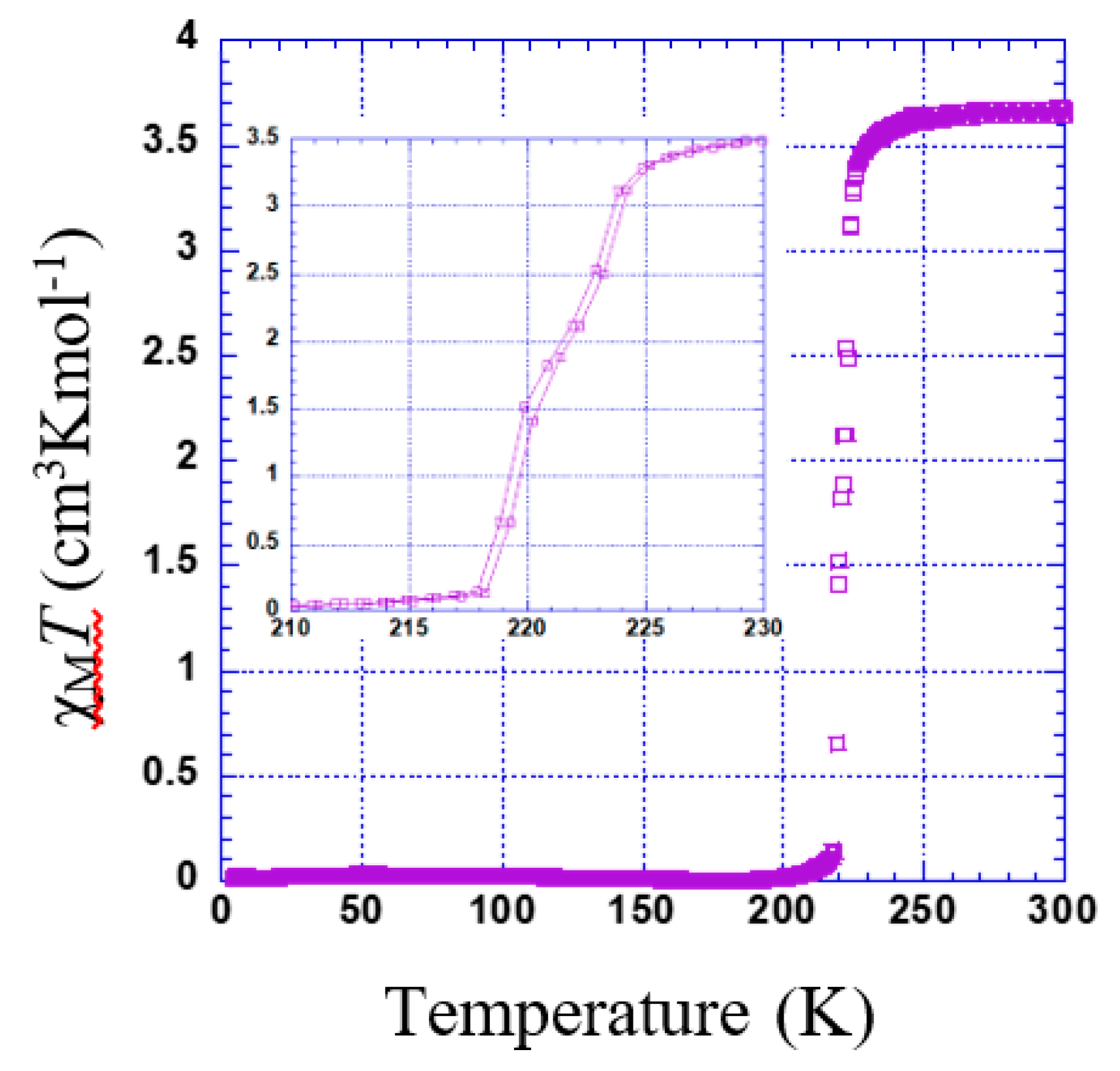
3.2.3. Thermal Dependence Magnetic Behavior of Compound 3
3.2.4. Thermal Dependence Magnetic Behavior of Compound 4
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Ariga, K.; Hill, J.P.; Lee, M.V.; Vinu, A.; Charvet, R.; Acharya, S. Challenges and breakthroughs in recent research on self-assembly. Sci. Technol. Adv. Mater. 2008, 9, 014109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, M.; Oguro, D.; Miyazawa, M.; Oka, H.; Yamaguchi, K.; Ogura, K.; Real, J.A.; Andrés, E.; Munoz, M.C.; Julve, M.; et al. Self-Assembly of ten molecules into nanometer Sized organic host framework. Nature 1995, 268, 265–268. [Google Scholar]
- Hofmann, K.A.; Kuspert, F.A. Verbindungen von Kohlenwasserstoffen mit Metallsalzen. Z. Anorg. Allg. Chem. 1897, 15, 204–207. [Google Scholar] [CrossRef]
- Kitazawa, T.; Gomi, Y.; Takahashi, M.; Takeda, M.; Enomoto, M.; Miyazaki, A.; Enoki, T. Spin-crossover behaviour of the coordination polymer FeII(C5H5N)2NiII(CN)4. J. Mater. Chem. 1996, 6, 119–121. [Google Scholar] [CrossRef]
- Galet, A.; Muñoz, M.C.; Martinez, V.; Real, J.A. supramolecular isomerism in spin crossover networks with aurophilic interactions. Chem. Commun. 2004, 2268–2269. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.C.; Gaspar, A.B.; Galet, A.; Real, J.A. Spin-crossover behavior in cyanide-bridged iron(II)–silver(I) bimetallic 2D Hofmann-like metal–organic frameworks. Inorg. Chem. 2007, 46, 8182–8192. [Google Scholar] [CrossRef] [PubMed]
- Agustí, G.; Muñoz, M.C.; Gaspar, A.B.; Real, J.A. Spin-crossover behavior in cyanide-bridged iron(II)–gold(I) bimetallic 2D Hofmann-like metal–organic frameworks. Inorg. Chem. 2008, 47, 2552–2561. [Google Scholar] [CrossRef] [PubMed]
- Kosone, T.; Kachi-Terajima, C.; Kanadani, C.; Saito, T.; Kitazawa, T. A two-step and hysteretic spin-crossover transition in new cyano-bridged hetero-metal FeIIAuI 2Dimensional assemblage. Chem. Lett. 2008, 37, 422–423. [Google Scholar] [CrossRef]
- Kosone, T.; Kanadani, C.; Saito, T.; Kitazawa, T. Synthesis, crystal structures, magnetic properties and fluorescent emissions of two-dimensional bimetallic coordination frameworks FeII(3-fluoropyridine)2[AuI(CN)2]2 and MnII(3-fluoropyridine)2[AuI(CN)2]2. Polyhedron 2009, 28, 1930–1934. [Google Scholar] [CrossRef]
- Kosone, T.; Kanadani, C.; Saito, T.; Kitazawa, T. Spin crossover behavior in two-dimensional bimetallic coordination polymer FeII(3-bromo-4-picoline)2[AuI(CN)2]2: Synthesis, crystal structures, and magnetic properties. Polyhedron 2009, 28, 1991–1995. [Google Scholar] [CrossRef]
- Kosone, T.; Tomori, I.; Kanadani, C.; Saito, T.; Mochida, T.; Kitazawa, T. Unprecedented three-step spin-crossover transition in new 2dimensional coordination polymer {FeII(4-methylpyridine)2[AuI(CN)2]2}. Dalton Trans. 2010, 39, 1719–1721. [Google Scholar] [CrossRef] [PubMed]
- Muñoz, M.C.; Real, J.A. Thermo-, piezo-, photo- and chemo-switchable spin crossover iron(II)-metallocyanate based coordination polymers. Coord. Chem. Rev. 2011, 255, 2068–2093. [Google Scholar] [CrossRef]
- Sugaya, A.; Ueno, S.; Okabayashi, J.; Kitazawa, T. Crystal structure and magnetic properties of the spin crossover complex Fe II (ethyl nicotinate) 2 [Au I (CN)2]2. New J. Chem. 2014, 38, 1955–1958. [Google Scholar] [CrossRef]
- Ueno, S.; Kawasaki, T.; Okabayashi, J.; Kitazawa, T. Structural, Electronic, and Magnetic Properties of Novel Spin-Crossover Complex: Fe(butyl nicotinate) 2 [Au(CN)2]2. Bull. Chem. Soc. Jpn. 2015, 88, 551–553. [Google Scholar] [CrossRef]
- Okabayashi, J.; Ueno, S.; Kawasaki, T.; Kitazawa, T. Ligand 4-X pyridine (X = Cl, Br, I) dependence in Hofmann-type spin crossover complexes: Fe(4-Xpyridine)2[Au(CN)2]2. Inorg. Chim. Acta 2016, 445, 17–21. [Google Scholar] [CrossRef]
- Kosone, T.; Kawasaki, T.; Tomori, I.; Okabayashi, J.; Kitazawa, T. Modification of cooperativity and critical temperatures on a hofmann-like template structure by modular substituent. Inorganics 2017, 5, 55. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SADABS, Program for Empirical Absorption Correction for Area Detector Data; University of Göttingen: Göttingen, Germany, 1996. [Google Scholar]
- Sheldrick, G.M. SHELXL, Program for the Solution of Crystal Structures; University of Göttingen: Göttingen, Germany, 1997. [Google Scholar]
- Kosone, T.; Kitazawa, T. Guest-dependent spin transition with long range intermediate state for 2dimensional Hofmann-like coordination polymer. Inorg. Chim. Acta 2016, 439, 159–163. [Google Scholar] [CrossRef]
- Hammett, L.P. The Effect of Structure upon the reactions of organic compounds. benzene derivatives. J. Am. Chem. Soc. 1937, 59, 96–103. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| X | 1 (296 K) | 1 (100 K) | 2 (250 K) | 2 (85 K) |
| Empirical formula | C18H12Ag2FeN8 | C18H12Ag2FeN8 | C22H18Ag2FeO4N6 | C22H18Ag2FeO4N6 |
| Formula weight | 611.95 | 611.95 | 702.09 | 702.09 |
| Crystal size (mm3) | 0.44 × 0.38 × 0.13 | 0.44 × 0.38 × 0.13 | 0.38 × 0.20 × 0.10 | 0.38 × 0.20 × 0.10 |
| Crystal system | Orthorhombic | Orthorhombic | Monoclinic | Monoclinic |
| A (Å) | 14.7200 (7) | 14.1147 (10) | 12.2912 (6) | 12.4008 (19) |
| B (Å) | 15.0169 (7) | 14.5991 (10) | 13.6582 (7) | 13.078 (2) |
| C (Å) | 20.5702 (10) | 20.3274 (15) | 15.9764 (9) | 15.512 (2) |
| V (Å3) | 4547.0 (4) | 4188.7 (5) | 2677.0 (2) | 2515.7 (7) |
| B (°) | 93.5210 (10) | 90.276 (2) | ||
| Space group | Pbca | Pbca | P21/c | P21/c |
| Z value | 8 | 8 | 4 | 4 |
| Dcalc | 1.788 | 1.941 | 1.742 | 1.857 |
| F(000) | 2368 | 2368 | 1512 | 1376 |
| Reflections collected | 33,414 | 28,412 | 20,225 | 18,877 |
| Independent reflections | 7037 | 6174 | 7881 | 7605 |
| Parameters | 264 | 264 | 316 | 316 |
| Final R1, Rw (I > 2s) | 0.0336, 0.0777 | 0.0317, 0.0629 | 0.0326, 0.0624 | 0.0409, 1.118 |
| Final R1, Rw (all data) | 0.0622, 0.0910 | 0.0540, 0.0754 | 0.0738, 0.0710 | 0.0618, 1.333 |
| Goodness-of-fit | 1.080 | 1.143 | 0.805 | 0.819 |
| 3 (250 K) | 3 (85 K) | 4 (275 K) | 4 (90 K) | |
| Empirical formula | C28H18Ag2FeN6O4 | C28H18Ag2FeN6O4 | C30H22Ag2Fe2N6O4 | C30H22Ag2Fe2N6O4 |
| Formula weight | 774.07 | 774.07 | 802.13 | 802.13 |
| Crystal size (mm3) | 0.24 × 0.23 × 0.20 | 0.24 × 0.23 × 0.20 | 0.28 × 0.24 × 0.22 | 0.28 × 0.24 × 0.22 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| A (Å) | 14.1172 (9) | 13.9673 (7) | 21.7109 (8) | 21.2491 (7) |
| B (Å) | 13.6365 (8) | 13.1675 (7) | 10.6183 (4) | 10.2384 (3) |
| C (Å) | 15.6922 (10) | 15.3684 (8) | 15.9222 (6) | 15.5439 (5) |
| V (Å3) | 3004.2 (3) | 2822.1 (3) | 3291.9 (2) | 3017.83 (16) |
| B (°) | 96.0340 (11) | 93.1900 (10) | 116.2570 (10) | 116.82 |
| Space group | P21/c | P21/c | C2/c | C2/c |
| Z value | 4 | 4 | 4 | 4 |
| Dcalc | 1.712 | 1.902 | 1.618 | 1.765 |
| F(000) | 1520 | 1608 | 1584 | 1584 |
| Reflections collected | 18,628 | 21,779 | 12,540 | 11,350 |
| Independent reflections | 6581 | 8529 | 4945 | 4517 |
| Parameters | 370 | 370 | 199 | 199 |
| Final R1, Rw (I > 2s) | 0.0329, 0.1028 | 0.0224, 0.0802 | 0.0262, 0.0415 | 0.0173, 0.0424 |
| Final R1, Rw (all data) | 0.0601, 0.1338 | 0.0299, 0.0935 | 0.0374, 0.0426 | 0.0186, 0.0429 |
| Goodness-of-fit | 0.813 | 0.702 | 1.776 | 1.091 |
| 1 (296 K) | 1 (100 K) | 2 (250 K) | 2 (85 K) |
| Fe(1)–N(1): 2.162(3) | Fe(1)–N(1): 1.941(3) | Fe(1)–N(1): 2.218(2) | Fe(1)–N(1): 1.997(3) |
| Fe(1)–N(2): 2.129(3) | Fe(1)–N(2): 1.940(3) | Fe(1)–N(2): 2.237(2) | Fe(1)–N(2): 2.009(3) |
| Fe(1)–N(3): 2.144(2) | Fe(1)–N(3): 1.932(3) | Fe(1)–N(3): 2.148(2) | Fe(1)–N(3): 1.938(3) |
| Fe(1)–N(4): 2.136(2) | Fe(1)–N(4): 1.944(3) | Fe(1)–N(4): 2.140(2) | Fe(1)–N(4): 1.941(3) |
| Fe(1)–N(5): 2.234(3) | Fe(1)–N(5): 2.003(3) | Fe(1)–N(5): 2.149(2) | Fe(1)–N(5): 1.939(3) |
| Fe(1)–N(6): 2.231(3) | Fe(1)–N(6): 1.994(3) | Fe(1)–N(6): 2.118(3) | Fe(1)–N(6): 1.937(3) |
| 3 (250 K) | 3 (85 K) | 4 (275 K) | 4 (90 K) |
| Fe(1)–N(1): 2.198(3) | Fe(1)–N(1): 2.017(2) | Fe(1)–N(1): 2.149(1) | Fe(1)–N(1): 2.003(1) |
| Fe(1)–N(2): 2.198(3) | Fe(1)–N(2): 1.995(2) | Fe(1)–N(2): 2.149(1) | Fe(1)–N(2): 1.926(2) |
| Fe(1)–N(3): 2.102(3) | Fe(1)–N(3): 1.934(2) | Fe(1)–N(3): 2.149(2) | Fe(1)–N(3): 1.934(1) |
| Fe(1)–N(4): 2.099(3) | Fe(1)–N(4): 1.932(2) | Fe(1)–N(4): 2.141(2) | Fe(1)–N(4): 1.914(1) |
| Fe(1)–N(5): 2.101(3) | Fe(1)–N(5): 1.929(2) | ||
| Fe(1)–N(6): 2.107(3) | Fe(1)–N(6): 1.934(2) |
| Substituents (X) | Cell Volume (Å3) | SCO Behavior Type | Critical Temperature Tc (K) | Reference |
|---|---|---|---|---|
| 3-F | 1934.1 | gradual (2-step) | 162(1st), 96 (2nd) | [6] |
| 3-Cl | 1959.7 | steep (1-step) | 106 | [6] |
| 3-Br | 1959.4 | none | None | [6] |
| 3-CN-4-Me (1) | 4547.0 * | gradual (1-step) | 182 | - |
| Allyl-Isonic (2) | 2677.0 | steep (1-step) | 221 | - |
| Ph-Isonic (3) | 3004.2 | gradual (1-step) | 227 | - |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosone, T.; Makido, Y.; Okuda, S.; Haigo, A.; Kawasaki, T.; Akahoshi, D.; Saito, T.; Kitazawa, T. Systematic Design of Crystal Structure for Hofmann-Like Spin Crossover Fe(L)2[Ag(CN)2]2 Complexes. Crystals 2019, 9, 370. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9070370
Kosone T, Makido Y, Okuda S, Haigo A, Kawasaki T, Akahoshi D, Saito T, Kitazawa T. Systematic Design of Crystal Structure for Hofmann-Like Spin Crossover Fe(L)2[Ag(CN)2]2 Complexes. Crystals. 2019; 9(7):370. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9070370
Chicago/Turabian StyleKosone, Takashi, Yoshinori Makido, Syogo Okuda, Ayaka Haigo, Takeshi Kawasaki, Daisuke Akahoshi, Toshiaki Saito, and Takafumi Kitazawa. 2019. "Systematic Design of Crystal Structure for Hofmann-Like Spin Crossover Fe(L)2[Ag(CN)2]2 Complexes" Crystals 9, no. 7: 370. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9070370