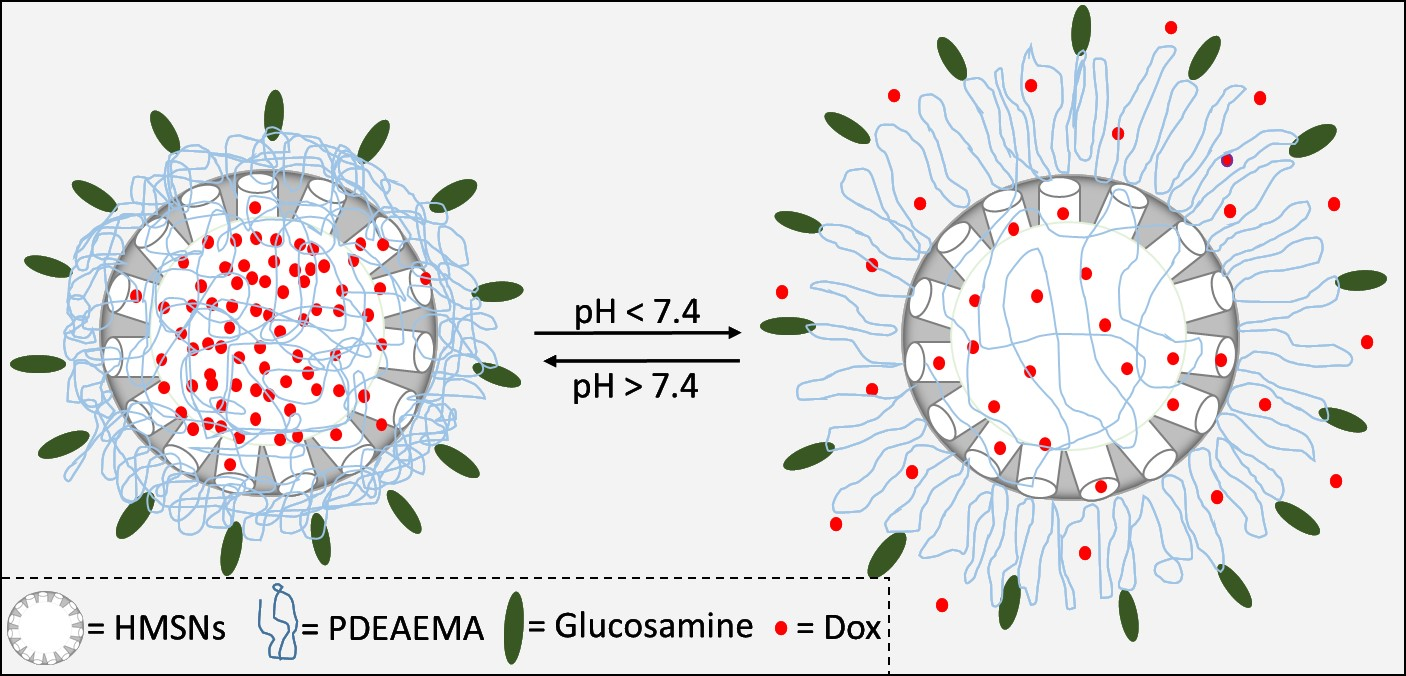
Glucosamine Modified the Surface of pH-Responsive Poly(2-(diethylamino)ethyl Methacrylate) Brushes Grafted on Hollow Mesoporous Silica Nanoparticles as Smart Nanocarrier

 , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
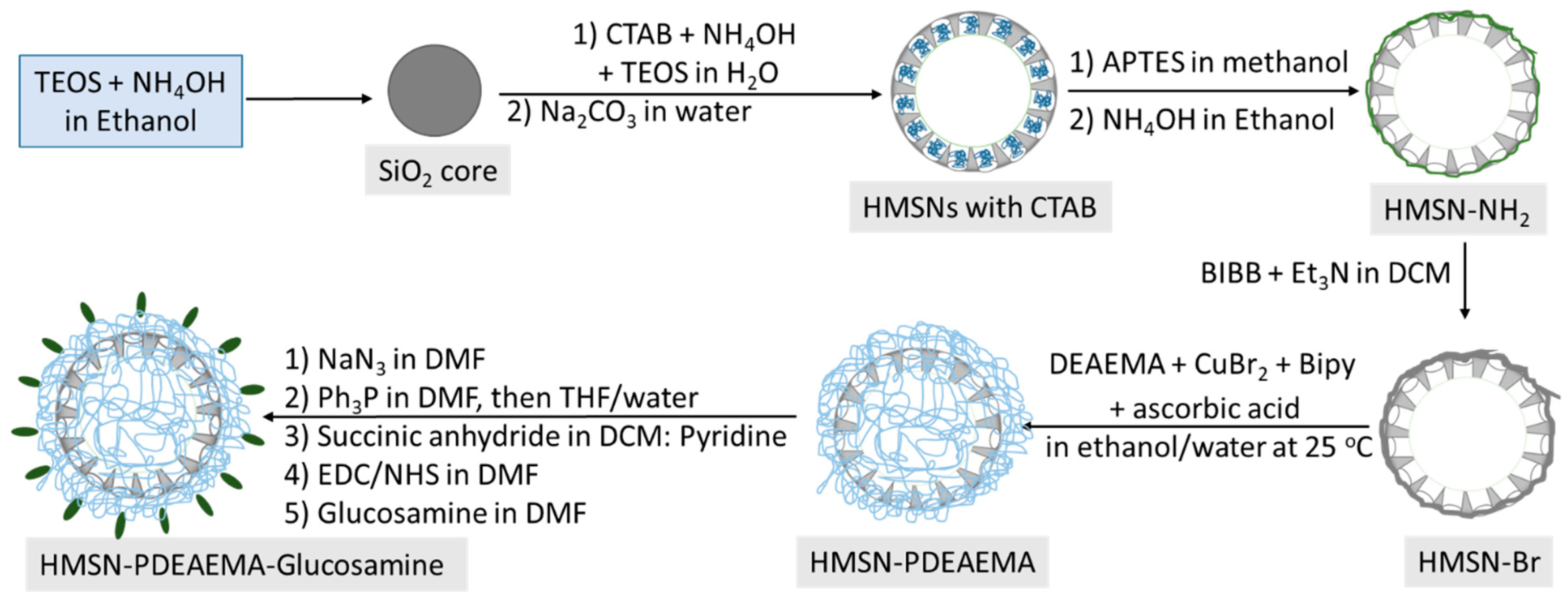
2.2.1. Synthesis of Uniform HMSNs
2.2.2. Synthesis of HMSNs Modified with Amine Groups (HMSN-NH2)
2.2.3. Formation of Channels of HMSNs
2.2.4. Immobilization of BIBB Initiator on HMSNs (HMSN-Br)
2.2.5. Synthesis of Poly(2-(diethylamino)ethyl Methacrylate) Brushes-Grafted HMSNs (HMSN-PDEAEMA)
2.2.6. Surface Coating of HMSNs with Amine Groups (HMSN-PDEAEMA-NH2)
2.2.7. Immobilization of Glucosamine on the Surface of HMSNs (HMSN-PDEAEMA-Glucosamine)
2.3. Measurement and Characterization
2.4. Drug Loading and Release
2.5. Cytotoxicity Assay
2.6. Data Analysis
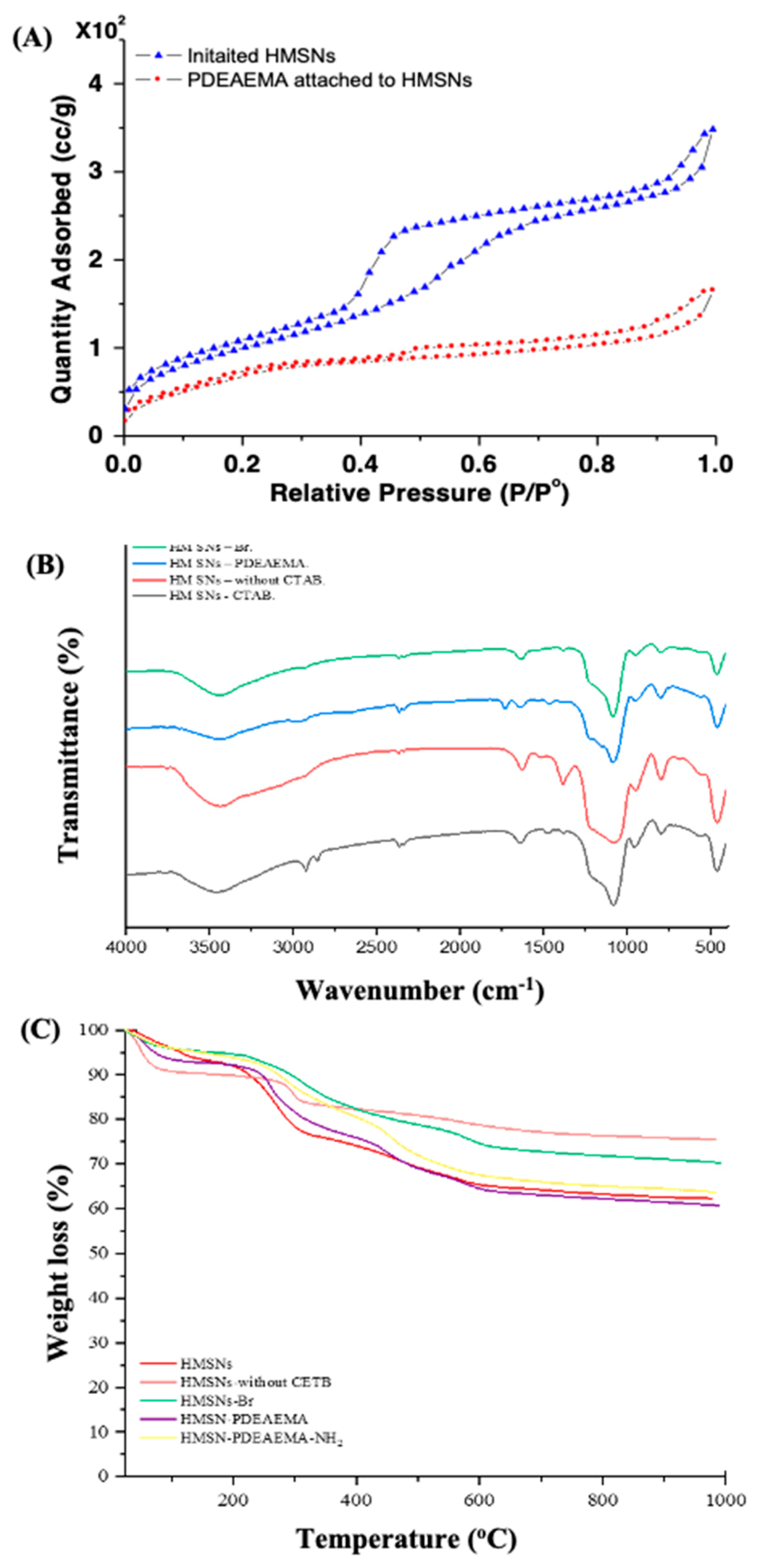
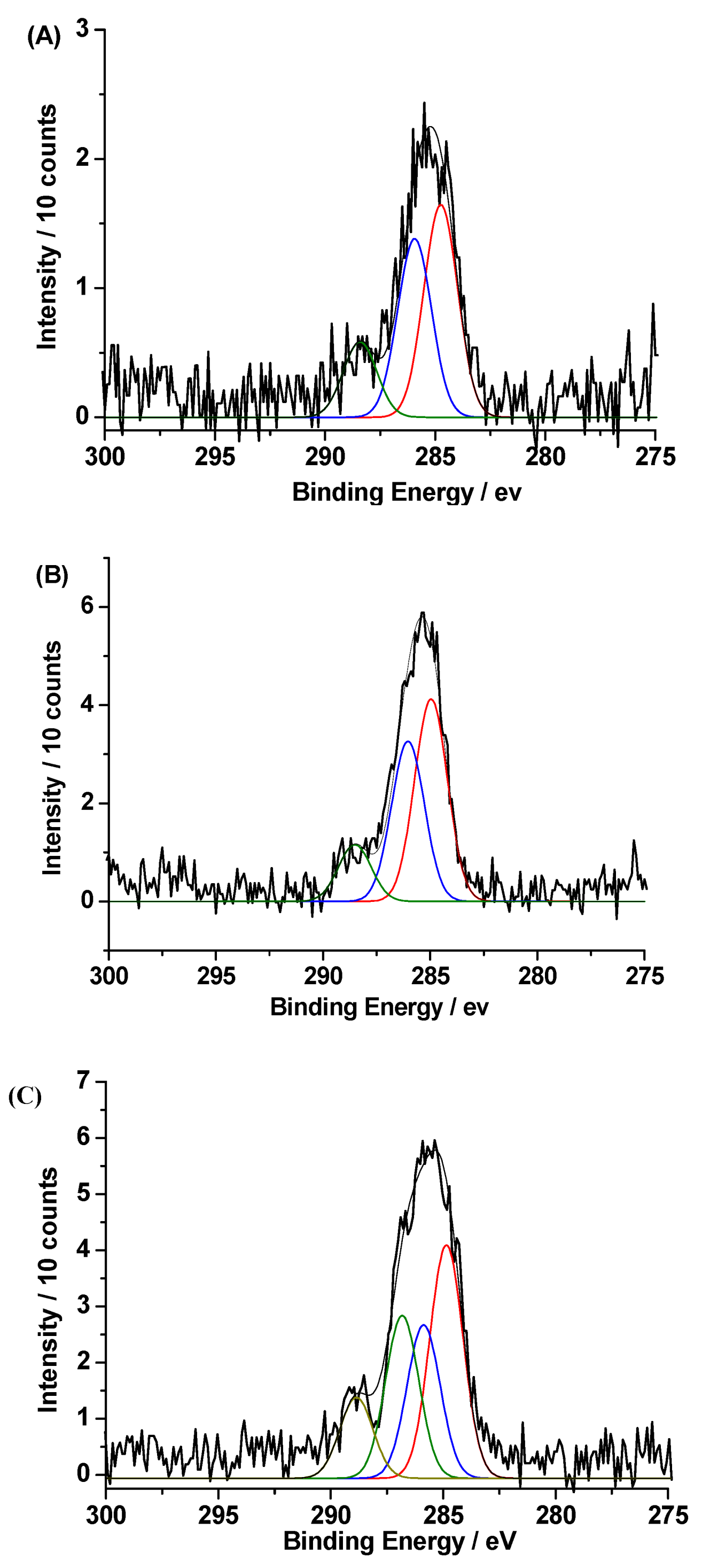
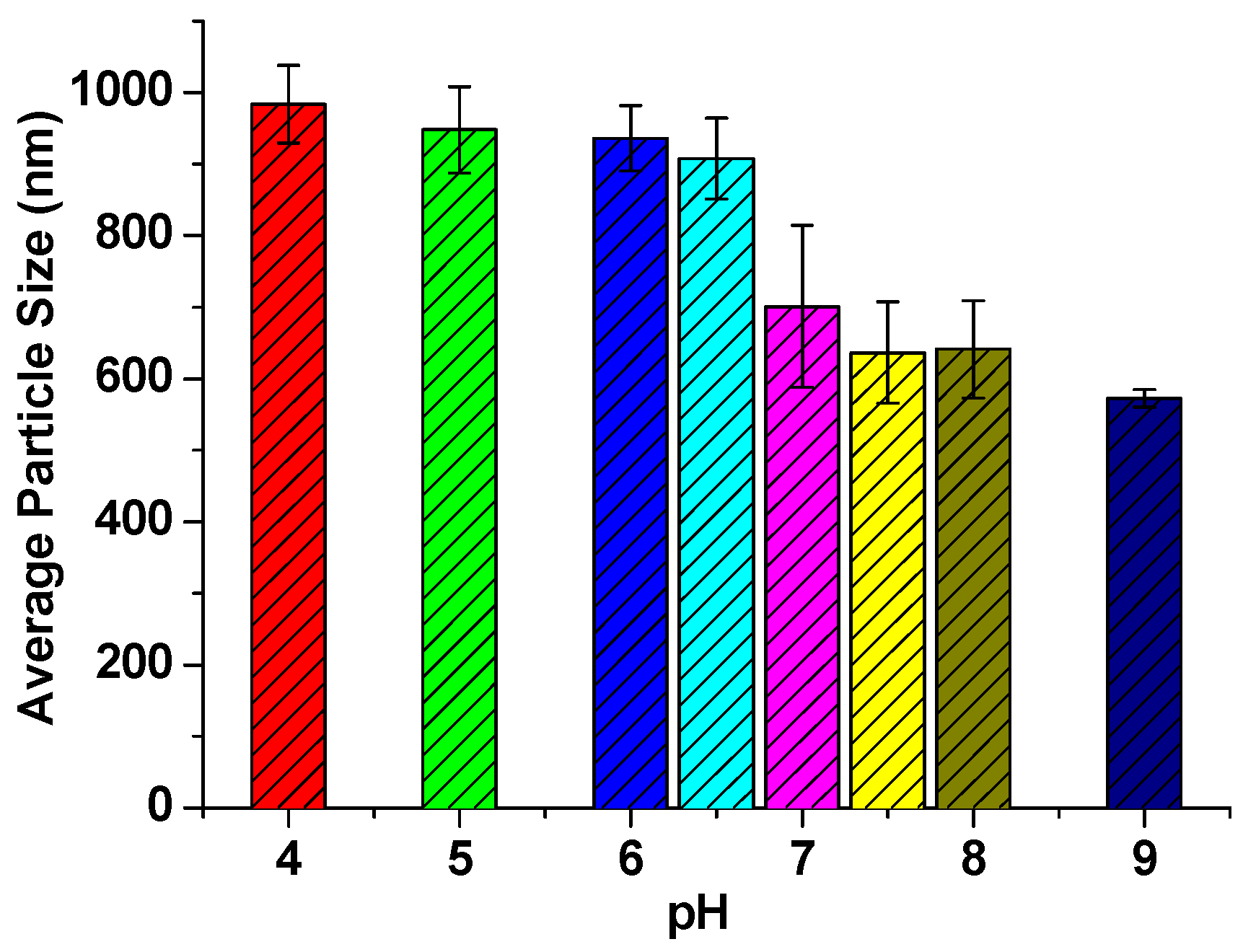
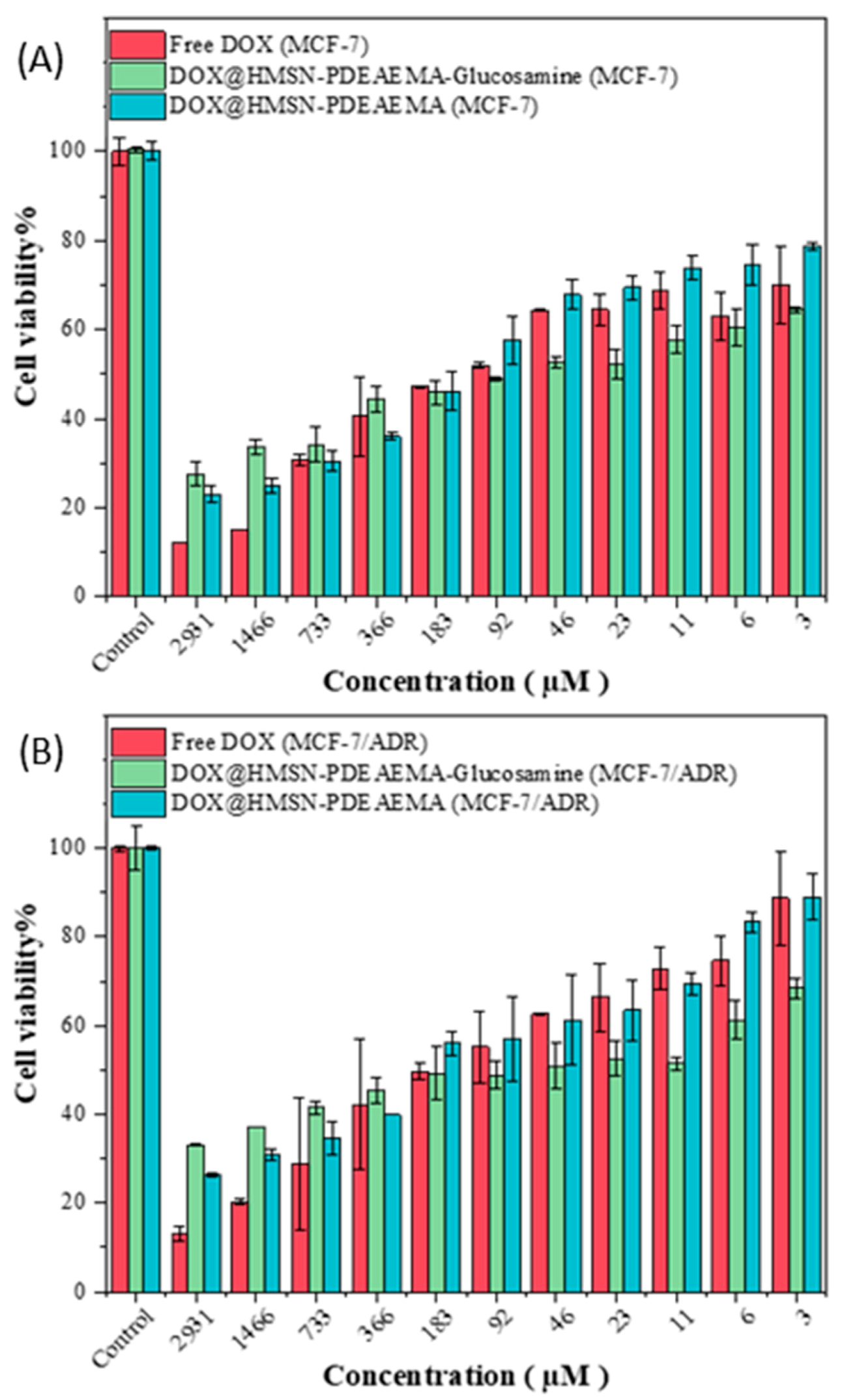
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Wen, H.; Guo, J.; Chang, B.; Yang, W. pH-responsive composite microspheres based on magnetic mesoporous silica nanoparticle for drug delivery. Eur. J. Pharm. Biopharm. 2013, 84, 91–98. [Google Scholar] [CrossRef] [PubMed]
- De Leo, V.; Milano, F.; Mancini, E.; Comparelli, R.; Giotta, L.; Nacci, A.; Longobardi, F.; Garbetta, A.; Agostiano, A.; Catucci, L. Encapsulation of Curcumin-Loaded Liposomes for Colonic Drug Delivery in a pH-Responsive Polymer Cluster Using a pH-Driven and Organic Solvent-Free Process. Molecules 2018, 23, 739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Wang, L.; Li, X.; Hu, X.; Han, Y.; Luo, Y.; Wang, Z.; Li, Q.; Aldalbahi, A.; Wang, L.; et al. Size-Dependent Regulation of Intracellular Trafficking of Polystyrene Nanoparticle-Based Drug-Delivery Systems. ACS Appl. Mater. Interfaces 2017, 9, 18619–18625. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lytton-Jean, A.K.; Chen, Y.; Love, K.T.; Park, A.I.; Karagiannis, E.D.; Sehgal, A.; Querbes, W.; Zurenko, C.S.; Jayaraman, M. Molecularly self-assembled nucleic acid nanoparticles for targeted in vivo siRNA delivery. Nat. Nanotechnol. 2012, 7, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wu, S.; Du, X. Gated mesoporous carbon nanoparticles as drug delivery system for stimuli-responsive controlled release. Carbon 2016, 101, 135–142. [Google Scholar] [CrossRef]
- Gao, Y.; Chen, Y.; Ji, X.; He, X.; Yin, Q.; Zhang, Z.; Chen, Y.; Li, Y.-P. Controlled Intracellular Release of Doxorubicin in Multidrug-Resistant Cancer Cells by Tuning the Shell-Pore Sizes of Mesoporous Silica Nanoparticles. ACS Nano 2011, 5, 9788–9798. [Google Scholar] [CrossRef]
- Abdo, G.G.; Zagho, M.M.; Khalil, A. Recent advances in stimuli-responsive drug release and targeting concepts using mesoporous silica nanoparticles. Emergent Mater. 2020, 3, 1–19. [Google Scholar] [CrossRef]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A new family of mesoporous molecular sieves prepared with liquid crystal templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar] [CrossRef]
- Wu, S.-H.; Mou, C.-Y.; Lin, H.-P. Synthesis of mesoporous silica nanoparticles. Chem. Soc. Rev. 2013, 42, 3862–3875. [Google Scholar] [CrossRef]
- Reich, S.-J.; Svidrytski, A.; Höltzel, A.; Florek, J.; Kleitz, F.; Wang, W.; Kübel, C.; Hlushkou, D.; Tallarek, U. Hindered Diffusion in Ordered Mesoporous Silicas: Insights from Pore-Scale Simulations in Physical Reconstructions of SBA-15 and KIT-6 Silica. J. Phys. Chem. C 2018, 122, 12350–12361. [Google Scholar] [CrossRef]
- Mal, N.K.; Fujiwara, M.; Tanaka, Y. Photocontrolled reversible release of guest molecules from coumarin-modified mesoporous silica. Nat. Cell Biol. 2003, 421, 350–353. [Google Scholar] [CrossRef]
- Burkett, S.L.; Sims, S.D.; Mann, S. Synthesis of hybrid inorganic–organic mesoporous silica by co-condensation of siloxane and organosiloxane precursors. Chem. Commun. 1996, 11, 1367–1368. [Google Scholar] [CrossRef]
- De Jong, W.H.; Borm, P.J. Drug delivery and nanoparticles: Applications and hazards. Int. J. Nanomedicine 2008, 3, 133. [Google Scholar] [CrossRef] [Green Version]
- Giret, S.; Wong Chi Man, M.; Carcel, C. Mesoporous Silica Functionalized Nanoparticles for Drug Delivery. Chem. Eur. J. 2015, 21, 13850–13865. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.; Chen, F.; Cai, W. Biomedical applications of functionalized hollow mesoporous silica nanoparticles: Focusing on molecular imaging. Nanomedicine 2013, 8, 2027–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Luo, Z.; Zhang, J.; Luo, T.; Zhou, J.; Zhao, X.; Cai, K. Hollow mesoporous silica nanoparticles facilitated drug delivery via cascade pH stimuli in tumor microenvironment for tumor therapy. Biomaterials 2016, 83, 51–65. [Google Scholar] [CrossRef]
- Zhu, Y.; Shi, J.; Chen, H.; Shen, W.; Dong, X. A facile method to synthesize novel hollow mesoporous silica spheres and advanced storage property. Microporous Mesoporous Mater. 2005, 84, 218–222. [Google Scholar] [CrossRef]
- Geng, H.; Zhao, Y.; Liu, J.; Cui, Y.; Wang, Y.; Zhao, Q.; Wang, S. Hollow mesoporous silica as a high drug loading carrier for regulation insoluble drug release. Int. J. Pharm. 2016, 510, 184–194. [Google Scholar] [CrossRef]
- Torchilin, V.P. Multifunctional nanocarriers. Adv. Drug Deliv. Rev. 2006, 58, 1532–1555. [Google Scholar] [CrossRef]
- Zhang, L.; Bei, H.P.; Piao, Y.; Wang, Y.; Yang, M.; Zhao, X. Polymer-Brush-Grafted Mesoporous Silica Nanoparticles for Triggered Drug Delivery. ChemPhysChem 2018, 19, 1956–1964. [Google Scholar] [CrossRef] [Green Version]
- Feng, C.; Huang, X. Polymer Brushes: Efficient Synthesis and Applications. Accounts Chem. Res. 2018, 51, 2314–2323. [Google Scholar] [CrossRef] [PubMed]
- Ayres, N. Polymer brushes: Applications in biomaterials and nanotechnology. Polym. Chem. 2010, 1, 769–777. [Google Scholar] [CrossRef]
- Conzatti, G.; Cavalie, S.; Combes, C.; Torrisani, J.; Carrere, N.; Tourrette, A. PNIPAM grafted surfaces through ATRP and RAFT polymerization: Chemistry and bioadhesion. Colloids Surf. B Biointerfaces 2017, 151, 143–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peralta, M.E.; Jadhav, S.A.; Magnacca, G.; Scalarone, D.; Mártire, D.O.; Parolo, M.E.; Carlos, L. Synthesis and in vitro testing of thermoresponsive polymer-grafted core-shell magnetic mesoporous silica nanoparticles for efficient controlled and targeted drug delivery. J. Colloid Interface Sci. 2019, 544, 198–205. [Google Scholar] [CrossRef]
- Nebhani, L.; Mishra, S.; Joshi, T. Polymer functionalization of mesoporous silica nanoparticles using controlled radical polymerization techniques. In Microporous and Mesoporous Materials; IntechOpen: London, UK, 2020. [Google Scholar]
- Xu, L.; Li, H.; Wang, L. PH-Sensitive, Polymer Functionalized, Nonporous Silica Nanoparticles for Quercetin Controlled Release. Polymers 2019, 11, 2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourjavadi, A.; Kohestanian, M.; Streb, C. pH and thermal dual-responsive poly(NIPAM-co-GMA)-coated magnetic nanoparticles via surface-initiated RAFT polymerization for controlled drug delivery. Mater. Sci. Eng. C 2020, 108, 110418. [Google Scholar] [CrossRef] [PubMed]
- Kaga, S.; Truong, N.P.; Esser, L.; Senyschyn, D.; Sanyal, A.; Sanyal, R.; Quinn, J.F.; Davis, T.P.; Kaminskas, L.M.; Whittaker, M.R. Influence of Size and Shape on the Biodistribution of Nanoparticles Prepared by Polymerization-Induced Self-Assembly. Biomacromolecules 2017, 18, 3963–3970. [Google Scholar] [CrossRef]
- Khor, S.Y.; Vu, M.N.; Pilkington, E.H.; Johnston, A.P.R.; Whittaker, M.R.; Quinn, J.F.; Truong, N.P.; Davis, T.P. Elucidating the Influences of Size, Surface Chemistry, and Dynamic Flow on Cellular Association of Nanoparticles Made by Polymerization-Induced Self-Assembly. Small 2018, 14, e1801702. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, P.; Dai, Y.; Ma, P.; Li, X.; Cheng, Z.; Hou, Z.; Kang, X.; Li, C.; Lin, J. Multifunctional Up-Converting Nanocomposites with Smart Polymer Brushes Gated Mesopores for Cell Imaging and Thermo/pH Dual-Responsive Drug Controlled Release. Adv. Funct. Mater. 2013, 23, 4067–4078. [Google Scholar] [CrossRef]
- Lee, N.-K.; Park, S.S.; Ha, C.-S. pH-Sensitive Drug Delivery System Based on Mesoporous Silica Modified with Poly-L-Lysine (PLL) as a Gatekeeper. J. Nanosci. Nanotechnol. 2020, 20, 6925–6934. [Google Scholar] [CrossRef]
- Alswieleh, A.M.; Beagan, A.M.; Alsheheri, B.M.; Alotaibi, K.M.; Alharthi, M.D.; Almeataq, M.S. Hybrid Mesoporous Silica Nanoparticles Grafted with 2-(tert-butylamino)ethyl Methacrylate-b-poly(ethylene Glycol) Methyl Ether Methacrylate Diblock Brushes as Drug Nanocarrier. Molecules 2020, 25, 195. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Li, L.; Zhao, F.; Han, H.; Wang, W.; Tian, Y.; Wang, Y.; Ye, Z.; Guo, X. Hollow silica–polyelectrolyte composite nanoparticles for controlled drug delivery. J. Mater. Sci 2019, 54, 2552–2565. [Google Scholar] [CrossRef]
- Yu, F.; Tang, X.; Pei, M. Facile synthesis of PDMAEMA-coated hollow mesoporous silica nanoparticles and their pH-responsive controlled release. Microporous Mesoporous Mater. 2013, 173, 64–69. [Google Scholar] [CrossRef]
- Zhang, Y.; Ang, C.Y.; Li, M.; Tan, S.Y.; Qu, Q.; Luo, Z.; Zhao, Y. Polymer-Coated Hollow Mesoporous Silica Nanoparticles for Triple-Responsive Drug Delivery. ACS Appl. Mater. Interfaces 2015, 7, 18179–18187. [Google Scholar] [CrossRef]
- Peng, W.; Zhang, Z.P.; Rong, M.; Zhang, M.Q. Core-Shell Structure Design of Hollow Mesoporous Silica Nanospheres Based on Thermo-Sensitive PNIPAM and pH-Responsive Catechol-Fe3+ Complex. Polymers 2019, 11, 1832. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Chen, C.; Liu, Z.; Liu, P.; Zheng, N. A cationic surfactant assisted selective etching strategy to hollow mesoporous silica spheres. Nanoscale 2011, 3, 1632–1639. [Google Scholar] [CrossRef]
- Alswieleh, A.M.; Alshahrani, M.M.; Alzahrani, K.E.; Alghamdi, H.S.; A Niazy, A.; Alsilme, A.S.; Beagan, A.M.; Alsheheri, B.M.; Alghamdi, A.A.; Almeataq, M.S. Surface modification of pH-responsive poly(2-(tert-butylamino)ethyl methacrylate) brushes grafted on mesoporous silica nanoparticles. Des. Monomers Polym. 2019, 22, 226–235. [Google Scholar] [CrossRef] [Green Version]
- Farr, T.D.; Lai, C.-H.; Grünstein, D.; Orts-Gil, G.; Wang, C.-C.; Boehm-Sturm, P.; Seeberger, P.H.; Harms, C. Imaging Early Endothelial Inflammation Following Stroke by Core Shell Silica Superparamagnetic Glyconanoparticles That Target Selectin. Nano Lett. 2014, 14, 2130–2134. [Google Scholar] [CrossRef]
- Bilalis, P.; Tziveleka, L.-A.; Varlas, S.; Iatrou, H. pH-Sensitive nanogates based on poly(l-histidine) for controlled drug release from mesoporous silica nanoparticles. Polym. Chem. 2016, 7, 1475–1485. [Google Scholar] [CrossRef]
- Boncler, M.; Różalski, M.; Krajewska, U.; Podsędek, A.; Watala, C. Comparison of PrestoBlue and MTT assays of cellular viability in the assessment of anti-proliferative effects of plant extracts on human endothelial cells. J. Pharmacol. Toxicol. Methods 2014, 69, 9–16. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beagan, A.; Lahmadi, S.; Alghamdi, A.; Halwani, M.; Almeataq, M.; Alhazaa, A.; Alotaibi, K.; Alswieleh, A. Glucosamine Modified the Surface of pH-Responsive Poly(2-(diethylamino)ethyl Methacrylate) Brushes Grafted on Hollow Mesoporous Silica Nanoparticles as Smart Nanocarrier. Polymers 2020, 12, 2749. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12112749
Beagan A, Lahmadi S, Alghamdi A, Halwani M, Almeataq M, Alhazaa A, Alotaibi K, Alswieleh A. Glucosamine Modified the Surface of pH-Responsive Poly(2-(diethylamino)ethyl Methacrylate) Brushes Grafted on Hollow Mesoporous Silica Nanoparticles as Smart Nanocarrier. Polymers. 2020; 12(11):2749. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12112749
Chicago/Turabian StyleBeagan, Abeer, Shatha Lahmadi, Ahlam Alghamdi, Majed Halwani, Mohammed Almeataq, Abdulaziz Alhazaa, Khalid Alotaibi, and Abdullah Alswieleh. 2020. "Glucosamine Modified the Surface of pH-Responsive Poly(2-(diethylamino)ethyl Methacrylate) Brushes Grafted on Hollow Mesoporous Silica Nanoparticles as Smart Nanocarrier" Polymers 12, no. 11: 2749. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12112749