Development of Coarse-Grained Models for Poly(4-vinylphenol) and Poly(2-vinylpyridine): Polymer Chemistries with Hydrogen Bonding


Abstract
:1. Introduction
2. Approach
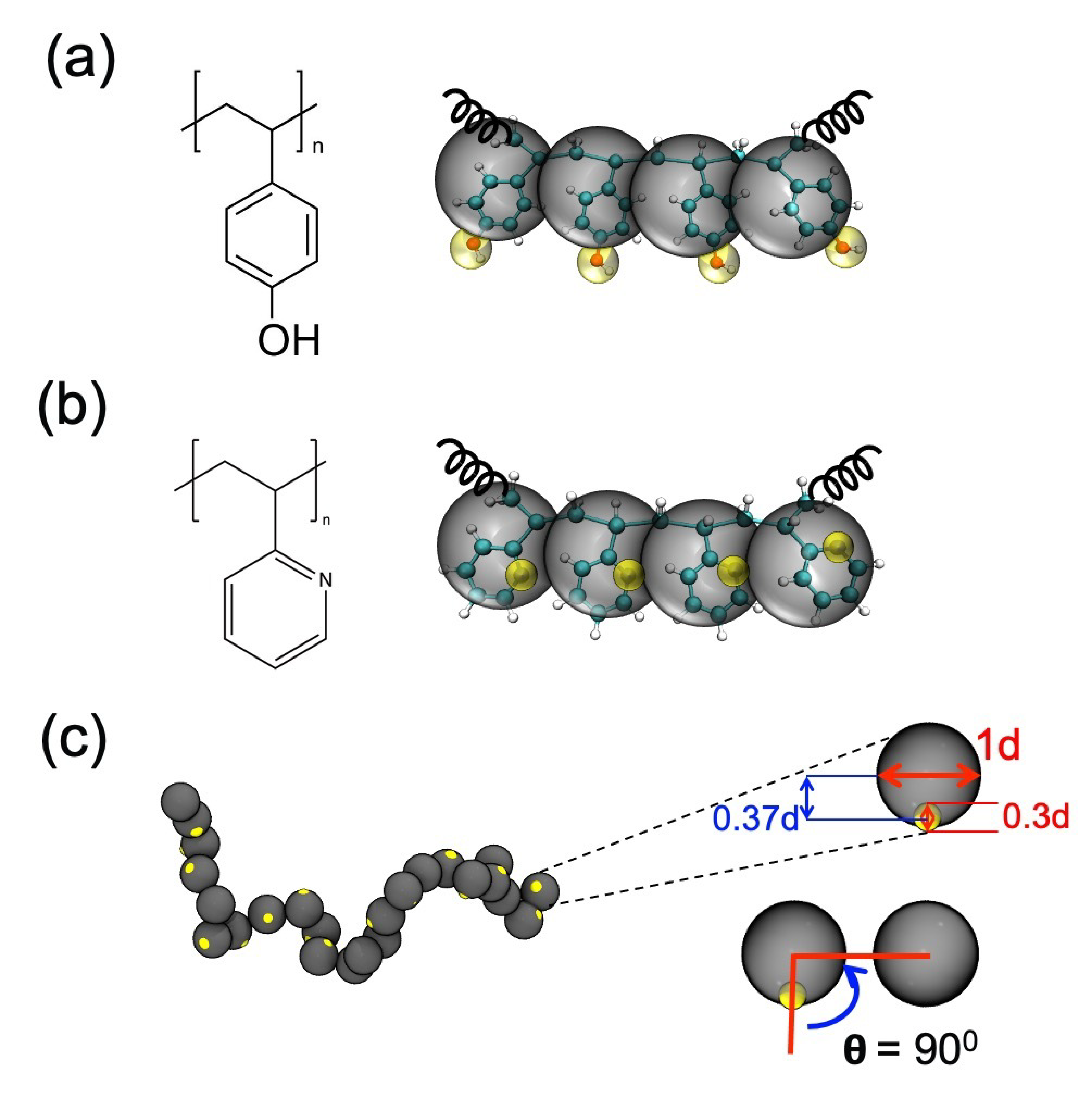
2.1. Atomistic Molecular Dynamics Simulation
2.2. Coarse-Grained (CG) Model
2.3. CG MD Simulation Details
2.4. Analyses
3. Results and Discussion
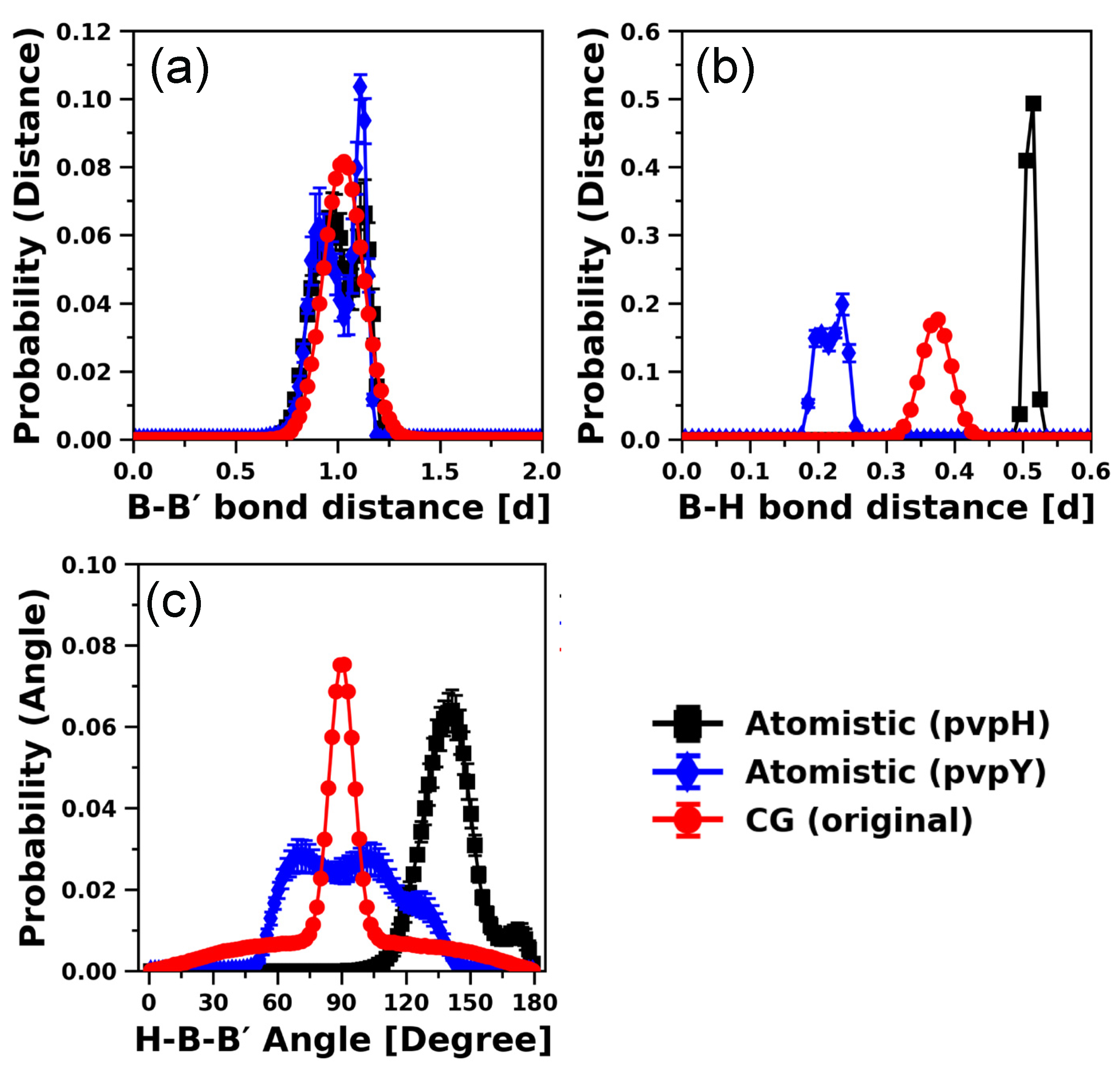
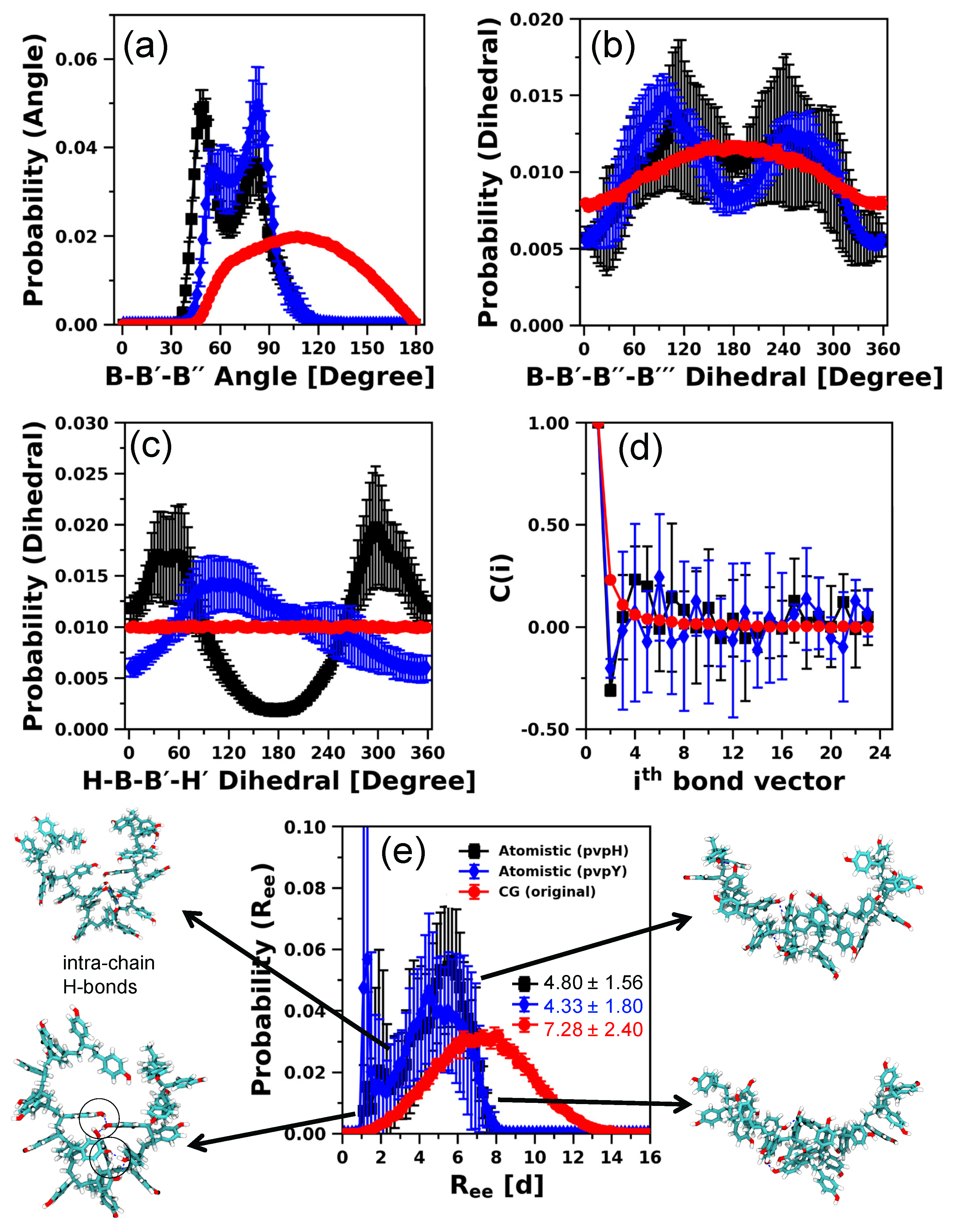
3.1. Development of CG Model Using 24-mer Chains
3.1.1. 24-mer pvpH Chain
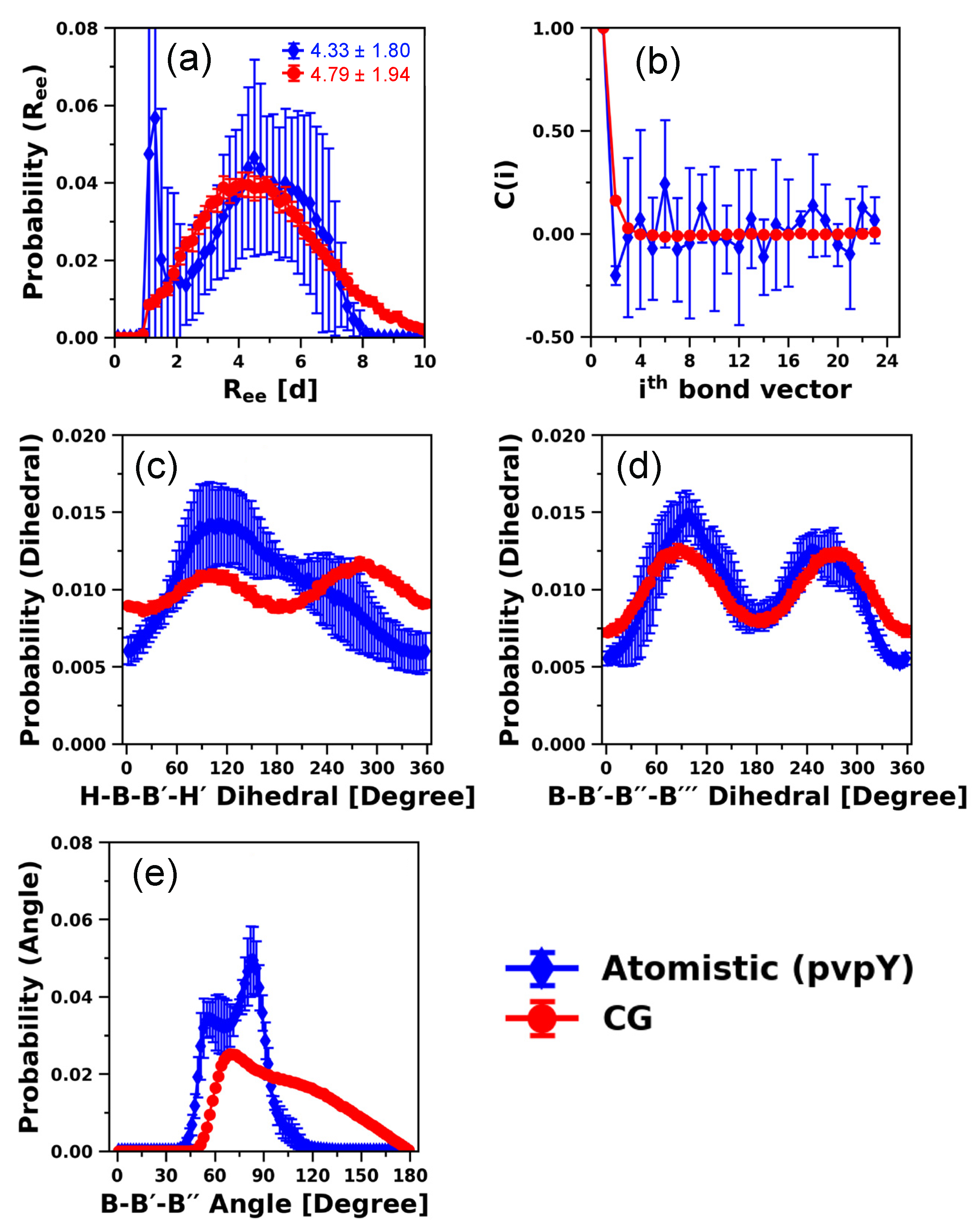
3.1.2. 24-mer pvpY Chain
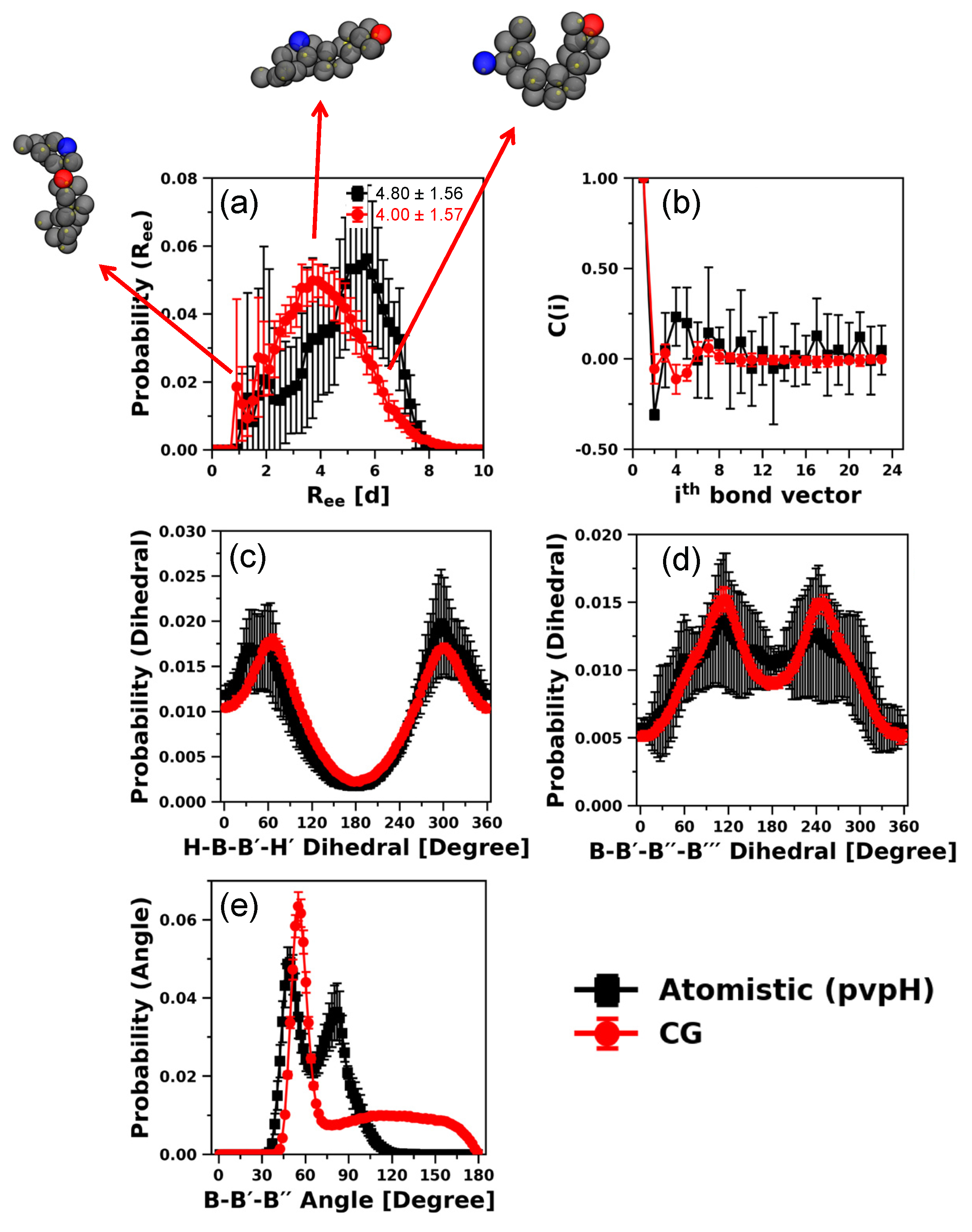
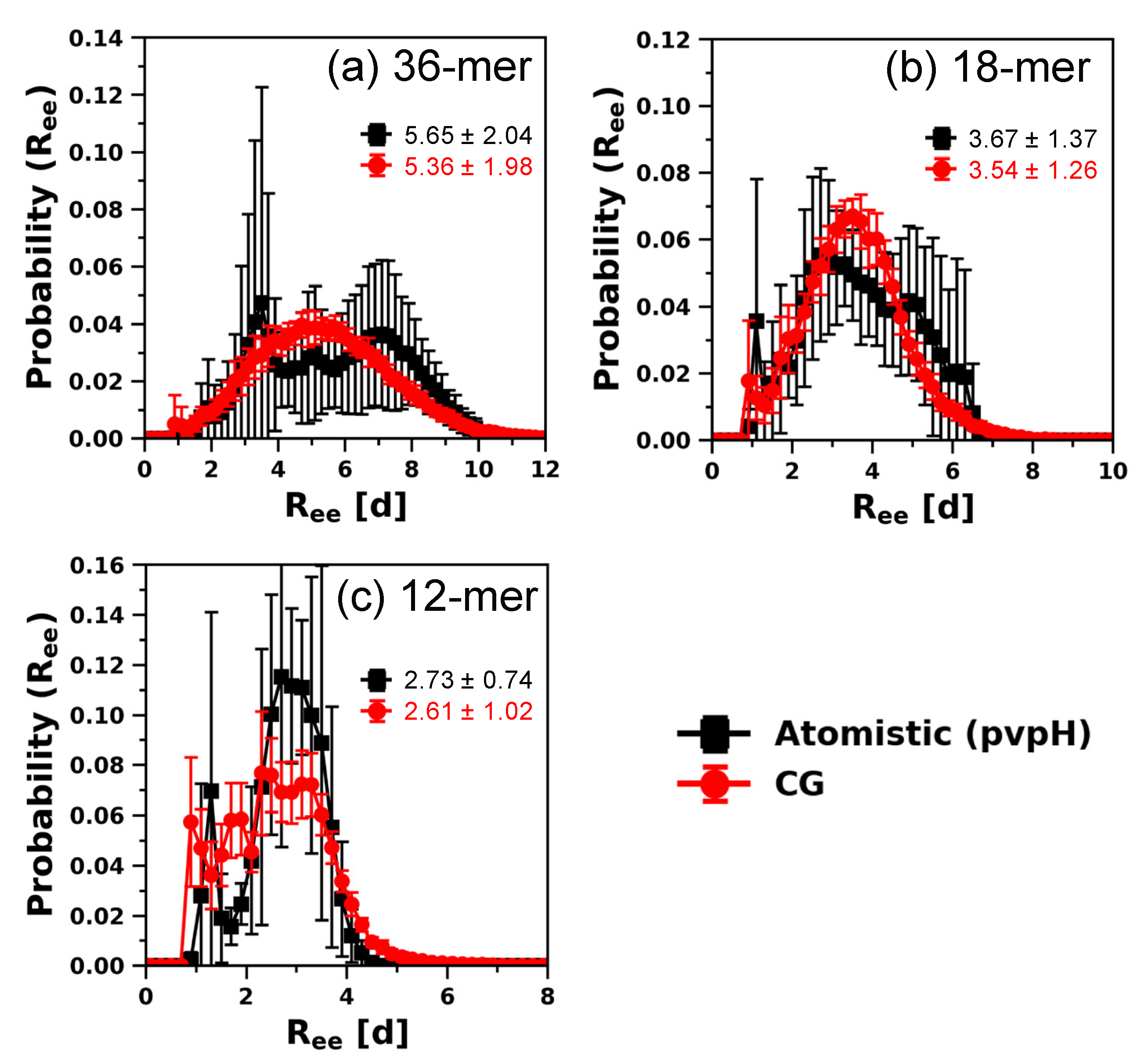
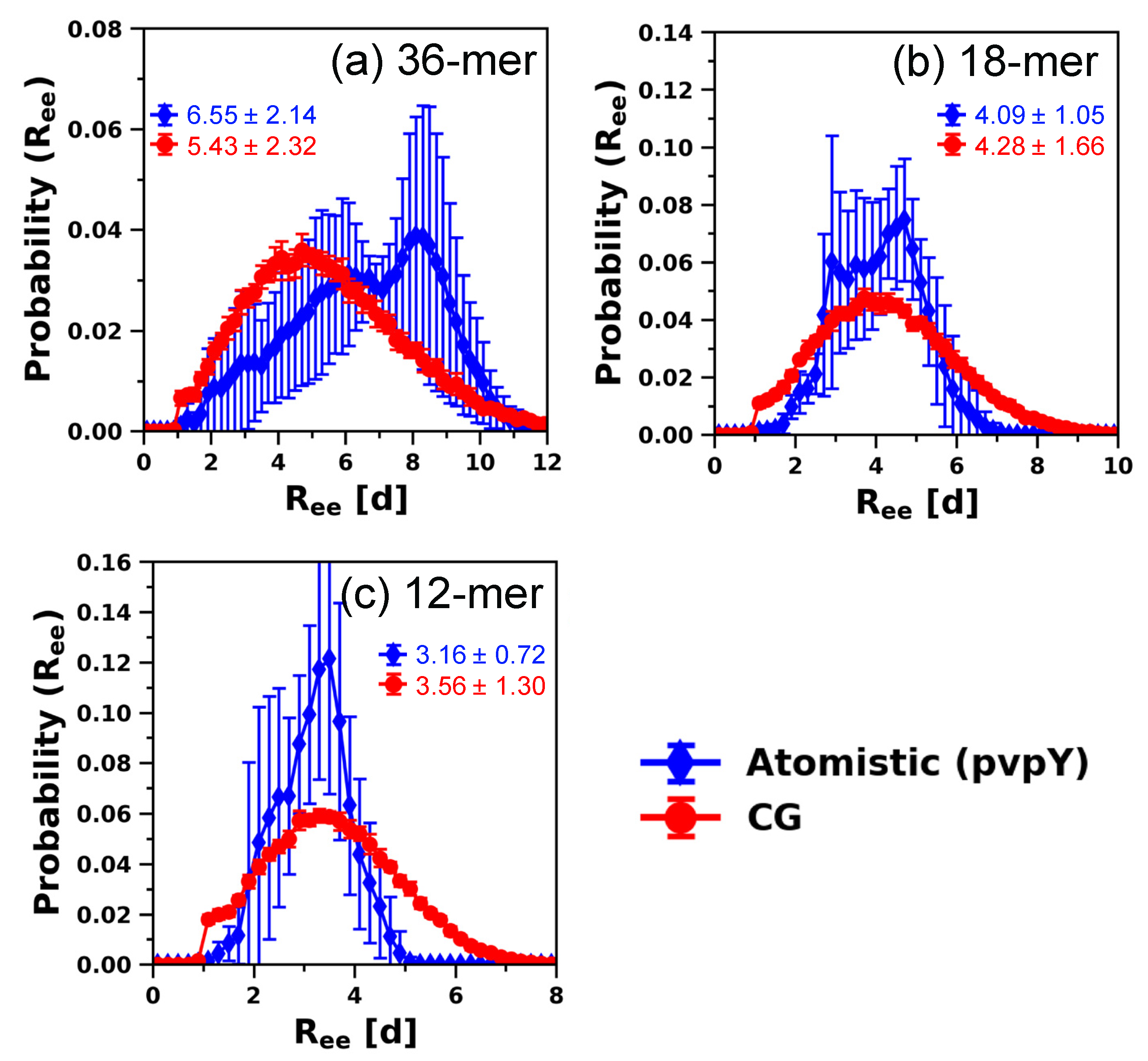
3.2. Testing the Transferability of the CG Model for Describing Structural Properties at Other Chain Lengths
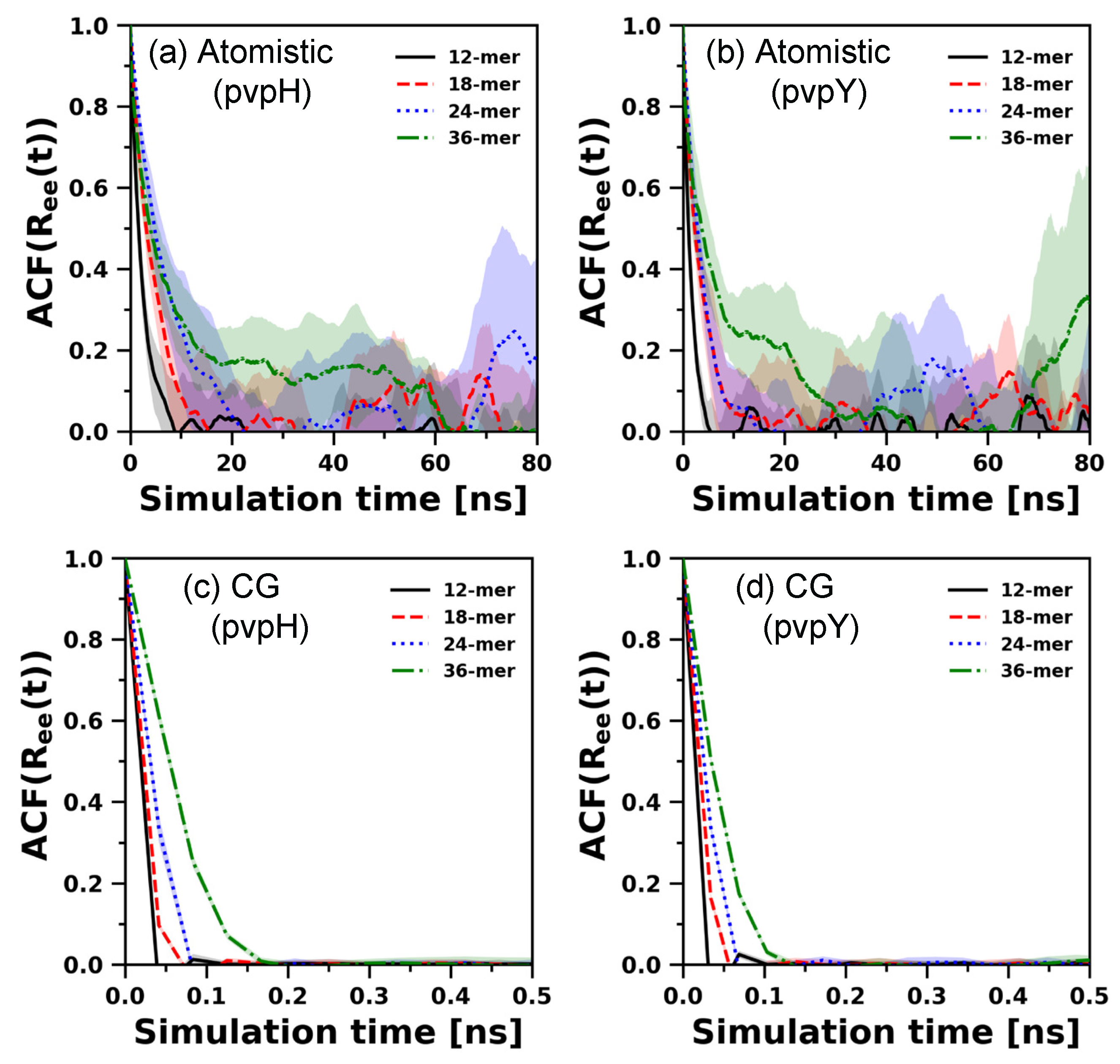
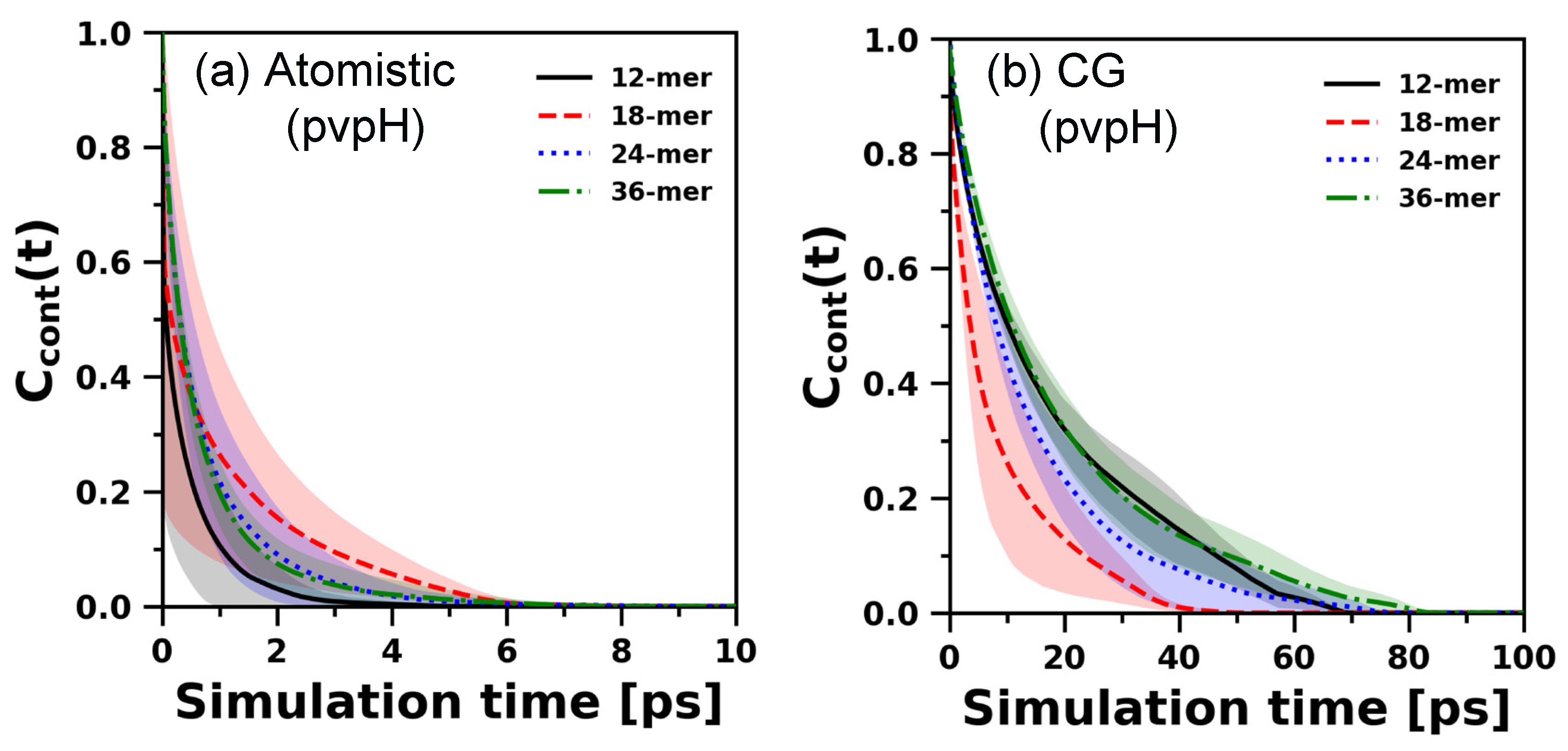
3.3. Chain Relaxation and Hydrogen Bonding Dynamics: CG Model versus Atomistic Model
3.4. Computational Performance
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Flory, P.J. Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, USA, 1953. [Google Scholar]
- De Gennes, P.G.; Witten, T.A. Scaling Concepts in Polymer Physics. Phys. Today 1980, 33, 51. [Google Scholar] [CrossRef]
- Roe, R.-J. Computer Simulation of Polymers; Prentice Hall: Englewood Cliffs, NJ, USA, 1991. [Google Scholar]
- Groot, R.D.; Warren, P.B. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Müller-Plathe, F. Scale-Hopping in Computer Simulations of Polymers. Soft Mater. 2002, 1, 1–31. [Google Scholar] [CrossRef]
- Kremer, K.; Müller-Plathe, F. Multiscale simulation in polymer science. Mol. Simul. 2002, 28, 729–750. [Google Scholar] [CrossRef]
- Theodorou, D.N. Hierarchical modelling of polymeric materials. Chem. Eng. Sci. 2007, 62, 5697–5714. [Google Scholar] [CrossRef]
- Praprotnik, M.; Site, L.D.; Kremer, K. Multiscale Simulation of Soft Matter: From Scale Bridging to Adaptive Resolution. Annu. Rev. Phys. Chem. 2008, 59, 545–571. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Abberton, B.C.; Kröger, M.; Liu, W.K. Challenges in Multiscale Modeling of Polymer Dynamics. Polymers 2013, 5, 751–832. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Strachan, A. Molecular scale simulations on thermoset polymers: A review. J. Polym. Sci. Part B Polym. Phys. 2015, 53, 103–122. [Google Scholar] [CrossRef]
- Karatrantos, A.; Clarke, N.; Kröger, M. Modeling of Polymer Structure and Conformations in Polymer Nanocomposites from Atomistic to Mesoscale: A Review. Polym. Rev. 2016, 56, 385–428. [Google Scholar] [CrossRef]
- Gartner, T.E.; Jayaraman, A. Modeling and Simulations of Polymers: A Roadmap. Macromolecules 2019, 52, 755–786. [Google Scholar] [CrossRef] [Green Version]
- Baschnagel, J.; Binder, K.; Doruker, P.; Gusev, A.A.; Hahn, O.; Kremer, K.; Mattice, W.L.; Müller-Plathe, F.; Murat, M.; Paul, W.; et al. Bridging the Gap Between Atomistic and Coarse-Grained Models of Polymers: Status and Perspectives. In Peptide Hybrid Polymers; Springer: Berlin/Heidelberg, Germany, 2007; pp. 41–156. [Google Scholar]
- Müller-Plathe, F. Coarse-graining in polymer simulation: From the atomistic to the mesoscopic scale and back. ChemPhysChem 2002, 3, 754–769. [Google Scholar] [CrossRef]
- Vettorel, T.; Besold, G.; Kremer, K. Fluctuating soft-sphere approach to coarse-graining of polymer models. Soft Matter 2010, 6, 2282–2292. [Google Scholar] [CrossRef]
- D’Adamo, G.; Pelissetto, A.; Pierleoni, C. Coarse-graining strategies in polymer solutions. Soft Matter 2012, 8, 5151. [Google Scholar] [CrossRef] [Green Version]
- Marzi, D.; Likos, C.N.; Capone, B. Coarse graining of star-polymer—Colloid nanocomposites. J. Chem. Phys. 2012, 137, 014902. [Google Scholar] [CrossRef]
- Narros, A.; Likos, C.N.; Moreno, A.J.; Capone, B. Multi-blob coarse graining for ring polymer solutions. Soft Matter 2014, 10, 9601–9614. [Google Scholar] [CrossRef] [Green Version]
- Kong, M.; Dalal, I.S.; Li, G.; Larson, R.G. Systematic Coarse-Graining of the Dynamics of Self-Attractive Semiflexible Polymers. Macromolecules 2014, 47, 1494–1502. [Google Scholar] [CrossRef]
- Nikoubashman, A.; Mahynski, N.A.; Capone, B.; Panagiotopoulos, A.Z.; Likos, C.N. Coarse-graining and phase behavior of model star polymer-colloid mixtures in solvents of varying quality. J. Chem. Phys. 2015, 143, 243108. [Google Scholar] [CrossRef]
- Zhang, W.; Gomez, E.D.; Milner, S.T. Predicting Flory-Huggins χ from Simulations. Phys. Rev. Lett. 2017, 119, 017801. [Google Scholar] [CrossRef] [Green Version]
- Morthomas, J.; Fusco, C.; Zhai, Z.; Lame, O.; Perez, M. Crystallization of finite-extensible nonlinear elastic Lennard-Jones coarse-grained polymers. Phys. Rev. E 2017, 96, 052502. [Google Scholar] [CrossRef]
- Dinpajooh, M.; Guenza, M. Coarse-graining simulation approaches for polymer melts: The effect of potential range on computational efficiency. Soft Matter 2018, 14, 7126–7144. [Google Scholar] [CrossRef] [Green Version]
- Hsu, H.-P.; Kremer, K. A coarse-grained polymer model for studying the glass transition. J. Chem. Phys. 2019, 150, 091101. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, Z.; Pan, W. Implicit-solvent coarse-grained modeling for polymer solutions via Mori-Zwanzig formalism. Soft Matter 2019, 15, 7567–7582. [Google Scholar] [CrossRef] [PubMed]
- Giuntoli, A.; Puosi, F.; Leporini, D.; Starr, F.W.; Douglas, J.F. Predictive relation for the α-relaxation time of a coarse-grained polymer melt under steady shear. Sci. Adv. 2020, 6, eaaz0777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rondina, G.G.; Böhm, M.C.; Müller-Plathe, F. Predicting the Mobility Increase of Coarse-Grained Polymer Models from Excess Entropy Differences. J. Chem. Theory Comput. 2020, 16, 1431–1447. [Google Scholar] [CrossRef] [PubMed]
- Milchev, A.; Binder, K. Semiflexible Polymers Interacting with Planar Surfaces: Weak versus Strong Adsorption. Polymers 2020, 12, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorichetti, V.; Hugouvieux, V.; Kob, W. Determining the Mesh Size of Polymer Solutions via the Pore Size Distribution. Macromolecules 2020, 53, 2568–2581. [Google Scholar] [CrossRef]
- Paul, W.; Binder, K.; Kremer, K.; Heermann, D.W. Structure-property correlation of polymers, a Monte Carlo approach. Macromolecules 1991, 24, 6332–6334. [Google Scholar] [CrossRef]
- Reith, D.; Meyer, H.; Müller-Plathe, F. Mapping Atomistic to Coarse-Grained Polymer Models Using Automatic Simplex Optimization To Fit Structural Properties. Macromolecules 2001, 34, 2335–2345. [Google Scholar] [CrossRef] [Green Version]
- Faller, R.; Müller-Plathe, F. Modeling of poly(isoprene) melts on different scales. Polymer 2002, 43, 621–628. [Google Scholar] [CrossRef]
- Padding, J.T.; Briels, W.J. Time and length scales of polymer melts studied by coarse-grained molecular dynamics simulations. J. Chem. Phys. 2002, 117, 925–943. [Google Scholar] [CrossRef] [Green Version]
- Milano, G.; Müller-Plathe, F. Mapping Atomistic Simulations to Mesoscopic Models: A Systematic Coarse-Graining Procedure for Vinyl Polymer Chains. J. Phys. Chem. B 2005, 109, 18609–18619. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Carbone, P.; Cavalcanti, W.L.; Milano, G.; Müller-Plathe, F. Viscosity and Structural Alteration of a Coarse-Grained Model of Polystyrene under Steady Shear Flow Studied by Reverse Nonequilibrium Molecular Dynamics. Macromolecules 2007, 40, 8087–8095. [Google Scholar] [CrossRef]
- Harmandaris, V.; Reith, D.; Van Der Vegt, N.F.A.; Kremer, K. Comparison Between Coarse-Graining Models for Polymer Systems: Two Mapping Schemes for Polystyrene. Macromol. Chem. Phys. 2007, 208, 2109–2120. [Google Scholar] [CrossRef]
- Chen, C.; Depa, P.; Maranas, J.; Sakai, V.G. Comparison of explicit atom, united atom, and coarse-grained simulations of poly(methyl methacrylate). J. Chem. Phys. 2008, 128, 124906. [Google Scholar] [CrossRef] [PubMed]
- Strauch, T.; Yelash, L.; Paul, W. A coarse-graining procedure for polymer melts applied to 1,4-polybutadiene. Phys. Chem. Chem. Phys. 2009, 11, 1942. [Google Scholar] [CrossRef]
- Fritz, D.; Koschke, K.; Harmandaris, V.A.; Van Der Vegt, N.F.A.; Kremer, K. Multiscale modeling of soft matter: Scaling of dynamics. Phys. Chem. Chem. Phys. 2011, 13, 10412–10420. [Google Scholar] [CrossRef]
- Karimi-Varzaneh, H.A.; Van Der Vegt, N.F.A.; Müller-Plathe, F.; Carbone, P. How Good Are Coarse-Grained Polymer Models? A Comparison for Atactic Polystyrene. ChemPhysChem 2012, 13, 3428–3439. [Google Scholar] [CrossRef]
- Wu, C. A Combined Scheme for Systematically Coarse-Graining of Stereoregular Polymer Blends. Macromolecules 2013, 46, 5751–5761. [Google Scholar] [CrossRef]
- Mccarty, J.; Clark, A.J.; Copperman, J.; Guenza, M. An analytical coarse-graining method which preserves the free energy, structural correlations, and thermodynamic state of polymer melts from the atomistic to the mesoscale. J. Chem. Phys. 2014, 140, 204913. [Google Scholar] [CrossRef] [Green Version]
- Maurel, G.; Goujon, F.; Schnell, B.; Malfreyt, P. Prediction of structural and thermomechanical properties of polymers from multiscale simulations. RSC Adv. 2015, 5, 14065–14073. [Google Scholar] [CrossRef]
- Salerno, K.M.; Agrawal, A.; Perahia, D.; Grest, G.S. Resolving Dynamic Properties of Polymers through Coarse-Grained Computational Studies. Phys. Rev. Lett. 2016, 116, 058302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, W.; Song, J.; Jeong, C.; Hsu, D.D.; Phelan, F.R.; Douglas, J.F.; Keten, S. Energy-Renormalization for Achieving Temperature Transferable Coarse-Graining of Polymer Dynamics. Macromolecules 2017, 50, 8787–8796. [Google Scholar] [CrossRef] [PubMed]
- De Silva, C.C.; Leophairatana, P.; Ohkuma, T.; Koberstein, J.T.; Kremer, K.; Mukherji, D. Sequence transferable coarse-grained model of amphiphilic copolymers. J. Chem. Phys. 2017, 147, 064904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, L.; Otter, W.K.D.; Briels, W.J. Coarse-Grained Simulations of Three-Armed Star Polymer Melts and Comparison with Linear Chains. J. Phys. Chem. B 2018, 122, 10210–10218. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Lu, T.; Guo, H. Developing a Transferable Coarse-Grained Model for the Prediction of Thermodynamic, Structural, and Mechanical Properties of Polyimides at Different Thermodynamic State Points. J. Chem. Inf. Model. 2019, 59, 2009–2025. [Google Scholar] [CrossRef] [PubMed]
- An, Y.; Singh, S.; Bejagam, K.K.; Deshmukh, S.A. Development of an Accurate Coarse-Grained Model of Poly(acrylic acid) in Explicit Solvents. Macromolecules 2019, 52, 4875–4887. [Google Scholar] [CrossRef]
- Miwatani, R.; Takahashi, K.Z.; Arai, N. Performance of Coarse Graining in Estimating Polymer Properties: Comparison with the Atomistic Model. Polymers 2020, 12, 382. [Google Scholar] [CrossRef] [Green Version]
- Szukalo, R.J.; Noid, W.G. Investigation of coarse-grained models across a glass transition. Soft Mater. 2020, 1–15. [Google Scholar] [CrossRef]
- Svaneborg, C.; Everaers, R. Characteristic Time and Length Scales in Melts of Kremer-Grest Bead-Spring Polymers with Wormlike Bending Stiffness. Macromolecules 2020, 53, 1917–1941. [Google Scholar] [CrossRef] [Green Version]
- Nébouy, M.; Morthomas, J.; Fusco, C.; Baeza, G.P.; Chazeau, L. Coarse-Grained Molecular Dynamics Modeling of Segmented Block Copolymers: Impact of the Chain Architecture on Crystallization and Morphology. Macromolecules 2020, 53, 3847–3860. [Google Scholar] [CrossRef]
- Pervaje, A.K.; Tilly, J.C.; Detwiler, A.T.; Spontak, R.J.; Khan, S.A.; Santiso, E.E. Molecular Simulations of Thermoset Polymers Implementing Theoretical Kinetics with Top-Down Coarse-Grained Models. Macromolecules 2020, 53, 2310–2322. [Google Scholar] [CrossRef]
- Sunday, D.F.; Chremos, A.; Martin, T.B.; Chang, A.B.; Burns, A.B.; Grubbs, R.H. Concentration Dependence of the Size and Symmetry of a Bottlebrush Polymer in a Good Solvent. Macromolecules 2020, 53, 7132–7140. [Google Scholar] [CrossRef]
- Bayramoglu, B.; Faller, R. Coarse-Grained Modeling of Polystyrene in Various Environments by Iterative Boltzmann Inversion. Macromolecules 2012, 45, 9205–9219. [Google Scholar] [CrossRef]
- Wang, Q.; Keffer, D.J.; Deng, S.; Mays, J. Multi-scale models for cross-linked sulfonated poly (1, 3-cyclohexadiene) polymer. Polymer 2012, 53, 1517–1528. [Google Scholar] [CrossRef]
- Hsu, D.D.; Xia, W.; Arturo, S.G.; Keten, S. Systematic Method for Thermomechanically Consistent Coarse-Graining: A Universal Model for Methacrylate-Based Polymers. J. Chem. Theory Comput. 2014, 10, 2514–2527. [Google Scholar] [CrossRef]
- Moore, T.C.; Iacovella, C.R.; McCabe, C. Derivation of coarse-grained potentials via multistate iterative Boltzmann inversion. J. Chem. Phys. 2014, 140, 224104. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, V.; Arya, G.; Oswald, J. Simultaneous Iterative Boltzmann Inversion for Coarse-Graining of Polyurea. Macromolecules 2014, 47, 3378–3389. [Google Scholar] [CrossRef] [Green Version]
- Boţan, V.; Ustach, V.D.; Leonhard, K.; Faller, R. Development and Application of a Coarse-Grained Model for PNIPAM by Iterative Boltzmann Inversion and Its Combination with Lattice Boltzmann Hydrodynamics. J. Phys. Chem. B 2017, 121, 10394–10406. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, P.; Roy, S.; Nair, N. Coarse-Grained Molecular Dynamics Force-Field for Polyacrylamide in Infinite Dilution Derived from Iterative Boltzmann Inversion and MARTINI Force-Field. J. Phys. Chem. B 2018, 122, 1516–1524. [Google Scholar] [CrossRef]
- Ohkuma, T.; Kremer, K. A composition transferable and time-scale consistent coarse-grained model for cis-polyisoprene and vinyl-polybutadiene oligomeric blends. J. Phys. Mater. 2020, 3, 034007. [Google Scholar] [CrossRef]
- Korolev, N.; Luo, D.; Lyubartsev, A.P.; Nordenskiöld, L. A Coarse-Grained DNA Model Parameterized from Atomistic Simulations by Inverse Monte Carlo. Polymers 2014, 6, 1655–1675. [Google Scholar] [CrossRef] [Green Version]
- Naômé, A.; Laaksonen, A.; Vercauteren, D.P. A Solvent-Mediated Coarse-Grained Model of DNA Derived with the Systematic Newton Inversion Method. J. Chem. Theory Comput. 2014, 10, 3541–3549. [Google Scholar] [CrossRef]
- Lyubartsev, A.P.; Naômé, A.; Vercauteren, D.P.; Laaksonen, A. Systematic hierarchical coarse-graining with the inverse Monte Carlo method. J. Chem. Phys. 2015, 143, 243120. [Google Scholar] [CrossRef] [PubMed]
- Shahidi, N.; Chazirakis, A.; Harmandaris, V.; Doxastakis, M. Coarse-graining of polyisoprene melts using inverse Monte Carlo and local density potentials. J. Chem. Phys. 2020, 152, 124902. [Google Scholar] [CrossRef]
- Das, A.; Andersen, H.C. The multiscale coarse-graining method. V. Isothermal-isobaric ensemble. J. Chem. Phys. 2010, 132, 164106. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Andersen, H.C. The multiscale coarse-graining method. IX. A general method for construction of three body coarse-grained force fields. J. Chem. Phys. 2012, 136, 194114. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Andersen, H.C. The multiscale coarse-graining method. VIII. Multiresolution hierarchical basis functions and basis function selection in the construction of coarse-grained force fields. J. Chem. Phys. 2012, 136, 194113. [Google Scholar] [CrossRef]
- Chaimovich, A.; Shell, M.S. Relative entropy as a universal metric for multiscale errors. Phys. Rev. E 2010, 81, 060104. [Google Scholar] [CrossRef]
- Chaimovich, A.; Shell, M.S. Coarse-graining errors and numerical optimization using a relative entropy framework. J. Chem. Phys. 2011, 134, 094112. [Google Scholar] [CrossRef]
- Foley, T.T.; Shell, M.S.; Noid, W.G. The impact of resolution upon entropy and information in coarse-grained models. J. Chem. Phys. 2015, 143, 243104. [Google Scholar] [CrossRef]
- Sanyal, T.; Shell, M.S. Coarse-grained models using local-density potentials optimized with the relative entropy: Application to implicit solvation. J. Chem. Phys. 2016, 145, 034109. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, T.; Shell, M.S. Transferable Coarse-Grained Models of Liquid–Liquid Equilibrium Using Local Density Potentials Optimized with the Relative Entropy. J. Phys. Chem. B 2018, 122, 5678–5693. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, T.; Mittal, J.; Shell, M.S. A hybrid, bottom-up, structurally accurate, Go-like coarse-grained protein model. J. Chem. Phys. 2019, 151, 044111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullinax, J.W.; Noid, W.G. A Generalized-Yvon−Born−Green Theory for Determining Coarse-Grained Interaction Potentials. J. Phys. Chem. C 2009, 114, 5661–5674. [Google Scholar] [CrossRef]
- Mullinax, J.W.; Noid, W.G. Reference state for the generalized Yvon–Born–Green theory: Application for coarse-grained model of hydrophobic hydration. J. Chem. Phys. 2010, 133, 124107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brini, E.; Marcon, V.; Van Der Vegt, N.F.A. Conditional reversible work method for molecular coarse graining applications. Phys. Chem. Chem. Phys. 2011, 13, 10468–10474. [Google Scholar] [CrossRef] [PubMed]
- Wu, C. Multiscale simulations of the structure and dynamics of stereoregular poly(methyl methacrylate)s. J. Mol. Model. 2014, 20. [Google Scholar] [CrossRef]
- Wu, C. Hydrogen bonding in stereoregular poly (methyl methacrylate)/poly (vinyl chloride) blends as studied by molecular dynamics simulations. Mol. Simul. 2015, 41, 547–554. [Google Scholar] [CrossRef]
- Wu, C. Melt-phase behavior of collapsed PMMA/PVC chains revealed by multiscale simulations. J. Mol. Model. 2016, 22, 99. [Google Scholar] [CrossRef]
- Shelley, J.C.; Shelley, M.Y.; Reeder, R.C.; Bandyopadhyay, S.; Klein, M.L. A Coarse Grain Model for Phospholipid Simulations. J. Phys. Chem. B 2001, 105, 4464–4470. [Google Scholar] [CrossRef]
- Marrink, S.J.; Tieleman, D.P. Perspective on the Martini model. Chem. Soc. Rev. 2013, 42, 6801–6822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinoda, W.; DeVane, R.; Klein, M.L. Multi-property fitting and parameterization of a coarse grained model for aqueous surfactants. Mol. Simul. 2007, 33, 27–36. [Google Scholar] [CrossRef]
- Müller, E.A.; Jackson, G. Force-Field Parameters from the SAFT-γ Equation of State for Use in Coarse-Grained Molecular Simulations. Annu. Rev. Chem. Biomol. Eng. 2014, 5, 405–427. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, A. 100th Anniversary of Macromolecular Science Viewpoint: Modeling and Simulation of Macromolecules with Hydrogen Bonds: Challenges, Successes, and Opportunities. ACS Macro Lett. 2020, 9, 656–665. [Google Scholar] [CrossRef]
- Coleman, M.M.; Serman, C.J.; Bhagwagar, D.E.; Painter, P.C. A practical guide to polymer miscibility. Polymer 1990, 31, 1187–1203. [Google Scholar] [CrossRef]
- Coleman, M.M.; Pehlert, G.J.; Painter, P.C. Functional Group Accessibility in Hydrogen Bonded Polymer Blends. Macromolecules 1996, 29, 6820–6831. [Google Scholar] [CrossRef]
- Radmard, B.; Dadmun, M. The accessibility of functional groups to intermolecular hydrogen bonding in polymer blends containing a liquid crystalline polymer. Polymer 2001, 42, 1591–1600. [Google Scholar] [CrossRef]
- Viswanathan, S.; Dadmun, M.D. Guidelines to Creating a True Molecular Composite: Inducing Miscibility in Blends by Optimizing Intermolecular Hydrogen Bonding. Macromolecules 2002, 35, 5049–5060. [Google Scholar] [CrossRef]
- Kuo, S.-W. Hydrogen-bonding in polymer blends. J. Polym. Res. 2008, 15, 459–486. [Google Scholar] [CrossRef]
- Coleman, M.M.; Graf, J.F.; Painter, P.C. Specific Interactions and the Miscibility of Polymer Blends; Informa UK Limited: Colchester, UK, 2017. [Google Scholar]
- Ikkala, O.; Ruokolainen, J.; Brinke, G.T.; Torkkeli, M.; Serimaa, R. Mesomorphic State of Poly(vinylpyridine)-Dodecylbenzenesulfonic Acid Complexes in Bulk and in Xylene Solution. Macromolecules 1995, 28, 7088–7094. [Google Scholar] [CrossRef] [Green Version]
- Ruokolainen, J.; Ten Brinke, G.; Ikkala, O.; Torkkeli, M.; Serimaa, R. Mesomorphic structures in flexible Polymer- Surfactant systems due to hydrogen Bonding: Poly (4-vinylpyridine)- pentadecylphenol. Macromolecules 1996, 29, 3409–3415. [Google Scholar] [CrossRef]
- Wang, S.-J.; Xu, Y.-S.; Yang, S.; Chen, E.-Q. Phase Behavior of a Hydrogen-Bonded Polymer with Lamella-to-Cylinder Transition: Complex of Poly(4-vinylpyridine) and Small Dendritic Benzoic Acid Derivative. Macromolecules 2012, 45, 8760–8769. [Google Scholar] [CrossRef]
- Dasgupta, S.; Hammond, A.W.B.; Iii, W.A.G. Crystal Structures and Properties of Nylon Polymers from Theory. J. Am. Chem. Soc. 1996, 118, 12291–12301. [Google Scholar] [CrossRef]
- Zhang, L.; Ruesch, M.; Zhang, X.; Bai, Z.; Liu, L. Tuning thermal conductivity of crystalline polymer nanofibers by interchain hydrogen bonding. RSC Adv. 2015, 5, 87981–87986. [Google Scholar] [CrossRef]
- Neikirk, C.C.; Chung, J.W.; Priestley, R.D. Thermomechanical behavior of hydrogen-bond based supramolecular poly(ε-caprolactone)-silica nanocomposites. RSC Adv. 2013, 3, 16686. [Google Scholar] [CrossRef]
- Heo, K.; Miesch, C.; Emrick, T.; Hayward, R.C. Thermally Reversible Aggregation of Gold Nanoparticles in Polymer Nanocomposites through Hydrogen Bonding. Nano Lett. 2013, 13, 5297–5302. [Google Scholar] [CrossRef]
- Yusa, S.-I.; Sakakibara, A.; Yamamoto, T.; Morishima, Y. Fluorescence Studies of pH-Responsive Unimolecular Micelles Formed from Amphiphilic Polysulfonates Possessing Long-Chain Alkyl Carboxyl Pendants. Macromolecules 2002, 35, 10182–10188. [Google Scholar] [CrossRef]
- O’Neal, J.T.; Bolen, M.J.; Dai, E.Y.; Lutkenhaus, J.L. Hydrogen-bonded polymer nanocomposites containing discrete layers of gold nanoparticles. J. Colloid Interface Sci. 2017, 485, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Taylor, P.A.; Jayaraman, A. Molecular Modeling and Simulations of Peptide-Polymer Conjugates. Annu. Rev. Chem. Biomol. Eng. 2020, 11, 257–276. [Google Scholar] [CrossRef]
- Coleman, M.; Painter, P. Hydrogen bonded polymer blends. Prog. Polym. Sci. 1995, 20, 1–59. [Google Scholar] [CrossRef]
- Karimi-Varzaneh, H.A.; Carbone, P.; Müller-Plathe, F. Fast dynamics in coarse-grained polymer models: The effect of the hydrogen bonds. J. Chem. Phys. 2008, 129, 154904. [Google Scholar] [CrossRef] [PubMed]
- Carbone, P.; Karimi-Varzaneh, H.A.; Chen, X.; Müller-Plathe, F. Transferability of coarse-grained force fields: The polymer case. J. Chem. Phys. 2008, 128, 064904. [Google Scholar] [CrossRef] [PubMed]
- Gowers, R.; Carbone, P. A multiscale approach to model hydrogen bonding: The case of polyamide. J. Chem. Phys. 2015, 142, 224907. [Google Scholar] [CrossRef] [PubMed]
- Di Pasquale, N.; Marchisio, D.L.; Carbone, P. Mixing atoms and coarse-grained beads in modelling polymer melts. J. Chem. Phys. 2012, 137, 164111. [Google Scholar] [CrossRef]
- Kulshreshtha, A.; Modica, K.J.; Jayaraman, A. Impact of Hydrogen Bonding Interactions on Graft–Matrix Wetting and Structure in Polymer Nanocomposites. Macromolecules 2019, 52, 2725–2735. [Google Scholar] [CrossRef]
- Arichi, S.; Tanimoto, Y.; Murata, H. Studies of Poly-2-vinylpyridine. VI. Thermodynamic Data on Solutions of Poly-2-vinylpyridine in Various Solvents. Bull. Chem. Soc. Jpn. 1968, 41, 1296–1301. [Google Scholar] [CrossRef]
- Hansen, C.M. The Universality of the Solubility Parameter. Ind. Eng. Chem. Prod. Res. Dev. 1969, 8, 2–11. [Google Scholar] [CrossRef]
- Arichi, S.; Sakamoto, N.; Yoshida, M.; Himuro, S. Dilute solution properties of poly(4-hydroxystyrene). Polymer 1986, 27, 1761–1767. [Google Scholar] [CrossRef]
- Arichi, S.; Himuro, S. Solubility parameters of poly(4-acetoxystyrene) and poly(4-hydroxystyrene). Polymer 1989, 30, 686–692. [Google Scholar] [CrossRef]
- Serman, C.J.; Xu, Y.; Painter, P.C.; Coleman, M.M. Poly(vinyl phenol)—Polyether blends. Polymer 1991, 32, 516–522. [Google Scholar] [CrossRef]
- Kuo, S.W.; Chang, F.C. Studies of Miscibility Behavior and Hydrogen Bonding in Blends of Poly(vinylphenol) and Poly(vinylpyrrolidone). Macromolecules 2001, 34, 5224–5228. [Google Scholar] [CrossRef]
- Qiu, Z.; Fujinami, S.; Komura, M.; Nakajima, K.; Ikehara, T.; Nishi, T. Miscibility and crystallization of poly (ethylene succinate)/poly (vinyl phenol) blends. Polymer 2004, 45, 4515–4521. [Google Scholar] [CrossRef]
- Antonietti, M.; Heinz, S.; Schmidt, M.; Rosenauer, C. Determination of the Micelle Architecture of Polystyrene/Poly(4-vinylpyridine) Block Copolymers in Dilute Solution. Macromolecules 1994, 27, 3276–3281. [Google Scholar] [CrossRef]
- Shen, H.; Zhang, L.; Eisenberg, A. Multiple pH-Induced Morphological Changes in Aggregates of Polystyrene-block-poly(4-vinylpyridine) in DMF/H2O Mixtures. J. Am. Chem. Soc. 1999, 121, 2728–2740. [Google Scholar] [CrossRef]
- Li, D.; He, Q.; Cui, A.Y.; Li, J. Fabrication of pH-Responsive Nanocomposites of Gold Nanoparticles/Poly(4-vinylpyridine). Chem. Mater. 2007, 19, 412–417. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; Van Der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Model. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; Van Der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Abraham, M.; Hess, B.; Van der Spoel, D.; Lindahl, E. GROMACS User Manual Version 5.1.; GROMACS Development Team; Royal Institute of Technology: Stockholm, Sweden; Uppsala University: Uppsala, Sweden, 2016. [Google Scholar]
- Jorgensen, W.L.; Severance, D.L. Aromatic-aromatic interactions: Free energy profiles for the benzene dimer in water, chloroform, and liquid benzene. J. Am. Chem. Soc. 1990, 112, 4768–4774. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Tirado-Rives, J. Potential energy functions for atomic-level simulations of water and organic and biomolecular systems. Proc. Natl. Acad. Sci. USA 2005, 102, 6665–6670. [Google Scholar] [CrossRef] [Green Version]
- Dahlgren, M.K.; Schyman, P.; Tirado-Rives, J.; Jorgensen, W.L. Characterization of Biaryl Torsional Energetics and its Treatment in OPLS All-Atom Force Fields. J. Chem. Inf. Model. 2013, 53, 1191–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, G.; Monticelli, L.; Puisto, S.R.; Vattulainen, I.; Ala-Nissila, T. Coarse-graining polymers with the MARTINI force-field: Polystyrene as a benchmark case. Soft Matter 2011, 7, 698–708. [Google Scholar] [CrossRef] [Green Version]
- Wu, C. Coarse-grained molecular dynamics simulations of stereoregular poly (methyl methacrylate)/poly (vinyl chloride) blends. J. Polym. Sci. Part B Polym. Phys. 2015, 53, 203–212. [Google Scholar] [CrossRef]
- Dodda, L.S.; De Vaca, I.C.; Tirado-Rives, J.; Jorgensen, W.L. LigParGen web server: An automatic OPLS-AA parameter generator for organic ligands. Nucleic Acids Res. 2017, 45, W331–W336. [Google Scholar] [CrossRef] [Green Version]
- Dodda, L.S.; Vilseck, J.Z.; Tirado-Rives, J.; Jorgensen, W.L. 1.14*CM1A-LBCC: Localized Bond-Charge Corrected CM1A Charges for Condensed-Phase Simulations. J. Phys. Chem. B 2017, 121, 3864–3870. [Google Scholar] [CrossRef] [Green Version]
- Jones, J.E. On the determination of molecular fields. II. From the equation of state of a gas. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1924, 106, 463–477. [Google Scholar]
- Allen, M.P.; Tildesley, D.J.; Banavar, J.R. Computer Simulation of Liquids. Phys. Today 1989, 42, 105–106. [Google Scholar] [CrossRef]
- Darden, T.A.; York, D.M.; Pedersen, L. Particle mesh Ewald: AnN⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Grest, G.S.; Kremer, K. Molecular dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A 1986, 33, 3628–3631. [Google Scholar] [CrossRef]
- Weeks, J.D.; Chandler, D.; Andersen, H.C. Role of Repulsive Forces in Determining the Equilibrium Structure of Simple Liquids. J. Chem. Phys. 1971, 54, 5237–5247. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Cifra, P. Differences and limits in estimates of persistence length for semi-flexible macromolecules. Polymer 2004, 45, 5995–6002. [Google Scholar] [CrossRef]
- Peter, C.; Kremer, K. Multiscale simulation of soft matter systems—From the atomistic to the coarse-grained level and back. Soft Matter 2009, 5, 4357–4366. [Google Scholar] [CrossRef]
- Padding, J.T.; Briels, W.J. Systematic coarse-graining of the dynamics of entangled polymer melts: The road from chemistry to rheology. J. Phys. Condens. Matter 2011, 23, 233101. [Google Scholar] [CrossRef] [Green Version]
- Brini, E.; Algaer, E.A.; Ganguly, P.; Li, C.; Rodríguez-Ropero, F.; Van Der Vegt, N.F.A. Systematic coarse-graining methods for soft matter simulation—A review. Soft Matter 2013, 9, 2108–2119. [Google Scholar] [CrossRef]
- Gooneie, A.; Schuschnigg, S.; Holzer, C. A Review of Multiscale Computational Methods in Polymeric Materials. Polymers 2017, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Caviness Community Cluster. Available online: https://sites.udel.edu/research-computing/caviness-cluster/ (accessed on 9 February 2020).










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Wall Time (Seconds) | Speedup | ||
|---|---|---|---|---|
| Atomistic for 10,000 Time Steps | CG for 10,000 Time Steps | |||
| pvpH | 12-mer | 4285 | 45 | 95 |
| 18-mer | 4317 | 55 | 79 | |
| 24-mer | 4336 | 108 | 40 | |
| 36-mer | 4421 | 133 | 33 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapoor, U.; Kulshreshtha, A.; Jayaraman, A. Development of Coarse-Grained Models for Poly(4-vinylphenol) and Poly(2-vinylpyridine): Polymer Chemistries with Hydrogen Bonding. Polymers 2020, 12, 2764. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12112764
Kapoor U, Kulshreshtha A, Jayaraman A. Development of Coarse-Grained Models for Poly(4-vinylphenol) and Poly(2-vinylpyridine): Polymer Chemistries with Hydrogen Bonding. Polymers. 2020; 12(11):2764. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12112764
Chicago/Turabian StyleKapoor, Utkarsh, Arjita Kulshreshtha, and Arthi Jayaraman. 2020. "Development of Coarse-Grained Models for Poly(4-vinylphenol) and Poly(2-vinylpyridine): Polymer Chemistries with Hydrogen Bonding" Polymers 12, no. 11: 2764. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12112764