Microwave-Assisted Fabrication of Mesoporous Silica-Calcium Phosphate Composites for Dental Application

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Synthesis of Mesoporous Silica SBA-15
2.2. Preparation of Coating Solutions
2.3. Preparation of SBA-15-CaP Composites
2.4. Mineralization Properties Assay of SBA-CaP Composites
2.5. Formulation of Pharmaceutical Forms
2.6. Characterization Methods
2.6.1. FTIR
2.6.2. XRD
2.6.3. SEM-EDX
2.6.4. Stereoscopic Microscopy
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

Appendix B

References
- Xu, X.; He, L.; Zhu, B.; Li, J.; Li, J. Advances in polymeric materials for dental applications. Polym. Chem. 2017, 8, 807–823. [Google Scholar] [CrossRef]
- Ferracane, J.L.; Stansbury, J.W.; Burke, F.J.T. Self-adhesive resin cements—Chemistry, properties and clinical considerations. J. Oral Rehabil. 2011, 38, 295–314. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Melo, M.; Antonucci, J.; Lin, N.; Lin-Gibson, S.; Bai, Y.; Xu, H. Novel dental cement to combat biofilms and reduce acids for orthodontic applications to avoid enamel demineralization. Materials 2016, 9, 413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Zhang, K.; Zhang, N.; Melo, M.A.S.; Weir, M.D.; Zhou, X.D.; Bai, Y.X.; Reynolds, M.A.; Xu, H.H.K. Developing a new generation of antimicrobial and bioactive dental resins. J. Dent. Res. 2017, 96, 855–863. [Google Scholar] [CrossRef] [PubMed]
- Encalada-Alayola, J.J.; Veranes-Pantoja, Y.; Uribe-Calderón, J.A.; Cauich-Rodríguez, J.V.; Cervantes-Uc, J.M. Effect of type and concentration of nanoclay on the mechanical and physicochemical properties of Bis-GMA/TTEGDMA dental resins. Polymers 2020, 12, 601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazayeri, H.E.; Lee, S.M.; Kuhn, L.; Fahimipour, F.; Tahriri, M.; Tayebi, L. Polymeric scaffolds for dental pulp tissue engineering: A review. Dent. Mater. 2020, 36, e47–e58. [Google Scholar] [CrossRef] [PubMed]
- Sprio, S.; Campodoni, E.; Sandri, M.; Preti, L.; Keppler, T.; Müller, F.; Pugno, N.; Tampieri, A. A graded Multifunctional Hybrid Scaffold with Superparamagnetic Ability for Periodontal Regeneration. Int. J. Mol. Sci. 2018, 19, 3604. [Google Scholar] [CrossRef] [Green Version]
- Sheikh, Z.; Qureshi, J.; Alshahrani, A.M.; Nassar, H.; Ikeda, Y.; Glogauer, M.; Ganss, B. Collagen based barrier membranes for periodontal guided bone regeneration applications. Odontology 2017, 105, 1–12. [Google Scholar] [CrossRef]
- Pouponneau, P.; Perrey, O.; Brunon, C.; Grossiord, C.; Courtois, N.; Salles, V.; Alves, A. Electrospun bioresorbable membrane eluting chlorhexidine for dental implants. Polymers 2020, 12, 66. [Google Scholar] [CrossRef] [Green Version]
- Fiorillo, L. Chlorhexidine gel use in the oral district: A systematic review. Gels 2019, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Akıncıbay, H.; Şenel, S.; Yetkin Ay, Z. Application of chitosan gel in the treatment of chronic periodontitis. J. Biomed. Mater. Res. Part B Appl. Biomater. 2007, 80, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M. Processing of collagen based biomaterials and the resulting materials properties. Biomed. Eng. Online 2019, 18, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, S.; Jiang, L.; Wu, J.; Su, C.; Huang, C.; Liu, X.; Shao, W. Flexible amoxicillin-grafted bacterial cellulose sponges for wound dressing: In vitro and in vivo evaluation. ACS Appl. Mater. Interfaces 2018, 10, 5862–5870. [Google Scholar] [CrossRef] [PubMed]
- Meireles, A.B.; Corrêa, D.K.; da Silveira, J.V.W.; Millás, A.L.G.; Bittencourt, E.; de Brito-Melo, G.E.A.; González-Torres, L.A. Trends in polymeric electrospun fibers and their use as oral biomaterials. Exp. Biol. Med. 2018, 243, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Scribante, A.; Massironi, S.; Pieraccini, G.; Vallittu, P.; Lassila, L.; Sfondrini, M.F.; Gandini, P. Effects of nanofillers on mechanical properties of fiber-reinforced composites polymerized with light-curing and additional postcuring. J. Appl. Biomater. Funct. Mater. 2015, 13. [Google Scholar] [CrossRef]
- Wang, R.; Habib, E.; Zhu, X.X. Synthesis of wrinkled mesoporous silica and its reinforcing effect for dental resin composites. Dent. Mater. 2017, 33, 1139–1148. [Google Scholar] [CrossRef]
- Canché-Escamilla, G.; Duarte-Aranda, S.; Toledano, M. Synthesis and characterization of hybrid silica/PMMA nanoparticles and their use as filler in dental composites. Mater. Sci. Eng. C 2014, 42, 161–167. [Google Scholar] [CrossRef]
- Samuel, S.P.; Li, S.; Mukherjee, I.; Guo, Y.; Patel, A.C.; Baran, G.; Wei, Y. Mechanical properties of experimental dental composites containing a combination of mesoporous and nonporous spherical silica as fillers. Dent. Mater. 2009, 25, 296–301. [Google Scholar] [CrossRef]
- Martim, G.C.; Kupfer, V.L.; Moisés, M.P.; dos Santos, A.; Buzzetti, P.H.M.; Rinaldi, A.W.; Rubira, A.F.; Girotto, E.M. Physical-chemical properties of dental composites and adhesives containing silane-modified SBA-15. J. Mech. Behav. Biomed. Mater. 2018, 80, 277–284. [Google Scholar] [CrossRef]
- Shin, K.; Acri, T.; Geary, S.; Salem, A.K. Biomimetic mineralization of biomaterials using simulated body fluids for bone tissue engineering and regenerative medicine. Tissue Eng. Part A 2017, 23, 1169–1180. [Google Scholar] [CrossRef]
- Lu, J.; Yu, H.; Chen, C. Biological properties of calcium phosphate biomaterials for bone repair: A review. RSC Adv. 2018, 8, 2015–2033. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Vega, J.M.; Hozumi, A.; Sugimura, H.; Takai, O. Ordered mesoporous silica coatings that induce apatite formation in vitro. Adv. Mater. 2001, 13, 822–825. [Google Scholar] [CrossRef]
- Chiang, Y.C.; Wang, Y.L.; Lin, P.Y.; Chen, Y.Y.; Chien, C.Y.; Lin, H.P.; Lin, C.P. A mesoporous biomaterial for biomimetic crystallization in dentinal tubules without impairing the bonding of a self-etch resin to dentin. J. Formos. Med. Assoc. 2016, 115, 455–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, Y.C.; Lin, H.P.; Chang, H.H.; Cheng, Y.W.; Tang, H.Y.; Yen, W.C.; Lin, P.Y.; Chang, K.-W.; Lin, C.P. A mesoporous silica biomaterial for dental biomimetic crystallization. ACS Nano 2014, 8, 12502–12513. [Google Scholar] [CrossRef]
- Canto, F.M.T.; Alexandria, A.K.; Vieira, T.I.; Justino, I.B.D.S.; Cabral, L.M.; Silva, R.F.d.; Maia, L.C. Comparative effect of calcium mesoporous silica versus calcium and/or fluoride products against dental erosion. Braz. Dent. J. 2020, 31, 164–170. [Google Scholar] [CrossRef]
- Canto, F.M.T.; Alexandria, A.K.; Justino, I.B.d.S.; Rocha, G.M.; Cabral, L.M.; Ferreira, R.d.S.; Pithon, M.M.; Maia, L.C. The use of a new calcium mesoporous silica nanoparticle versus calcium and/or fluoride products in reducing the progression of dental erosion. J. Appl. Oral Sci. 2020, 28. [Google Scholar] [CrossRef]
- Szewczyk, A.; Prokopowicz, M.; Sawicki, W.; Majda, D.; Walker, G. Aminopropyl-functionalized mesoporous silica SBA-15 as drug carrier for cefazolin: Adsorption profiles, release studies, and mineralization potential. Microporous Mesoporous Mater. 2019, 274, 113–126. [Google Scholar] [CrossRef]
- Izquierdo-Barba, I.; Ruiz-González, L.; Doadrio, J.C.; González-Calbet, J.M.; Vallet-Regí, M. Tissue regeneration: A new property of mesoporous materials. Solid State Sci. 2005, 7, 983–989. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, X.; He, C. Mesoporous silica nanoparticles for tissue-engineering applications. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2019, 11. [Google Scholar] [CrossRef]
- Hertz, A.; Bruce, I.J. Inorganic materials for bone repair or replacement applications. Nanomedicine 2007, 2, 899–918. [Google Scholar] [CrossRef]
- Shuai, C.; Xu, Y.; Feng, P.; Xu, L.; Peng, S.; Deng, Y. Co-enhance bioactive of polymer scaffold with mesoporous silica and nano-hydroxyapatite. J. Biomater. Sci. Polym. Ed. 2019, 30, 1097–1113. [Google Scholar] [CrossRef] [PubMed]
- Szcześ, A.; Hołysz, L.; Chibowski, E. Synthesis of hydroxyapatite for biomedical applications. Adv. Colloid Interface Sci. 2017, 249, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Pajor, K.; Pajchel, L.; Kolmas, J. Hydroxyapatite and fluorapatite in conservative dentistry and oral implantology—A review. Materials 2019, 12, 2683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, L.; Zhang, H.; Ren, A.; Li, Y.; Fu, G.; Cannon, R.D.; Ji, P.; Wu, X.; Yang, S. Nano-hydroxyapatite mineralized silk fibroin porous scaffold for tooth extraction site preservation. Dent. Mater. 2019, 35, 1397–1407. [Google Scholar] [CrossRef]
- Nosrat, A.; Kolahdouzan, A.; Khatibi, A.H.; Verma, P.; Jamshidi, D.; Nevins, A.J.; Torabinejad, M. Clinical, radiographic, and histologic outcome of regenerative endodontic treatment in human teeth using a novel collagen-hydroxyapatite scaffold. J. Endod. 2019, 45, 136–143. [Google Scholar] [CrossRef]
- Scribante, A.; Dermenaki Farahani, M.R.; Marino, G.; Matera, C.; Rodriguez y Baena, R.; Lanteri, V.; Butera, A. Biomimetic effect of nano-hydroxyapatite in demineralized enamel before orthodontic bonding of brackets and attachments: Visual, adhesion strength, and hardness in in vitro tests. Biomed Res. Int. 2020, 2020, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Van den Vreken, N.M.F.F.; De Canck, E.; Ide, M.; Lamote, K.; Van Der Voort, P.; Verbeeck, R.M.H.H. Calcium phosphate cements modified with pore expanded SBA-15 materials. J. Mater. Chem. 2012, 22, 14502–14509. [Google Scholar] [CrossRef]
- Qian, X.F.; Kamegawa, T.; Mori, K.; Li, H.X.; Yamashita, H. Calcium phosphate coatings incorporated in mesoporous TiO2/SBA-15 by a facile inner-pore sol–gel process toward enhanced adsorption-photocatalysis performances. J. Phys. Chem. C 2013, 117, 19544–19551. [Google Scholar] [CrossRef]
- Díaz, A.; López, T.; Manjarrez, J.; Basaldella, E.; Martínez-Blanes, J.M.; Odriozola, J.A. Growth of hydroxyapatite in a biocompatible mesoporous ordered silica. Acta Biomater. 2006, 2, 173–179. [Google Scholar] [CrossRef]
- Hassan, M.N.; Mahmoud, M.M.; El-Fattah, A.A.; Kandil, S. Microwave-assisted preparation of nano-hydroxyapatite for bone substitutes. Ceram. Int. 2016, 42, 3725–3744. [Google Scholar] [CrossRef]
- Liu, J.; Li, K.; Wang, H.; Zhu, M.; Yan, H. Rapid formation of hydroxyapatite nanostructures by microwave irradiation. Chem. Phys. Lett. 2004, 396, 429–432. [Google Scholar] [CrossRef]
- Cao, J.M.; Feng, J.; Deng, S.G.; Chang, X.; Wang, J.; Liu, J.S.; Lu, P.; Lu, H.X.; Zheng, M.B.; Zhang, F.; et al. Microwave-assisted solid-state synthesis of hydroxyapatite nanorods at room temperature. J. Mater. Sci. 2005, 40, 6311–6313. [Google Scholar] [CrossRef]
- Stanislavov, A.S.; Sukhodub, L.F.; Sukhodub, L.B.; Kuznetsov, V.N.; Bychkov, K.L.; Kravchenko, M.I. Structural features of hydroxyapatite and carbonated apatite formed under the influence of ultrasound and microwave radiation and their effect on the bioactivity of the nanomaterials. Ultrason. Sonochem. 2018, 42, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Attik, N.; Hallay, F.; Bois, L.; Brioude, A.; Grosgogeat, B.; Colon, P. Mesoporous silica fillers and resin composition effect on dental composites cytocompatibility. Dent. Mater. 2017, 33, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Surintanasarn, A.; Siralertmukul, K.; Thamrongananskul, N. Synthesized mesoporous silica and calcium aluminate cement fillers increased the fluoride recharge and lactic acid neutralizing ability of a resin-based pit and fissure sealant. Dent. Mater. J. 2017, 36, 706–713. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.F.; Wu, R.; Fan, Y.; Liao, S.; Wang, Y.; Wen, Z.T.; Xu, X. Antibacterial dental composites with chlorhexidine and mesoporous silica. J. Dent. Res. 2014, 93, 1283–1289. [Google Scholar] [CrossRef]
- Wang, Y.; Hua, H.; Li, W.; Wang, R.; Jiang, X.; Zhu, M. Strong antibacterial dental resin composites containing cellulose nanocrystal/zinc oxide nanohybrids. J. Dent. 2019, 80, 23–29. [Google Scholar] [CrossRef]
- Roh, J.; Han, M.; Kim, K.N.; Kim, K.M. The in vitro and in vivo effects of a fast-dissolving mucoadhesive bi-layered strip as topical anesthetics. Dent. Mater. J. 2016, 35, 601–605. [Google Scholar] [CrossRef] [Green Version]
- Mai, H.-N.; Kim, D.-Y.; Hyun, D.C.; Park, J.H.; Lee, S.M.; Lee, D.-H. A new antibacterial agent-releasing polydimethylsiloxane coating for polymethyl methacrylate dental restorations. J. Clin. Med. 2019, 8, 1831. [Google Scholar] [CrossRef] [Green Version]
- Zhao, D.; Huo, Q.; Feng, J.; Chmelka, B.F.; Stucky, G.D. Nonionic triblock and star diblock copolymer and oligomeric surfactant syntheses of highly ordered, hydrothermally stable, mesoporous silica structures. J. Am. Chem. Soc. 1998, 120, 6024–6036. [Google Scholar] [CrossRef]
- Chou, Y.F.; Chiou, W.A.; Xu, Y.; Dunn, J.C.Y.; Wu, B.M. The effect of pH on the structural evolution of accelerated biomimetic apatite. Biomaterials 2004, 25, 5323–5331. [Google Scholar] [CrossRef] [PubMed]
- Tas, A.C.; Bhaduri, S.B. Rapid coating of Ti6Al4V at room temperature with a calcium phosphate solution similar to 10× simulated body fluid. J. Mater. Res. 2004, 19, 2742–2749. [Google Scholar] [CrossRef]
- Tolga Demirtaş, T.; Kaynak, G.; Gümüşderelioğlu, M. Bone-like hydroxyapatite precipitated from 10× SBF-like solution by microwave irradiation. Mater. Sci. Eng. C 2015, 49, 713–719. [Google Scholar] [CrossRef] [PubMed]
- Ciobanu, G.; Ilisei, S.; Luca, C.; Carja, G.; Ciobanu, O. The effect of vitamins to hydroxyapatite growth on porous polyurethane substrate. Prog. Org. Coat. 2012, 74, 648–653. [Google Scholar] [CrossRef]
- Kokubo, T.; Takadama, H. How useful is SBF in predicting in vivo bone bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef] [PubMed]
- Skwira, A.; Szewczyk, A.; Prokopowicz, M. The effect of polydimethylsiloxane-ethylcellulose coating blends on the surface characterization and drug release of ciprofloxacin loaded mesoporous silica. Polymers 2019, 11, 1450. [Google Scholar] [CrossRef] [Green Version]
- Skwira, A.; Szewczyk, A.; Konopacka, A.; Górska, M.; Majda, D.; Sądej, R.; Prokopowicz, M. Silica-polymer composites as the novel antibiotic delivery systems for bone tissue infection. Pharmaceutics 2019, 12, 28. [Google Scholar] [CrossRef] [Green Version]
- Prokopowicz, M.; Żeglinski, J.; Szewczyk, A.; Skwira, A.; Walker, G. Surface-activated fibre-like SBA-15 as drug carriers for bone diseases. AAPS PharmSciTech 2019, 20, 17. [Google Scholar] [CrossRef]
- Sekar, C.; Kanchana, P.; Nithyaselvi, R.; Girija, E.K. Effect of fluorides (KF and NaF) on the growth of dicalcium phosphate dihydrate (DCPD) crystal. Mater. Chem. Phys. 2009, 115, 21–27. [Google Scholar] [CrossRef]
- Dorozhkin, S.V. Calcium orthophosphates (CaPO4): Occurrence and properties. Prog. Biomater. 2016, 5, 9–70. [Google Scholar] [CrossRef] [Green Version]
- Rabadjieva, D.; Sezanova, K.; Gergulova, R.; Titorenkova, R.; Tepavitcharova, S. Precipitation and phase transformation of dicalcium phosphate dihydrate in electrolyte solutions of simulated body fluids: Thermodynamic modeling and kinetic studies. J. Biomed. Mater. Res. Part A 2020, 108, 1607–1616. [Google Scholar] [CrossRef]
- Montastruc, L.; Azzaro-Pantel, C.; Biscans, B.; Cabassud, M.; Domenech, S. A thermochemical approach for calcium phosphate precipitation modeling in a pellet reactor. Chem. Eng. J. 2003, 94, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Leng, Y. Theoretical analysis of calcium phosphate precipitation in simulated body fluid. Biomaterials 2005, 26, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Casciani, F.; Condrate, R.A. The vibrational spectra of brushite, CaHPO4·2H2O. Spectrosc. Lett. 1979, 12, 699–713. [Google Scholar] [CrossRef]
- Prokopowicz, M.; Żegliński, J.; Gandhi, A.; Sawicki, W.; Tofail, S.A.M. Bioactive silica-based drug delivery systems containing doxorubicin hydrochloride: In vitro studies. Colloids Surf. B Biointerfaces 2012, 93, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Kaynak Bayrak, G.; Demirtaş, T.T.; Gümüşderelioğlu, M. Microwave-induced biomimetic approach for hydroxyapatite coatings of chitosan scaffolds. Carbohydr. Polym. 2017, 157, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Ghorbani, F.; Zamanian, A.; Behnamghader, A.; Joupari, M.D. Microwave-induced rapid formation of biomimetic hydroxyapatite coating on gelatin-siloxane hybrid microspheres in 10×-SBF solution. e-Polymers 2018, 18, 247–255. [Google Scholar] [CrossRef]
- Vueva, Y.; Connell, L.S.; Chayanun, S.; Wang, D.; McPhail, D.S.; Romer, F.; Hanna, J.V.; Jones, J.R. Silica/alginate hybrid biomaterials and assessment of their covalent coupling. Appl. Mater. Today 2018, 11, 1–12. [Google Scholar] [CrossRef]
- Dorozhkin, S. Self-setting calcium orthophosphate formulations. J. Funct. Biomater. 2013, 4, 209–311. [Google Scholar] [CrossRef] [Green Version]
- Tas, A.C. X-ray-amorphous calcium phosphate (ACP) synthesis in a simple biomineralization medium. J. Mater. Chem. B 2013, 1, 4511. [Google Scholar] [CrossRef]
- Chavan, P.N.; Bahir, M.M.; Mene, R.U.; Mahabole, M.P.; Khairnar, R.S. Study of nanobiomaterial hydroxyapatite in simulated body fluid: Formation and growth of apatite. Mater. Sci. Eng. B 2010, 168, 224–230. [Google Scholar] [CrossRef]
- Xiong, Y.; Li, H.; Zhou, C.; Yang, X.; Song, Y.; Qing, Y.; Yan, Y. Evaluation of biomechanical strength, stability, bioactivity, and in vivo biocompatibility of a novel calcium deficient hydroxyapatite/poly(amino acid) composite cervical vertebra cage. J. Biomater. Sci. Polym. Ed. 2014, 25, 1842–1855. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, Y.; Wang, M.; Chen, X.; Xiao, Y.; Zhang, X. Stabilization of Ca-deficient hydroxyapatite in biphasic calcium phosphate ceramics by adding alginate to enhance their biological performances. J. Mater. Chem. B 2018, 6, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.W.; Zi, Y.P.; Xu, W.; Zhou, R.; Cai, Z.Y.; Zheng, W.J.; Chen, F.; Qian, Q.R. Electrospinning of calcium phosphate-poly(D,L-lactic acid) nanofibers for sustained release of water-soluble drug and fast mineralization. Int. J. Nanomed. 2016, 11, 5087–5097. [Google Scholar] [CrossRef] [Green Version]
- Zhou, R.; Ni, H.J.; Peng, J.H.; Liu, N.; Chen, S.; Shao, J.H.; Fu, Q.W.; Liu, J.J.; Chen, F.; Qian, Q.R. The mineralization, drug release and in vivo bone defect repair properties of calcium phosphates/PLA modified tantalum scaffolds. RSC Adv. 2020, 10, 7708–7717. [Google Scholar] [CrossRef] [Green Version]
- Al-Oweini, R.; El-Rassy, H. Synthesis and characterization by FTIR spectroscopy of silica aerogels prepared using several Si(OR)4 and R″Si(OR′)3 precursors. J. Mol. Struct. 2009, 919, 140–145. [Google Scholar] [CrossRef]
- Szewczyk, A.; Skwira, A.; Konopacka, A.; Sądej, R.; Walker, G.; Prokopowicz, M. Mesoporous silica pellets as bifunctional bone drug delivery system for cefazolin. Int. J. Pharm. 2020, 588, 119718. [Google Scholar] [CrossRef]
- Prokopowicz, M.; Szewczyk, A.; Skwira, A.; Sądej, R.; Walker, G. Biphasic composite of calcium phosphate-based mesoporous silica as a novel bone drug delivery system. Drug Deliv. Transl. Res. 2020, 10, 455–470. [Google Scholar] [CrossRef] [Green Version]
- Hokmabad, V.R.; Davaran, S.; Aghazadeh, M.; Rahbarghazi, R.; Salehi, R.; Ramazani, A. Fabrication and characterization of novel ethyl cellulose-grafted-poly (ɛ-caprolactone)/alginate nanofibrous/macroporous scaffolds incorporated with nano-hydroxyapatite for bone tissue engineering. J. Biomater. Appl. 2019, 33, 1128–1144. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, C.; Chang, J. Ca-Doped mesoporous SiO2/dental resin composites with enhanced mechanical properties, bioactivity and antibacterial properties. J. Mater. Chem. B 2018, 6, 477–486. [Google Scholar] [CrossRef]
- Zhang, W.; Luo, X.; Niu, L.; Yang, H.; Yiu, C.K.Y.; Wang, T.; Zhou, L.; Mao, J.; Huang, C.; Pashley, D.H.; et al. Biomimetic intrafibrillar mineralization of type I collagen with intermediate precursors-loaded mesoporous carriers. Sci. Rep. 2015, 5, 11199. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
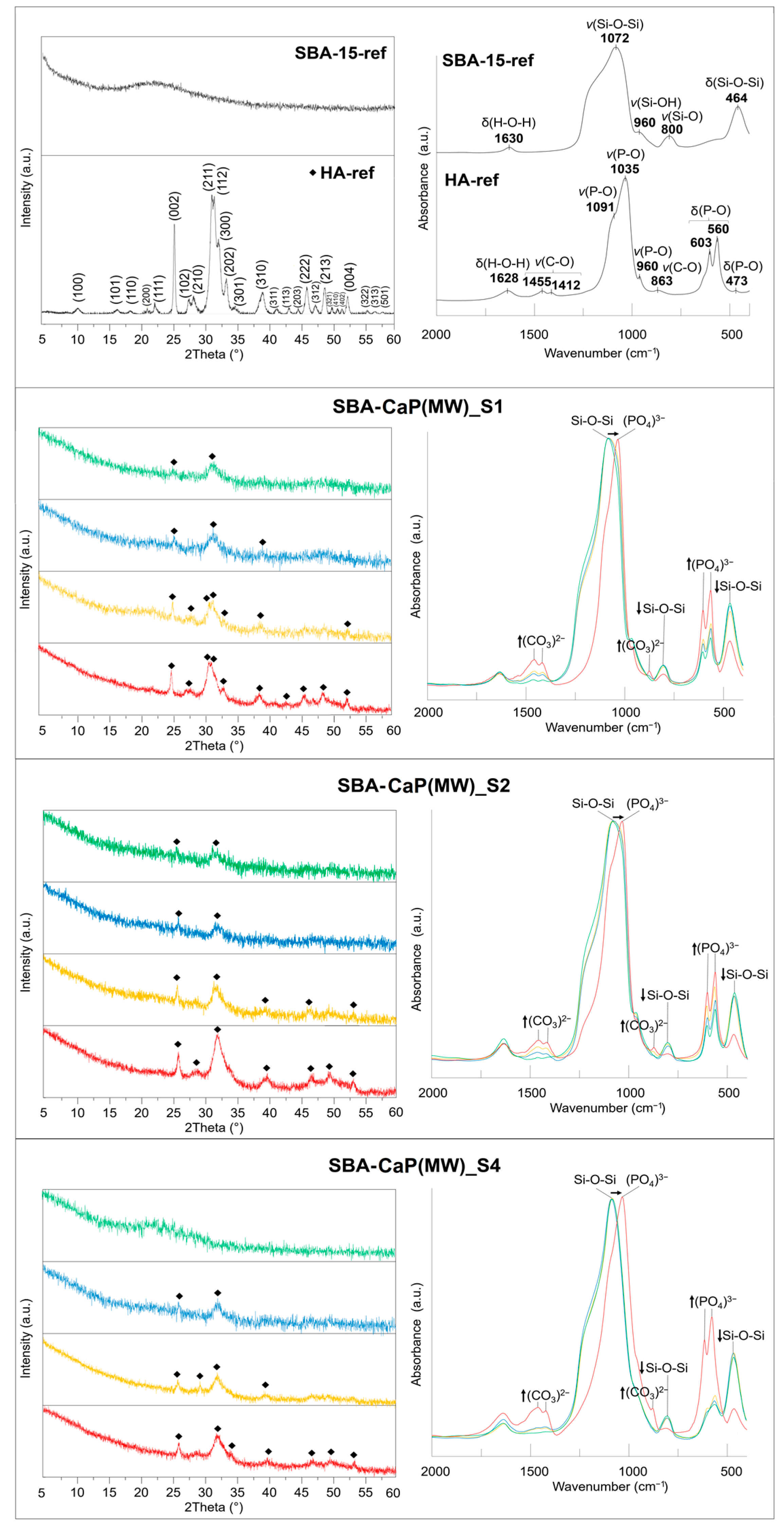{kind=link}
{kind=link}
{kind=link}
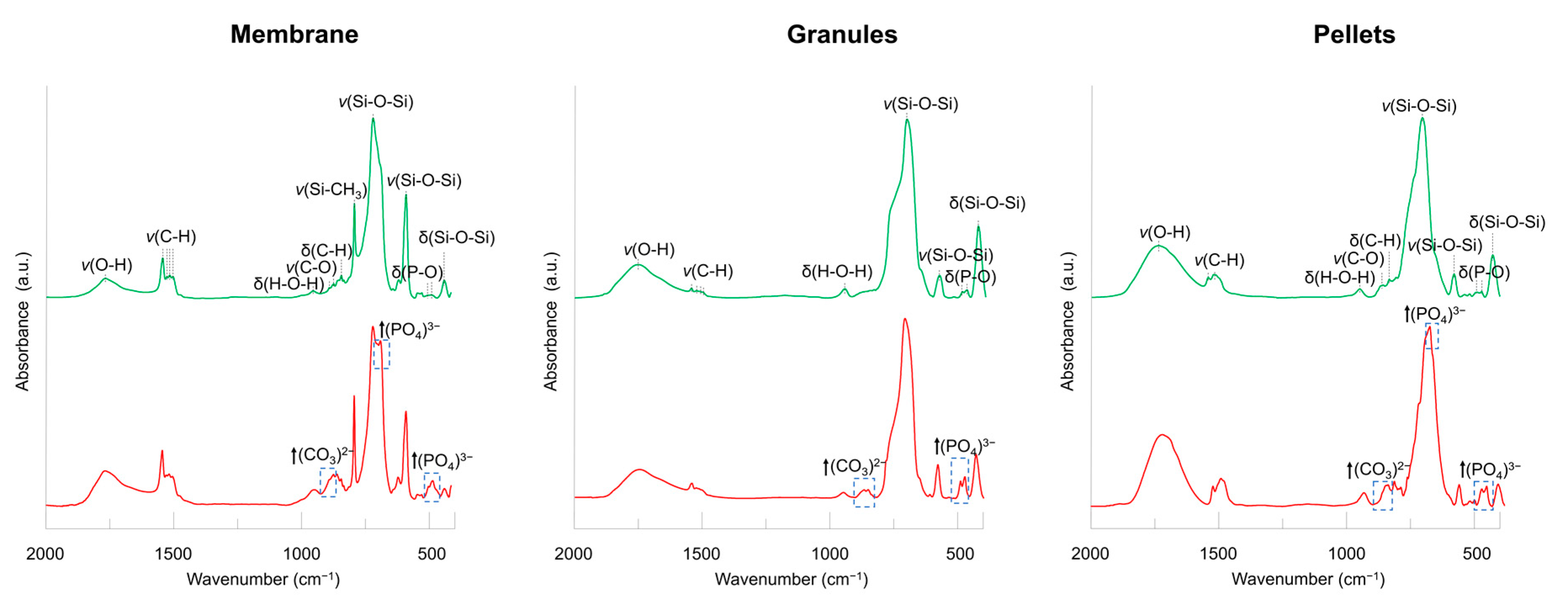{kind=link}
| Solution Number | Ion Concentration (mmol/L) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Na+ | K+ | Mg2+ | Ca2+ | HPO42− | H2PO4− | Cl− | HCO3− | SO42− | |
| S1 a [53] | 1013.6 | 5 | 5 | 25 | - | 3.6 | 1065 | 10 | - |
| S2 a [52] | 1010 | 5 | 5 | 25 | - | 10 | 1065 | 10 | - |
| S3 [54] | 4 | - | - | 5 | - | 2.5 | 10 | 1.5 | - |
| S4 b [51] | 710 | 25 | 7.5 | 12.5 | 5 | - | 740 | 21 | 2.5 |
| S5 c [55] | 142 | 5 | 1.5 | 2.5 | 1 | - | 103 | 27 | 0.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szewczyk, A.; Skwira, A.; Ginter, M.; Tajer, D.; Prokopowicz, M. Microwave-Assisted Fabrication of Mesoporous Silica-Calcium Phosphate Composites for Dental Application. Polymers 2021, 13, 53. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13010053
Szewczyk A, Skwira A, Ginter M, Tajer D, Prokopowicz M. Microwave-Assisted Fabrication of Mesoporous Silica-Calcium Phosphate Composites for Dental Application. Polymers. 2021; 13(1):53. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13010053
Chicago/Turabian StyleSzewczyk, Adrian, Adrianna Skwira, Marta Ginter, Donata Tajer, and Magdalena Prokopowicz. 2021. "Microwave-Assisted Fabrication of Mesoporous Silica-Calcium Phosphate Composites for Dental Application" Polymers 13, no. 1: 53. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13010053