Crystallization and Crystallographic Analysis of a Bradyrhizobium Elkanii USDA94 Haloalkane Dehalogenase Variant with an Eliminated Halide-Binding Site

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Gene Synthesis, Cloning, Expression and Protein Purification
2.2. Crystallization
2.3. Data Collection, Processing and Structure Solution
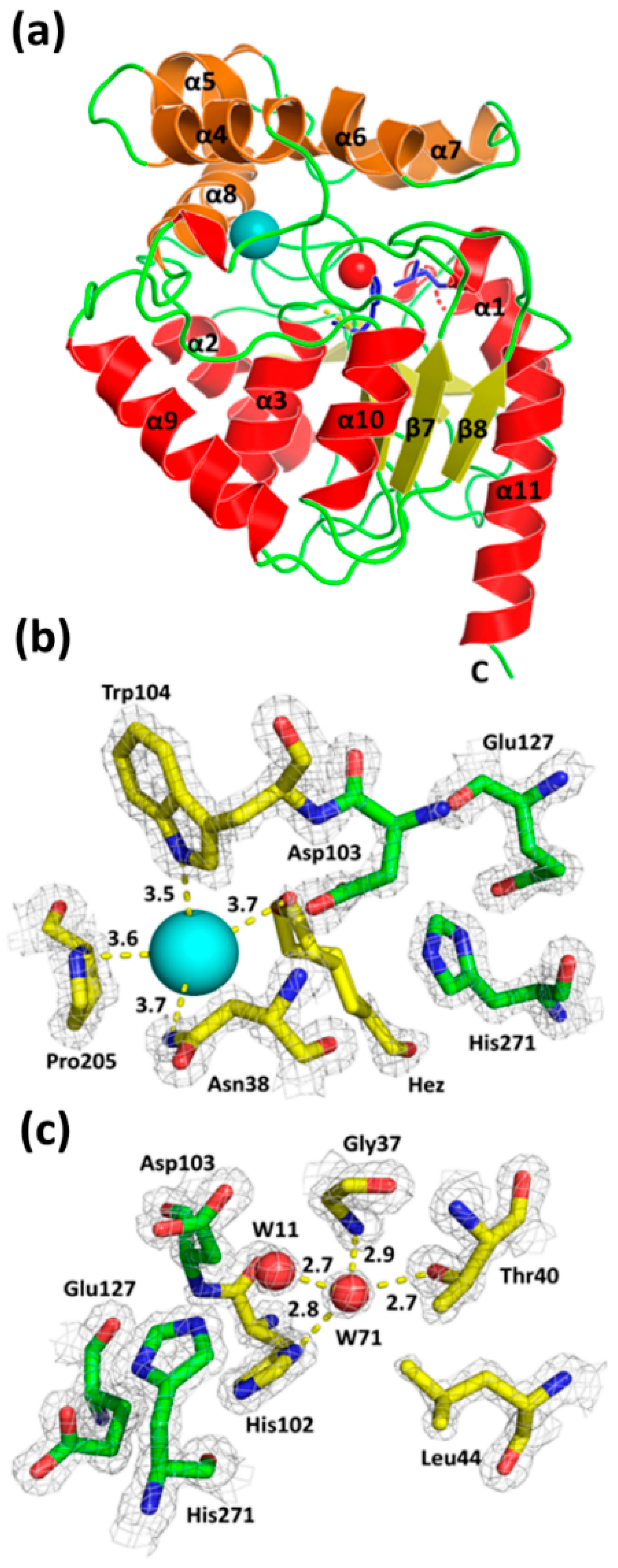
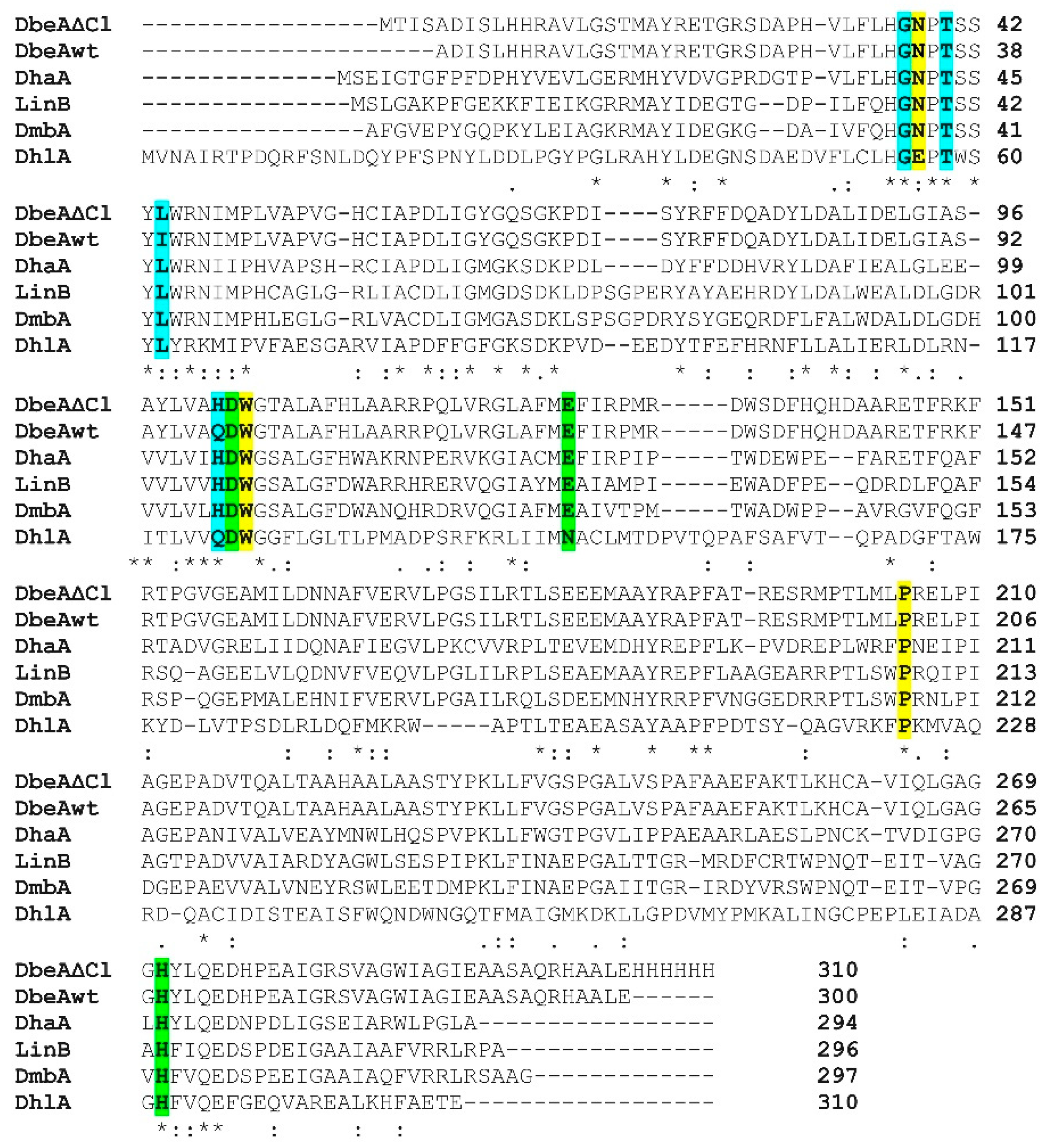
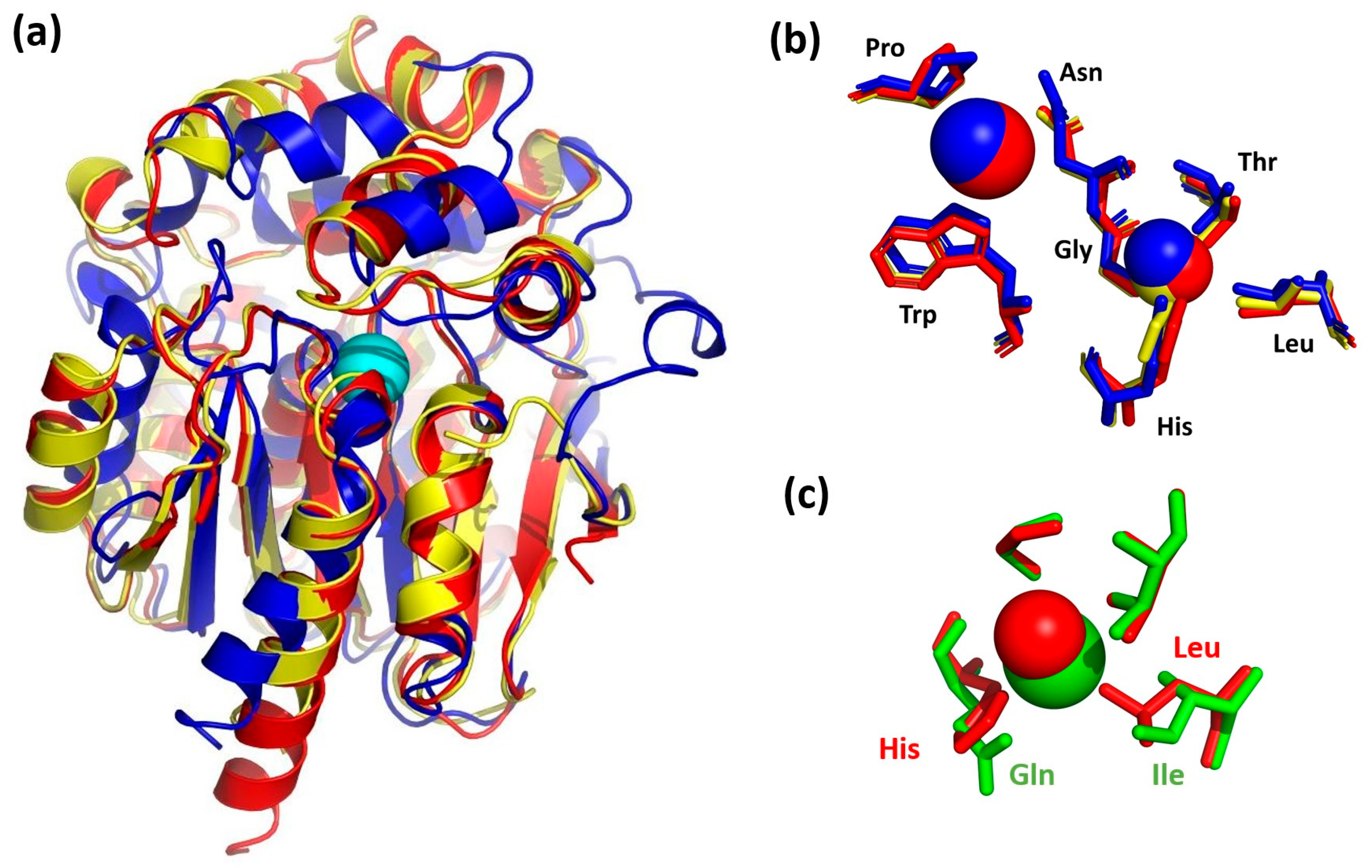
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Newman, J.; Peat, T.S.; Richard, R.; Kan, L.; Swanson, P.E.; Affholter, J.A.; Holmes, I.H.; Schindler, J.F.; Unkefer, C.J.; Terwilliger, T.C. Haloalkane dehalogenases: Structure of a Rhodococcus enzyme. Biochemistry 1999, 38, 16105–16114. [Google Scholar] [CrossRef] [PubMed]
- Janssen, D.B.; Dinkla, I.J.T.; Poelarends, G.J.; Terpstra, P. Bacterial degradation of xenobiotic compounds: Evolution and distribution of novel enzyme activities. Environ. Microbiol. 2005, 7, 1868–1882. [Google Scholar] [CrossRef] [PubMed]
- Prokop, Z.; Sato, Y.; Brezovsky, J.; Mozga, T.; Chaloupkova, R.; Koudelakova, T.; Jerabek, P.; Stepankova, V.; Natsume, R.; van Leeuwen, J.G.; et al. Enantioselectivity of haloalkane dehalogenases and its modulation by surface loop engineering. Angew. Chem. Int. Ed. Engl. 2010, 49, 6111–6115. [Google Scholar] [CrossRef] [PubMed]
- Chovancova, E.; Kosinski, J.; Bujnicki, J.M.; Damborsky, J. Phylogenetic analysis of haloalkane dehalogenases. Proteins 2007, 62, 305–306. [Google Scholar] [CrossRef] [PubMed]
- Holmquist, M. Alpha/beta-hydrolase fold enzymes: Structures, functions and mechanisms. Curr. Protein Pept. Sci. 2000, 1, 209–235. [Google Scholar] [CrossRef] [PubMed]
- Prokop, Z.; Oplustil, F.; DeFrank, J.; Damborsky, J. Enzymes fight chemical weapons. Biotechnol. J. 2006, 1, 1370–1380. [Google Scholar] [CrossRef]
- Chaloupkova, R.; Prudnikova, T.; Rezacova, P.; Prokop, Z.; Koudelakova, T.; Daniel, L.; Brezovsky, J.; Ikeda-Ohtsubo, W.; Sato, Y.; Kuty, M.; et al. Structural and functional analysis of a novel haloalkane dehalogenase with two halide-binding sites. Acta Cryst. 2014, 70, 1884–1897. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Ducruix, A.; Giegé, R. Crystallization of Nucleic Acids and Proteins; Oxford University Press: Oxford, UK, 1999. [Google Scholar] [CrossRef]
- Chayen, N.E.J. Crystallization with oils: A new dimension in macromolecular crystal growth. J. Cryst. Growth 1999, 196, 434–441. [Google Scholar] [CrossRef]
- Bergfors, T.M. Protein Crystallization: Techniques, Strategies and Tips; International University Line: La Jolla, CA, USA, 1999. [Google Scholar] [CrossRef]
- Gavira, J.A.; Jesus, W.; Camara-Artigas, A.; Lopez-Garriga, J.; Garcia-Ruiz, J.M. Crystallization and diffraction patterns of the oxy and cyano forms of the Lucina pectinata haemoglobins complex. Acta Cryst. 2006, 62, 196–199. [Google Scholar] [CrossRef]
- Gerlach, M.; Mueller, U.; Weiss, M.S. The MX beamlines BL14.1-3 at BESSY II. JLSRF 2016, 2, 1–6. [Google Scholar] [CrossRef]
- Sparta, K.M.; Krug, M.; Heinemann, U.; Mueller, U.; Weiss, M.S. XDSAPP2.0. J. Appl. Cryst. 2016, 49, 1085–1092. [Google Scholar] [CrossRef]
- Kabsch, W. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Cryst. 1993, 26, 795–800. [Google Scholar] [CrossRef]
- Vagin, A.; Teplyakov, A. MOLREP: An automated program for Molecular Replacement. J. Appl. Cryst. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst. 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Cryst. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Cryst. 2011, 67, 235–242. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Cryst. 2010, 66, 12–21. [Google Scholar] [CrossRef]
- Hintze, B.J.; Lewis, S.M.; Richardson, J.S.; Richardson, D.C. Molprobity’s ultimate rotamer-library distributions for model validation. Proteins 2016, 84, 1177–1189. [Google Scholar] [CrossRef]
- Gore, S.; Velankar, S.; Kleywegt, G.J. Implementing an X-ray validation pipeline for the Protein Data Bank. Acta Cryst. 2012, 68, 478–483. [Google Scholar] [CrossRef] [Green Version]
- Schrodinger, L.L.C. The PyMOL Molecular Graphics System, Version 2.0. 2019. Available online: https://pymol.org/2/ (accessed on 23 July 2019).
- Prudnikova, T.; Mozga, T.; Rezacova, P.; Chaloupkova, R.; Sato, Y.; Nagata, Y.; Brynda, J.; Kuty, M.; Damborsky, J.; Kuta-Smatanova, I. Crystallization and Preliminary X-ray Analysis of a Novel Haloalkane Dehalogenase DbeA from Bradyrhizobium elkani USDA94. Acta Cryst. 2009, 65, 353–356. [Google Scholar] [CrossRef]
- Shaw Stewart, P.D.; Kolek, S.A.; Briggs, R.A.; Chayen, N.E.; Baldock, P.F.M. Random Microseeding: A Theoretical and Practical Exploration of Seed Stability and Seeding Techniques for Successful Protein Crystallization. Cryst. Growth Des. 2011, 11, 3432–3441. [Google Scholar] [CrossRef]
- Ollis, D.L.; Cheah, E.; Cygler, M.; Dijkstra, B.; Frolow, F.; Franken, S.M.; Harel, M.; Remington, S.J.; Silman, I.; Schrag, J.; et al. The alpha/beta hydrolase fold. Protein Eng. 1992, 5, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Pikkemaat, M.G.; Ridder, I.S.; Rozeboom, H.J.; Kalk, K.H.; Dijkstra, B.W.; Janssen, D.B. Crystallographic and kinetic evidence of a collision complex formed during halide import in haloalkane dehalogenase. Biochemistry 1999, 38, 12052–12061. [Google Scholar] [CrossRef] [PubMed]
- Marek, J.; Vevodova, J.; Smatanova, I.K.; Nagata, Y.; Svensson, L.A.; Newman, J.; Takagi, M.; Damborsky, J. Crystal structure of the haloalkane dehalogenase from Sphingomonas paucimobilis UT26. Biochemistry 2000, 39, 14082–14086. [Google Scholar] [CrossRef]
- Mazumdar, P.A.; Hulecki, J.C.; Cherney, M.M.; Garen, C.R.; James, M.N.G. X-ray crystal structure of Mycobacterium tuberculosis haloalkane dehalogenase Rv2579. Biochim. Biophys. Acta. 2008, 1784, 351–362. [Google Scholar] [CrossRef]
- Koudelakova, T.; Bidmanova, S.; Dvorak, P.; Pavelka, A.; Chaloupkova, R.; Prokop, Z.; Damborsky, J. Haloalkane dehalogenases: Biotechnological applications. Biotechnol. J. 2013, 8, 32–45. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source Organism | Bradyrhizobium Elkanii USDA94 |
|---|---|
| DNA source | Artificially synthesized DNA |
| Transport vector | pMA |
| Expression vector | pET-21b |
| Expression host | E. coli BL21(DE3) |
| Complete amino acid sequence of DbeAΔCl | MTISADISLHHRAVLGSTMAYRETGRSDAPHVLFLHGNPTSSYL WRNIMPLVAPVGHCIAPDLIGYGQSGKPDISYRFFDQADY LDALIDELGIASAYLVAHDWGTALAFHLAARRPQLVRGLA FMEFIRPMRDWSDFHQHDAARETFRKFRTPGVGEAMILDN NAFVERVLPGSILRTLSEEEMAAYRAPFATRESRMPTLML PRELPIAGEPADVTQALTAAHAALAASTYPKLLFVGSPGA LVSPAFAAEFAKTLKHCAVIQLGAGGHYLQEDHPEAIGRS VAGWIAGIEAASAQRHAALEHHHHHH |
| X-ray Diffraction Data Collection Statistics | |
| Space group | C121 |
| Cell parameters (Å, °) | a = 128.95, b = 63.95, c = 46.05; α = γ = 90, β = 106.27 |
| Wavelength (Å) | 0.918 |
| Resolution (Å) | 1.39 |
| Number of unique reflections | 68,322 |
| Redundancy | 2.18 (2.20) |
| Completeness (%) | 96.13 (92.17) |
| Rmerge # | 4.9 (26.3) |
| Average I/σ(I) | 12.72 (3.19) |
| Wilson B (Å2) | 20.8 |
| Refinement Statistics | |
| Resolution range (Å) | 41.96–1.4 (1.43–1.39) |
| No. of reflections in working set | 64,907 (4,609) |
| R value (%) ## | 13.98 |
| Rfree value (%) ### | 15.22 |
| RMSD bond length (Å) | 0.006 |
| RMSD angle (°) | 1.635 |
| No. of atoms in AU | 2,823 |
| No. of protein atoms in AU | 2,333 |
| No. of water molecules in AU | 470 |
| No. of iodide ions in AU | 8 |
| No. of chloride ions in AU | 3 |
| Mean B value (Å2) | 13.42 |
| Ramachandran Plot Statistics | |
| - Residues in favoured regions (%) | 97.2 |
| - Residues in allowed regions (%) | 100 |
| PDB code | 6s42 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prudnikova, T.; Kascakova, B.; Mesters, J.R.; Grinkevich, P.; Havlickova, P.; Mazur, A.; Shaposhnikova, A.; Chaloupkova, R.; Damborsky, J.; Kuty, M.; et al. Crystallization and Crystallographic Analysis of a Bradyrhizobium Elkanii USDA94 Haloalkane Dehalogenase Variant with an Eliminated Halide-Binding Site. Crystals 2019, 9, 375. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9070375
Prudnikova T, Kascakova B, Mesters JR, Grinkevich P, Havlickova P, Mazur A, Shaposhnikova A, Chaloupkova R, Damborsky J, Kuty M, et al. Crystallization and Crystallographic Analysis of a Bradyrhizobium Elkanii USDA94 Haloalkane Dehalogenase Variant with an Eliminated Halide-Binding Site. Crystals. 2019; 9(7):375. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9070375
Chicago/Turabian StylePrudnikova, Tatyana, Barbora Kascakova, Jeroen R. Mesters, Pavel Grinkevich, Petra Havlickova, Andrii Mazur, Anastasiia Shaposhnikova, Radka Chaloupkova, Jiri Damborsky, Michal Kuty, and et al. 2019. "Crystallization and Crystallographic Analysis of a Bradyrhizobium Elkanii USDA94 Haloalkane Dehalogenase Variant with an Eliminated Halide-Binding Site" Crystals 9, no. 7: 375. https://0-doi-org.brum.beds.ac.uk/10.3390/cryst9070375