Hyperbranched Poly(ether-siloxane)s Containing Ammonium Groups: Synthesis, Characterization and Catalytic Activity

 , , ,
, , , 
Abstract
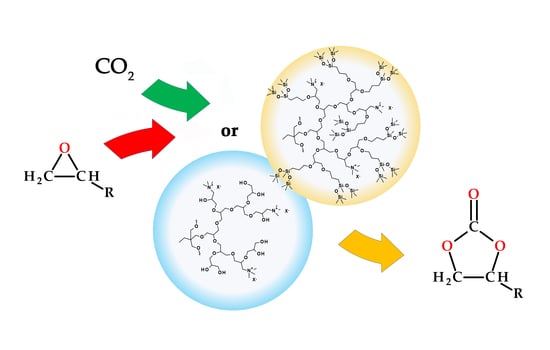
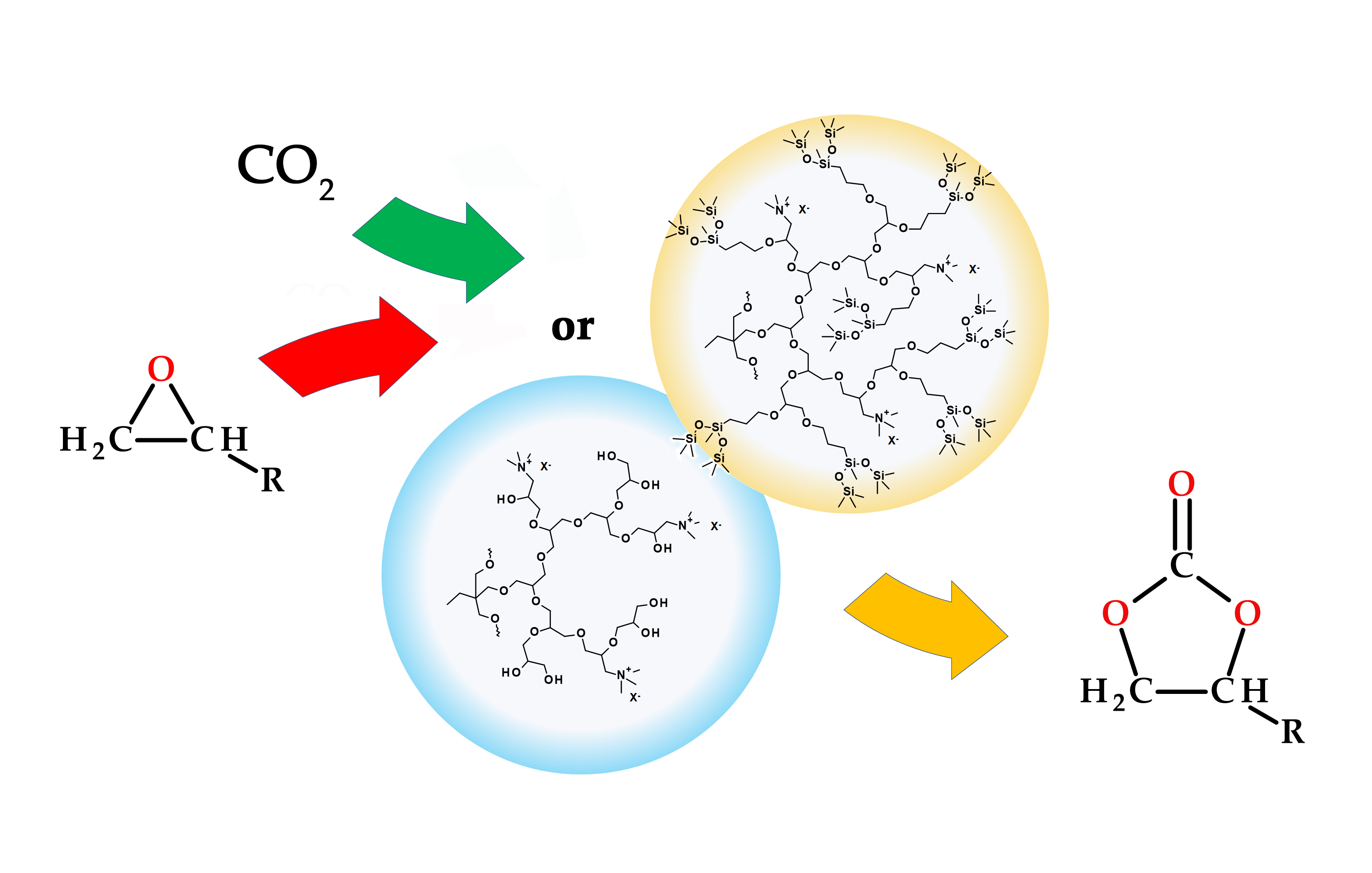
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instrumentation
2.3. Syntheses
2.3.1. ROP Polymerization of Epoxy Phthalimide and Glycidol
2.3.2. Alkylation of 1 with Allyl Bromide
2.3.3. Hydrosililation of 2 with Heptamethyltrisiloxane
2.3.4. Procedure of Hydrazinolysis of Phthalimide Groups
2.3.5. Procedure of Quaternization of Amine Groups
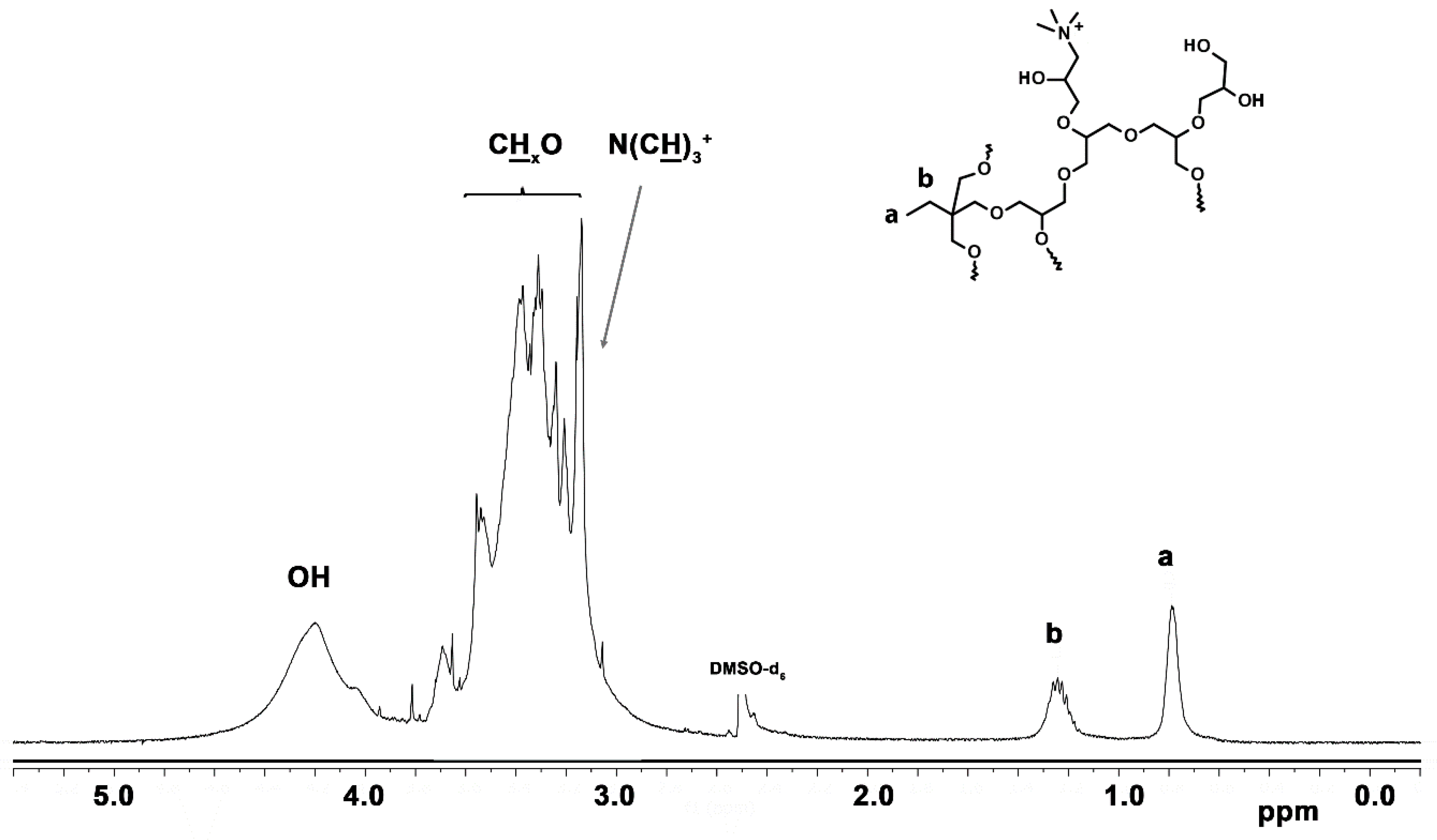
2.3.6. Anion Exchange Procedure
2.3.7. Synthesis of Ethylene Carbonate
3. Results and Discussion
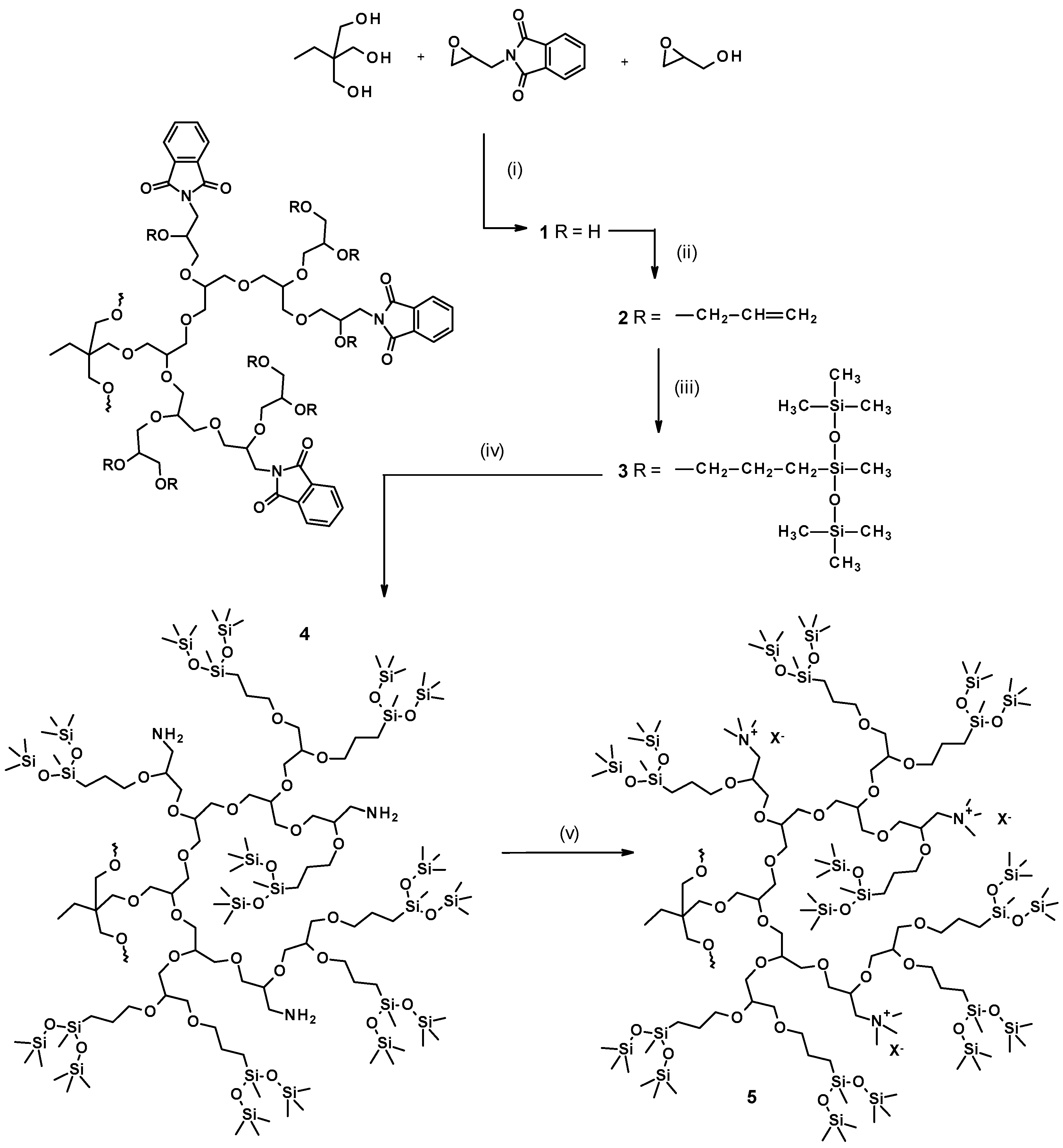
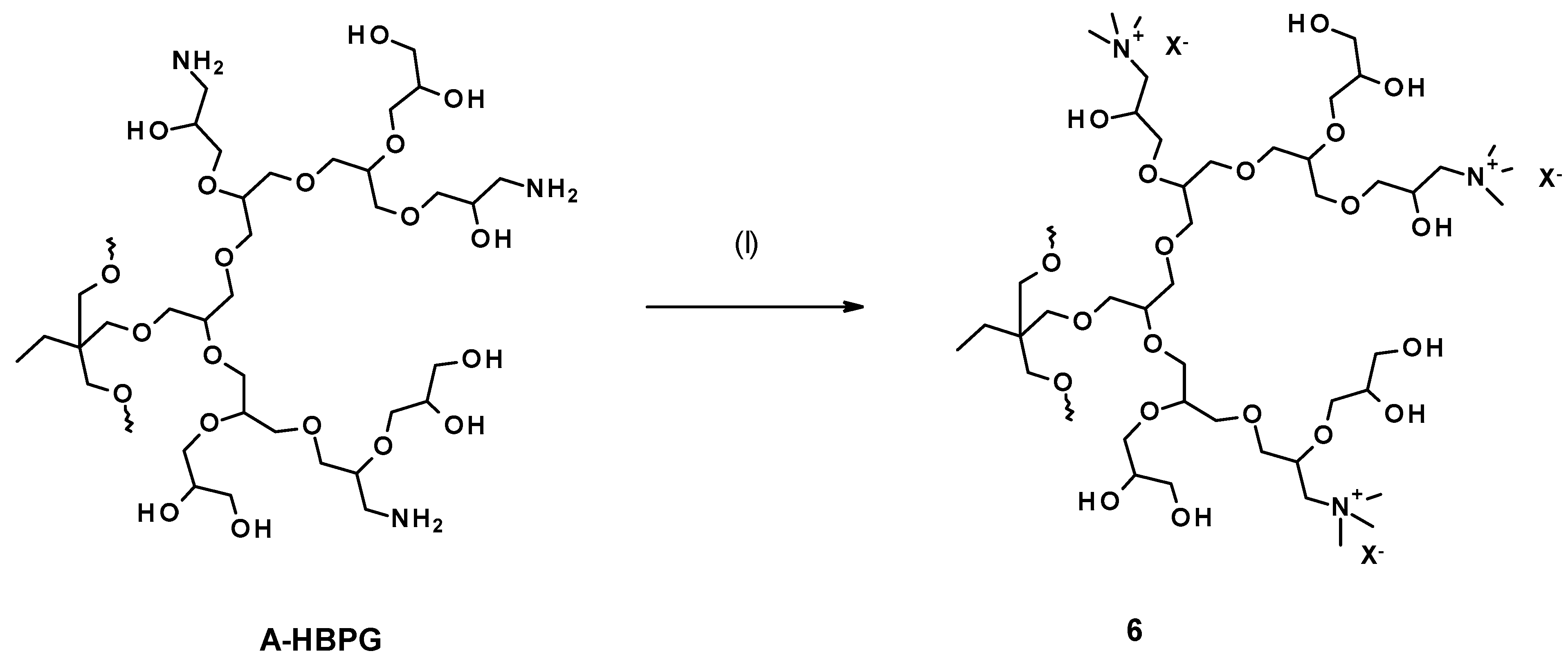
3.1. Syntheses and Structure Analysis
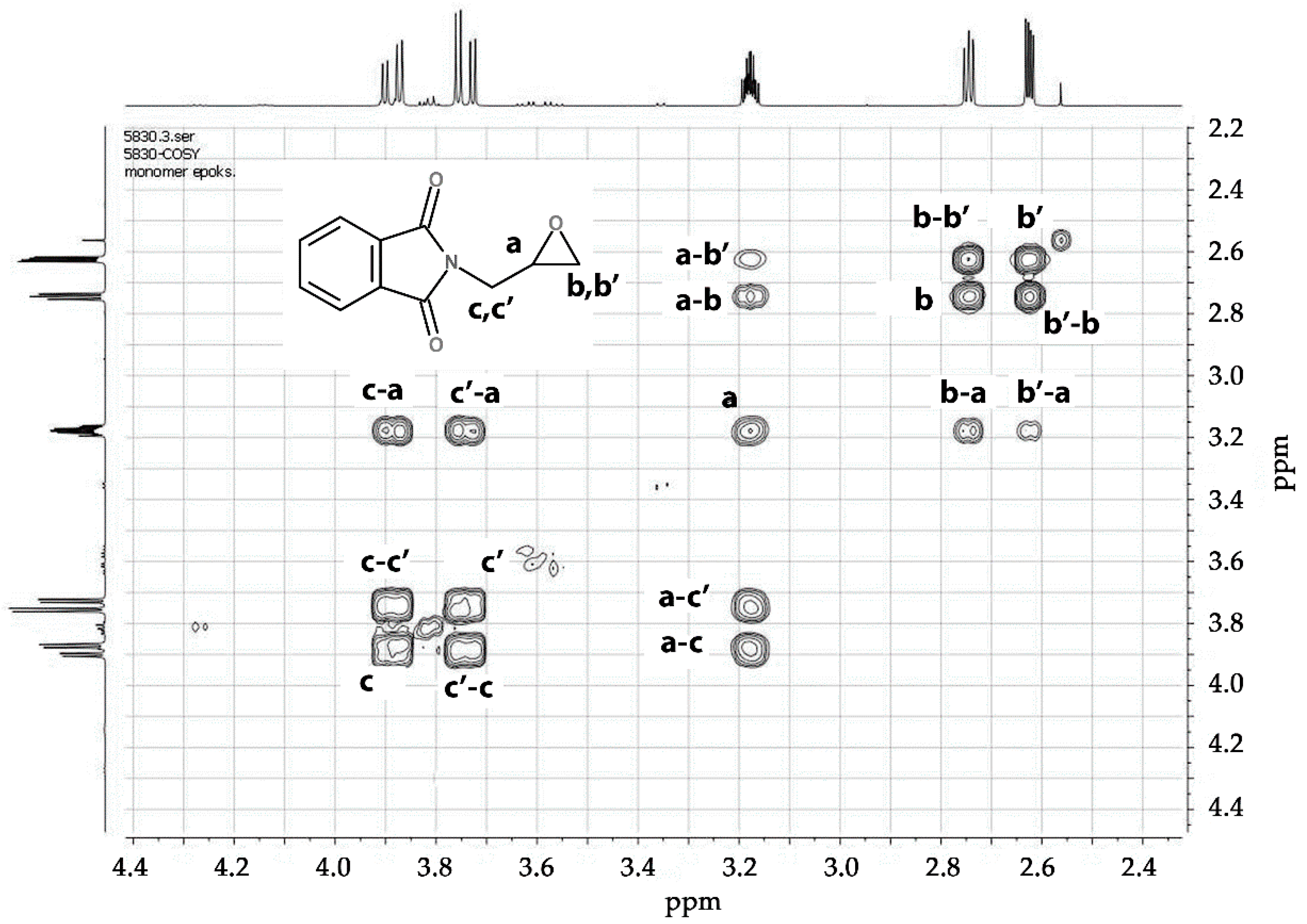
3.1.1. Monomer
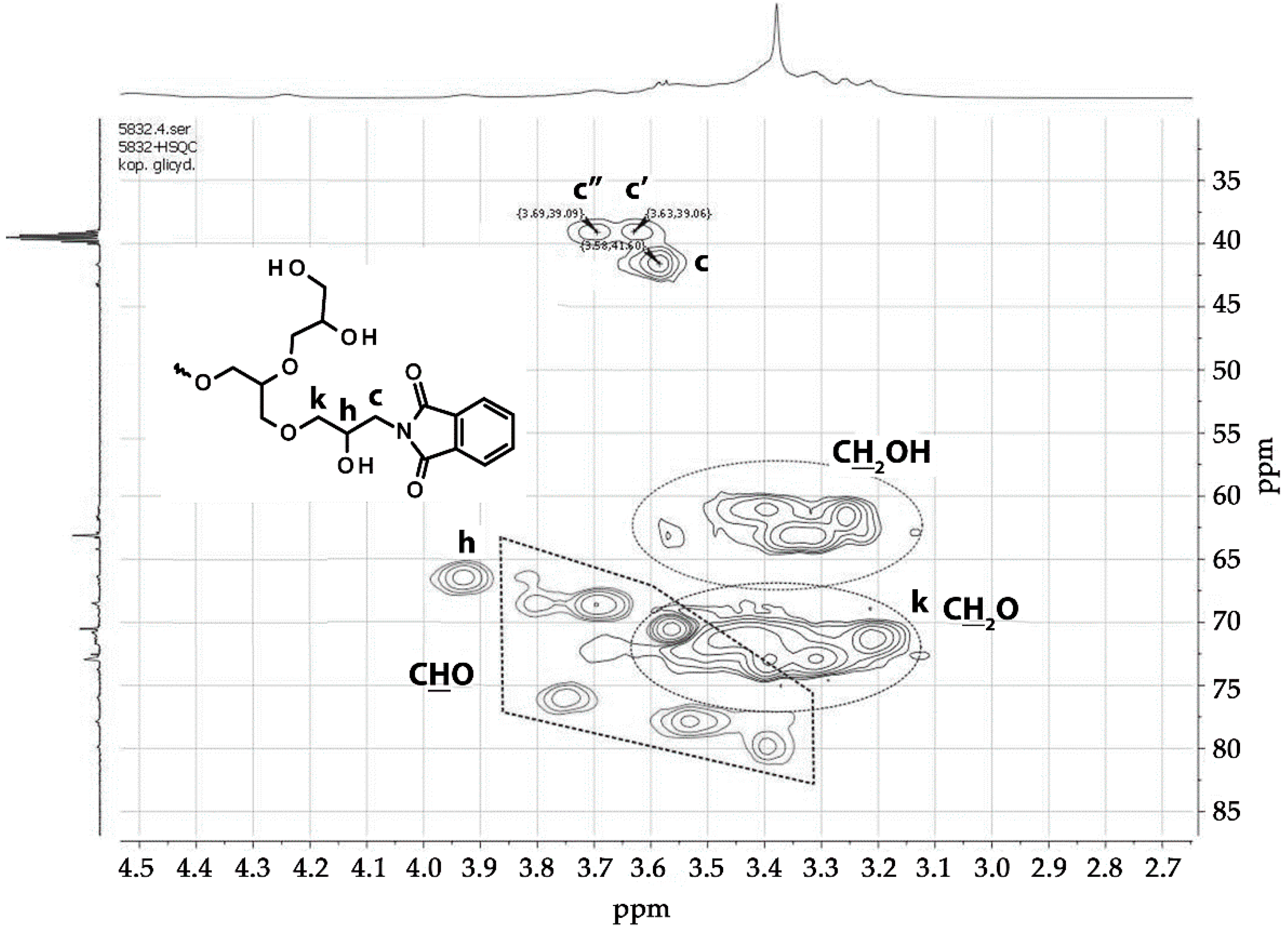
3.1.2. Copolymerization
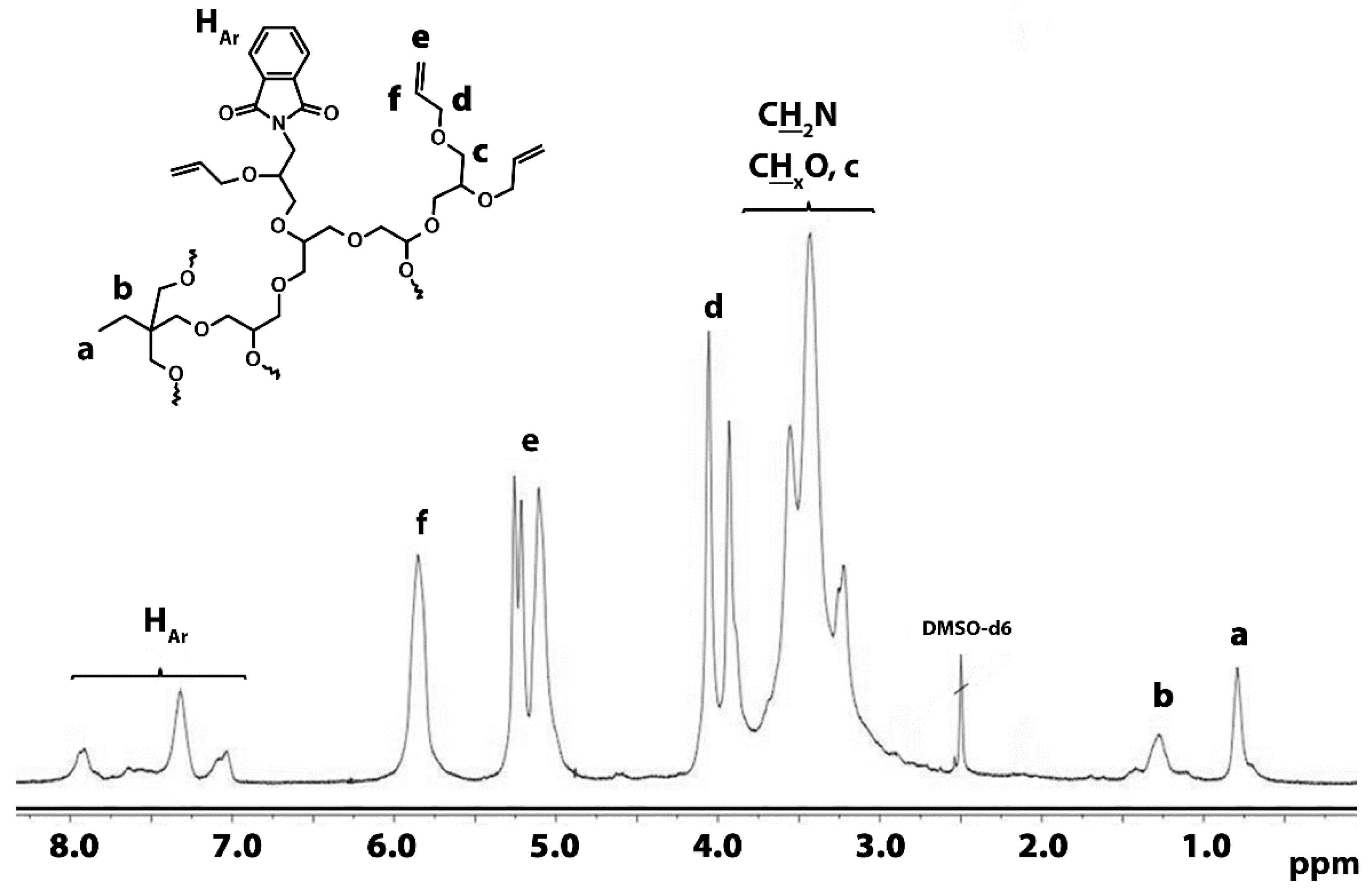
3.1.3. Introduction of Allyl Groups
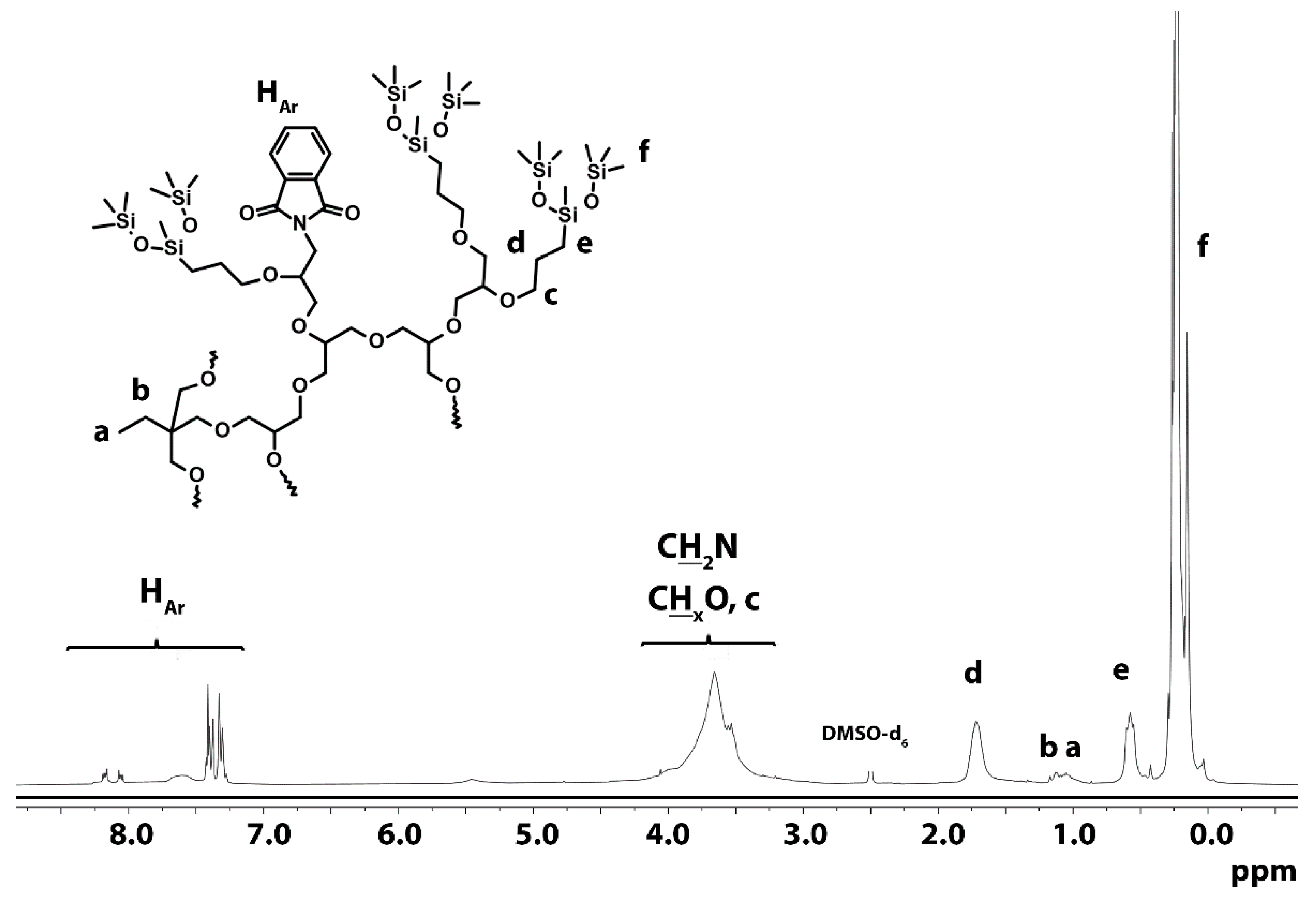
3.1.4. Hydrosililation of Allyl Groups with Heptamethyltrisiloxane
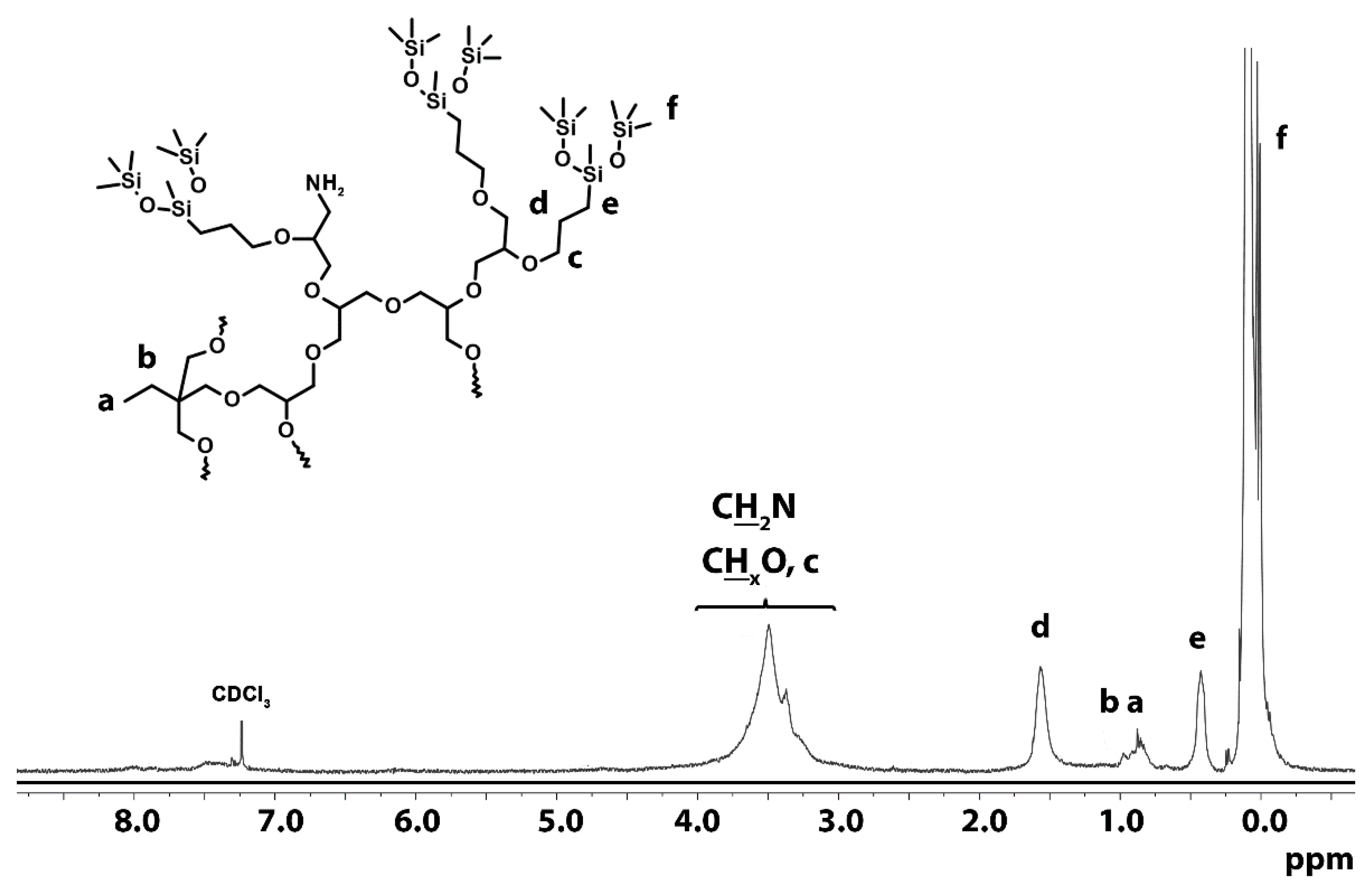
3.1.5. Hydrazinolysis
3.1.6. Quaternization of Amine Groups with Methyl Iodide
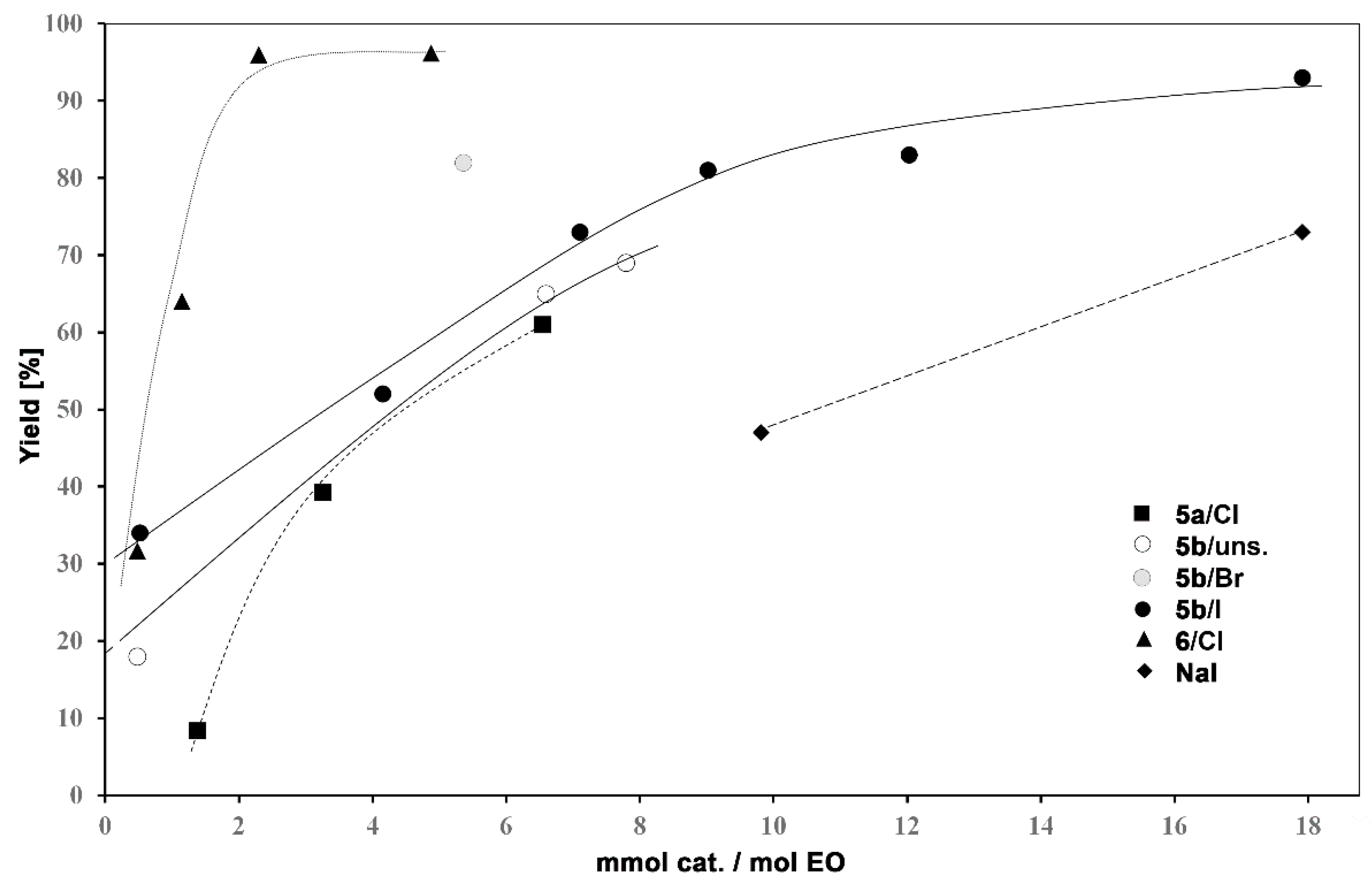
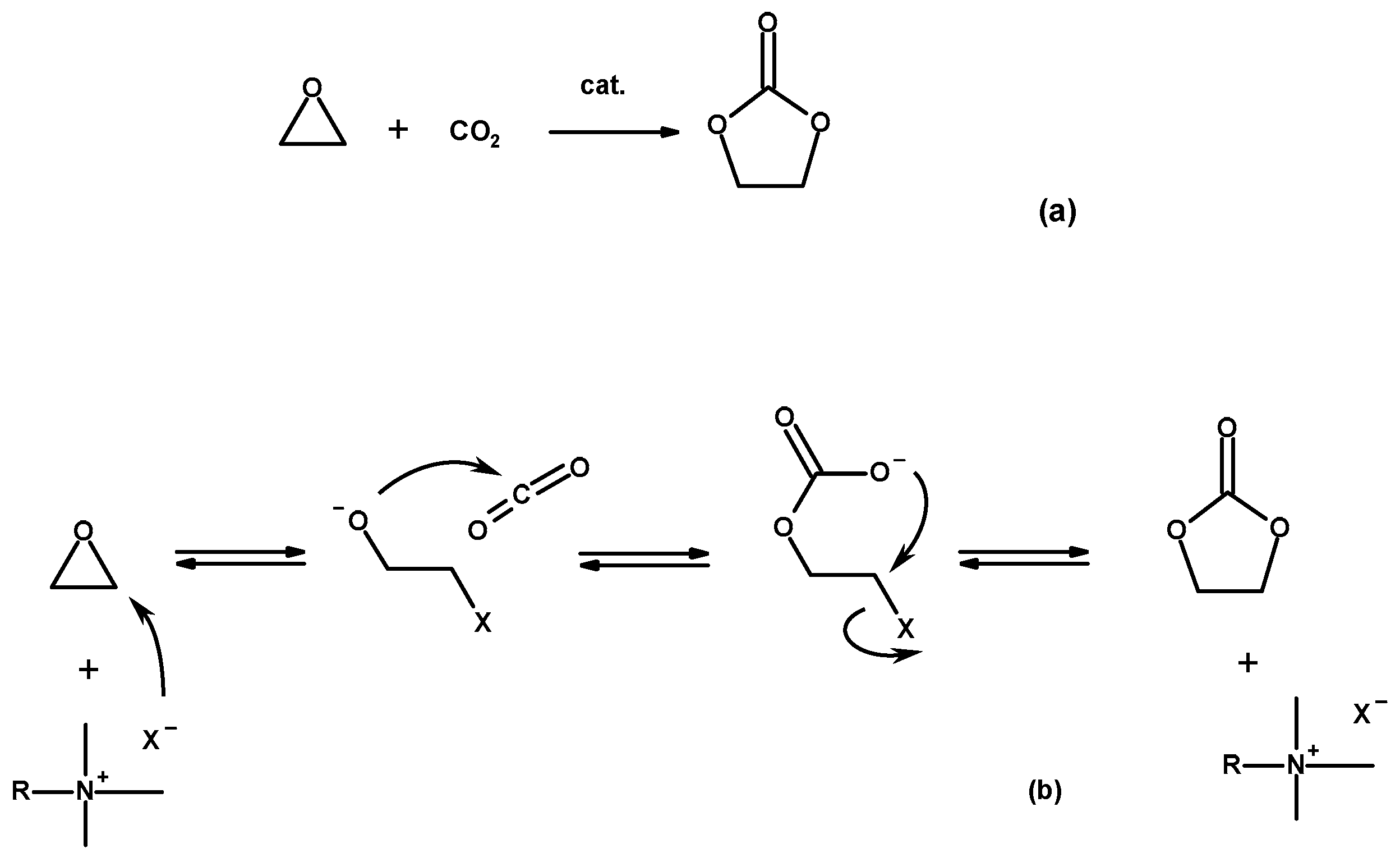
3.2. Catalytic Activity of Hyperbranched Polymers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Carbon dioxide direct measurements. Available online: https://climate.nasa.gov (accessed on 1 March 2020).
- Chen, C.; Kim, J.; Ahn, W.-S. CO2 capture by amine-functionalized nanoporous materials: A review. Korean J. Chem. Eng. 2014, 31, 1919–1934. [Google Scholar] [CrossRef]
- Boot-Handford, M.E.; Abanades, J.C.; Anthony, E.J.; Blunt, M.J.; Brandani, S.; Mac Dowell, N.; Fernández, J.R.; Ferrari, M.-C.; Gross, R.; Hallett, J.P. Carbon capture and storage update. Energy Environ. Sci. 2014, 7, 130–189. [Google Scholar] [CrossRef]
- D’Alessandro, D.M.; Smit, B.; Long, J.R. Carbon dioxide capture: Prospects for new materials. Angew. Chem. Int. Ed. 2010, 49, 6058–6082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, K.M.K.; Curcic, I.; Gabriel, J.; Tsang, S.C.E. Recent advances in CO2 capture and utilization. ChemSusChem 2008, 1, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Artz, J.; Müller, T.E.; Thenert, K.; Kleinekorte, J.; Meys, R.; Sternberg, A.; Bardow, A.; Leitner, W. Sustainable conversion of carbon dioxide: An integrated review of catalysis and life cycle assessment. Chem. Rev. 2018, 118, 434–504. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Yu, J.; Sun, Y.; Xie, W. Study of POSS on the Properties of Novel Inorganic Dental Composite Resin. Polymers 2020, 12, 478. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Wu, X.; Sun, Y.; Xie, W. POSS Dental Nanocomposite Resin: Synthesis, Shrinkage, Double Bond Conversion, Hardness, and Resistance Properties. Polymers 2018, 10, 369. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hao, Z.; Yu, J.; Zhou, X.; Lee, P.S.; Sun, Y.; Mu, Z.; Zeng, F. A high-performance soft actuator based on a poly (vinylidene fluoride) piezoelectric bimorph. Smart Mater. Struct. 2019, 28, 055011. [Google Scholar] [CrossRef]
- Rokicki, G.; Parzuchowski, P.G.; Mazurek, M. Non-isocyanate polyurethanes-synthesis, properties and applications. Polym. Adv. Technol. 2015, 26, 707–761. [Google Scholar] [CrossRef]
- Blattmann, H.; Fleischer, M.; Bähr, M.; Mülhaupt, R. Isocyanate-and phosgene-free routes to polyfunctional cyclic carbonates and green polyurethanes by fixation of carbon dioxide. Macromol. Rapid Commun. 2014, 35, 1238–1254. [Google Scholar] [CrossRef]
- Rokicki, G.; Parzuchowski, P. ROP of Cyclic Carbonates and ROP of Macrocycles–Latest Developments. In Reference Module in Materials Science and Materials Engineering; Hashmi, S., Ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 1–92. [Google Scholar]
- Razali, N.A.M.; Lee, K.T.; Bhatia, S.; Mohamed, A.R. Heterogeneous catalysts for production of chemicals using carbon dioxide as raw material: A review. Renew. Sustain. Energy Rev. 2012, 16, 4951–4964. [Google Scholar] [CrossRef]
- North, M.; Pasquale, R.; Young, C. Synthesis of cyclic carbonates from epoxides and CO2. Green Chem. 2010, 12, 1514–1539. [Google Scholar] [CrossRef]
- Rokicki, G.; Parzuchowski, P.G. ROP of Cyclic Carbonates and ROP of Macrocycles. In Polymer Science: A Comprehensive Reference; Elsevier: Amsterdam, The Netherlands, 2012; Volume 4, pp. 247–308. [Google Scholar]
- Beattie, C.; North, M.; Villuendas, P.; Young, C. Influence of temperature and pressure on cyclic carbonate synthesis catalyzed by bimetallic aluminum complexes and application to overall syn-bis-hydroxylation of alkenes. J. Org. Chem. 2013, 78, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Metcalfe, I.S.; North, M.; Villuendas, P. Influence of reactor design on cyclic carbonate synthesis catalysed by a bimetallic aluminium (salen) complex. J. CO2 Util. 2013, 2, 24–28. [Google Scholar] [CrossRef] [Green Version]
- Kilic, A.; Ulusoy, M.; Durgun, M.; Aytar, E. The multinuclear cobaloxime complexes-based catalysts for direct synthesis of cyclic carbonate from of epichlorohydrin using carbon dioxide: Synthesis and characterization. Inorg. Chim. Acta 2014, 411, 17–25. [Google Scholar] [CrossRef]
- Tian, D.; Liu, B.; Gan, Q.; Li, H.; Darensbourg, D.J. Formation of Cyclic Carbonates from Carbon Dioxide and Epoxides Coupling Reactions Efficiently Catalyzed by Robust, Recyclable One-Component Aluminum-Salen Complexes. ACS Catal. 2012, 2, 2029–2035. [Google Scholar] [CrossRef]
- Ren, W.-M.; Liu, Y.; Lu, X.-B. Bifunctional Aluminum Catalyst for CO2 Fixation: Regioselective Ring Opening of Three-Membered Heterocyclic Compounds. J. Org. Chem. 2014, 79, 9771–9777. [Google Scholar] [CrossRef]
- Dworak, A.; Slomkowski, S.; Basinska, T.; Gosecka, M.; Walach, W.; Trzebicka, B. Polyglycidol—How is it synthesized and what is it used for. Polimery 2013, 58, 641–649. [Google Scholar] [CrossRef]
- Frey, H.; Haag, R. Dendritic polyglycerol: A new versatile biocompatible material. Rev. Mol. Biotechnol. 2002, 90, 257–267. [Google Scholar] [CrossRef]
- Gosecki, M.; Gadzinowski, M.; Gosecka, M.; Basinska, T.; Slomkowski, S. Polyglycidol, Its Derivatives, and Polyglycidol-Containing Copolymers—synthesis and Medical Applications. Polymers 2016, 8, 227. [Google Scholar] [CrossRef]
- Sunder, A.; Hanselmann, R.; Frey, H.; Mulhaupt, R. Controlled synthesis of hyperbranched polyglycerols by ring-opening multibranching polymerization. Macromolecules 1999, 32, 4240–4246. [Google Scholar] [CrossRef]
- Rokicki, G.; Rakoczy, P.; Parzuchowski, P.; Sobiecki, M. Hyperbranched aliphatic polyethers obtained from environmentally benign monomer: Glycerol carbonate. Green Chem. 2005, 7, 529–539. [Google Scholar] [CrossRef]
- Parzuchowski, P.G.; Gregorowicz, J.; Fraś, Z.; Wawrzyńska, E.P.; Brudzyńska, E.; Rokicki, G. Hyperbranched poly (ether-siloxane) amphiphiles of surprisingly high solubility in supercritical carbon dioxide. J. Supercrit. Fluids 2014, 95, 222–227. [Google Scholar] [CrossRef]
- Gregorowicz, J.; Fras, Z.; Parzuchowski, P.; Rokicki, G.; Kusznerczuk, M.; Dziewulski, S. Phase behaviour of hyperbranched polyesters and polyethers with modified terminal OH groups in supercritical solvents. J. Supercrit. Fluids 2010, 55, 786–796. [Google Scholar] [CrossRef]
- Parzuchowski, P.G.; Stefańska, M.; Świderska, A.; Roguszewska, M.; Zybert, M. Hyperbranched polyglycerols containing amine groups—Synthesis, characterization and carbon dioxide capture. J. CO2 Util. 2018, 27, 145–160. [Google Scholar] [CrossRef]
- Zheng, Y.C.; Li, S.P.; Weng, Z.L.; Gao, C. Hyperbranched polymers: Advances from synthesis to applications. Chem. Soc. Rev. 2015, 44, 4091–4130. [Google Scholar] [CrossRef]
- Galina, H.; Lechowicz, J.B.; Walczak, M. Methods of narrowing the molecular size distribution in hyperbranched polymerization involving AB(2) and B-2 monomers. J. Macromol. Sci. Phys. 2005, 44, 925–940. [Google Scholar] [CrossRef]
- Galina, H.; Walczak, M. A theoretical model of hyperbranched polymerization involving an AB(f) monomer—Part II. The average polymerization degree and dispersity index. Polimery 2005, 50, 713–717. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, Y.; Kayatani, T.; Sugimoto, H.; Suzuki, M.; Inomata, K.; Uehara, A.; Mizutani, Y.; Kitagawa, T.; Maeda, Y. Synthesis, characterization, and reversible oxygenation of Mu-alkoxo diiron(II) complexes with the dinucleating ligand N,N,N’,N’-tetrakis((6-methyl-2-pyridyl)methyl)-1,3-diaminopropan-2-olate. J. Am. Chem. Soc. 1995, 117, 11220–11229. [Google Scholar] [CrossRef]
- Han, L.; Choi, H.-J.; Choi, S.-J.; Liu, B.; Park, D.-W. Ionic liquids containing carboxyl acid moieties grafted onto silica: Synthesis and application as heterogeneous catalysts for cycloaddition reactions of epoxide and carbon dioxide. Green Chem. 2011, 13, 1023. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer | TMP/G/P * | TMP | G.* | Pht.* | K | THF | Yield | Yield | |
|---|---|---|---|---|---|---|---|---|---|
| Molar Ratio | g | g | mol % | g | g | mL | g | % | |
| 1a | 1/7.5/2.5 | 6.02 | 24.79 | 22.75 | 22.72 | 0.71 | 125 | 45.5 | 84 |
| 1b | 1/37.5/12.5 | 6.00 | 124.2 | 25.0 | 113.6 | 0.58 | 240 | 232.2 | 95 |
| Polymer | OH groups | 1 | NaH | All. Bromide* | DMF | Yield | Yield |
|---|---|---|---|---|---|---|---|
| mmol | g | g | g | ml | g | % | |
| 2a | 266.0 | 30.34 | 13.0 | 38.8 | 450 | 28.54 | 70 |
| 2b | 225.4 | 30.34 | 10.88 | 32.65 | 450 | 33.65 | 86 |
| Polymer | Allyl groups | 2 | 1,1,1,3,5,5,5-Heptamethyltrisiloxane | Toluene | Yield | Yield |
|---|---|---|---|---|---|---|
| mmol | g | g | mL | g | % | |
| 3a | 118.1 | 18.2 | 31.6 | 300 | 26.44 | 59 |
| 3b | 192.1 | 33.55 | 59.1 | 550 | 58.44 | 77 |
| Polymer | Phthalimide Groups | 3 | 65% Hydrazine Hydrate w.s.* | THF | Yield | Yield |
|---|---|---|---|---|---|---|
| mmol | g | g | mL | g | % | |
| 4a | 29 | 46.3 | 1.16 | 150 | 41.96 | 99 |
| 4b | 37 | 48.0 | 5.1 | 250 | 41.22 | 96 |
| Polymer | Amine Groups | 4 | K2CO3 | MeI | Chloroform | Yield | Yield |
|---|---|---|---|---|---|---|---|
| mmol | g | g | g | mL | g | % | |
| 5a | 16 | 23.4 | 12.1 | 4.4 | 450 | 23.3 | 93 |
| 5b | 9 | 10.6 | 6.3 | 2.5 | 300 | 10.5 | 91 |
| 6 | 75 | 24 (A-HBPG) | 52.1 | 20.8 | 250 (methanol) | 22.5 | 75 |
| Polymer | TMP/G/P * Theoret. Molar Ratio | Theoretical Mol. Mass g/mol | Theoret. % N | Elem. Analysis %N | TMP/G/P * NMR Molar Ratio | NMR Mol. Mass g/mol | NMR %N |
|---|---|---|---|---|---|---|---|
| 1a | 1/7.5/2.5 | 1200 | 2.92 | 2.67 | 1/7.4/2.48 | 1190 | 2.92 |
| 2a | 1/7.5/2.5 | 1620 | 2.16 | 1.65 | 1/7.2/2 | 1490 | 1.88 |
| 3a | 1/7.5/2.5 | 3950 | 0.88 | 0.69 | 1/7.2/1.75 | 3680 | 0.67 |
| 4a | 1/7.5/2.5 | 3630 | 0.96 | 0.85 | 1/7.3/2.0 | 3500 | 0.80 |
| 5a | 1/7.5/2.5 | 3890 | 0.92 | 0.46 (Cl−) | 1/7.3/2.0 | 3660 | 0.76 |
| 1b | 1/37.5/12.5 | 5450 | 3.21 | - | -** | -** | -** |
| 2b | 1/37.5/12.5 | 7070 | 2.47 | - | -** | -** | -** |
| 3b | 1/37.5/12.5 | 16,080 | 1.09 | 0.87 | -** | -** | -** |
| 4b | 1/37.5/12.5 | 14,460 | 1.11 | 1.04 | -** | -** | -** |
| 5b | 1/37.5/12.5 | 15,750 | 1.06 | 0.71 (I−) | -** | ||
| 5b | 1/37.5/12.5 | 1.09 | 0.83 (Br−) | -** | -** | -** | |
| 5b | 1/37.5/12.5 | - | 0.74 (uns.) | -** | |||
| 6 | 1/7.1/2.7 | 1070 | 3.53 | 2.51 (Cl−) | 1/7.1/2.7 | 924 | 2.65 |
| Catalyst | Ethylene Oxide | Carbon Dioxide | CO2/EO | N/EO | Yield | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cat./anion | [g] | [%wt. N] | [mmol N] | [g] | [mmol] | [g] | [mmol] | [mol/mol] | [mmol/mol] | [%] | |
| 1 | 5a/Cl | 0.63 | 0.46 | 0.21 | 6.0 | 150 | 7.4 | 168.2 | 1.1 | 1.4 | 8 |
| 2 | 5a/Cl | 1.26 | 0.46 | 0.41 | 5.1 | 127.5 | 5.8 | 131.8 | 1.0 | 3.2 | 39 |
| 3 | 5a/ClR1 | 2.54 | 0.46 | 0.83 | 5.1 | 127.5 | 5.5 | 125.0 | 1.0 | 6.5 | 61 |
| 4 | 5a/ClR2 | 2.54 | 0.46 | 0.83 | 5.8 | 145 | 5.9 | 134.1 | 0.9 | 5.8 | 59 |
| 5 | 5a/ClR3 | 2.54 | 0.46 | 0.83 | 5.0 | 125 | 5.7 | 129.5 | 1.0 | 6.7 | 57 |
| 6 | 5a/ClR4 | 2.53 | 0.46 | 0.83 | 4.7 | 117.5 | 5.7 | 129.5 | 1.1 | 7.1 | 57 |
| 7 | 5a/ClR5 | 2.54 | 0.46 | 0.83 | 4.9 | 122.5 | 5.8 | 131.8 | 1.1 | 6.8 | 57 |
| 8 | 5b/uns. | 0.13 | 0.74 | 0.07 | 5.5 | 137.5 | 5.8 | 131.8 | 1.0 | 0.5 | 18 |
| 9 | 5b/uns. | 1.34 | 0.74 | 0.71 | 4.3 | 107.5 | 4.5 | 102.3 | 1.0 | 6.6 | 65 |
| 10 | 5b/uns. | 1.58 | 0.74 | 0.84 | 4.3 | 107.5 | 5.9 | 134.1 | 1.2 | 7.8 | 69 |
| 11 | 5b/I | 0.13 | 0.71 | 0.06 | 4.9 | 122.5 | 4.8 | 109.1 | 0.9 | 0.5 | 34 |
| 12 | 5b/I | 1.00 | 0.71 | 0.51 | 4.9 | 122.5 | 5.2 | 118.2 | 1.0 | 4.2 | 52 |
| 13 | 5b/IC1 | 1.65 | 0.71 | 0.83 | 4.7 | 117.5 | 5.7 | 129.5 | 1.1 | 7.1 | 73 |
| 14 | 5b/I | 2.00 | 0.71 | 1.01 | 4.5 | 112.5 | 4.6 | 104.5 | 0.9 | 9.0 | 81 |
| 15 | 5b/I | 2.55 | 0.71 | 1.29 | 4.3 | 107.5 | 4.7 | 106.8 | 1.0 | 12.0 | 83 |
| 16 | 5b/I | 3.00 | 0.71 | 1.52 | 3.4 | 85 | 4.8 | 109.1 | 1.3 | 17.9 | 93 |
| 17 | 5b/IC2 | 1.59 | 0.71 | 0.81 | 4.8 | 120 | 6.6 | 150.0 | 1.3 | 6.7 | 53 |
| 18 | 5b/IC3 | 1.52 | 0.71 | 0.77 | 4.1 | 102.5 | 5.9 | 134.1 | 1.3 | 7.5 | 52 |
| 19 | NaI | 0.13 | - | 0.831 | 3.4 | 85 | 4.1 | 93.2 | 1.1 | 9.8 | 47 |
| 20 | 5b/Br | 1.40 | 0.83 | 0.83 | 6.2 | 155 | 6.8 | 154.5 | 1.0 | 5.4 | 82 |
| 21 | 5b/IP | 1.00 | 0.71 | 0.51 | 6.4 | 160 | 5.2 | 118.2 | 0.7 | 3.2 | 50 |
| 22 | 6/Cl | 0.04 | 2.51 | 0.07 | 6 | 150 | 7.3 | 165.9 | 1.1 | 0.5 | 32 |
| 23 | 6/Cl | 0.08 | 2.51 | 0.14 | 5 | 125 | 5.6 | 127.3 | 1.0 | 1.1 | 64 |
| 24 | 6/Cl | 0.16 | 2.51 | 0.29 | 5 | 125 | 6.2 | 140.9 | 1.1 | 2.3 | 96 |
| 25 | 6/ClR6 | 0.34 | 2.51 | 0.61 | 5.2 | 130 | 5.7 | 129.5 | 1.0 | 4.7 | 96 |
| 26 | 6/ClR7 | 0.34 | 2.51 | 0.61 | 5 | 125 | 6.2 | 140.9 | 1.1 | 4.9 | 95 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parzuchowski, P.G.; Świderska, A.; Roguszewska, M.; Rolińska, K.; Wołosz, D.; Mamiński, M. Hyperbranched Poly(ether-siloxane)s Containing Ammonium Groups: Synthesis, Characterization and Catalytic Activity. Polymers 2020, 12, 856. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12040856
Parzuchowski PG, Świderska A, Roguszewska M, Rolińska K, Wołosz D, Mamiński M. Hyperbranched Poly(ether-siloxane)s Containing Ammonium Groups: Synthesis, Characterization and Catalytic Activity. Polymers. 2020; 12(4):856. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12040856
Chicago/Turabian StyleParzuchowski, Paweł G., Aleksandra Świderska, Marlena Roguszewska, Karolina Rolińska, Dominik Wołosz, and Mariusz Mamiński. 2020. "Hyperbranched Poly(ether-siloxane)s Containing Ammonium Groups: Synthesis, Characterization and Catalytic Activity" Polymers 12, no. 4: 856. https://0-doi-org.brum.beds.ac.uk/10.3390/polym12040856