
Phospholipid-Conjugated PEG-b-PCL Copolymers as Precursors of Micellar Vehicles for Amphotericin B

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
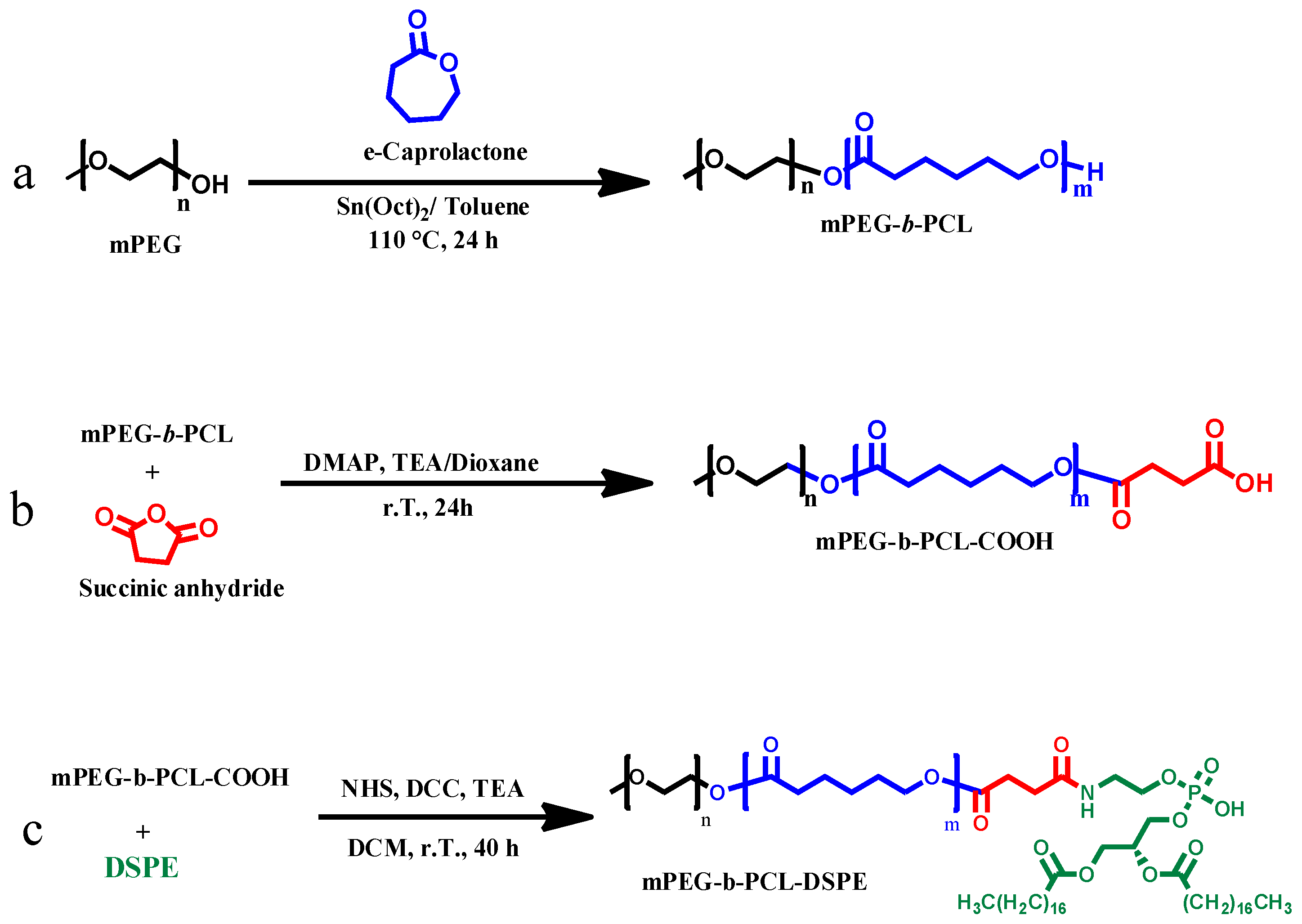
2.1. Synthesis of PEG-b-PCL Copolymers
2.2. Synthesis of mPEG-b-PCL-COOH
2.3. Conjugation of PCL-DSPE Copolymers
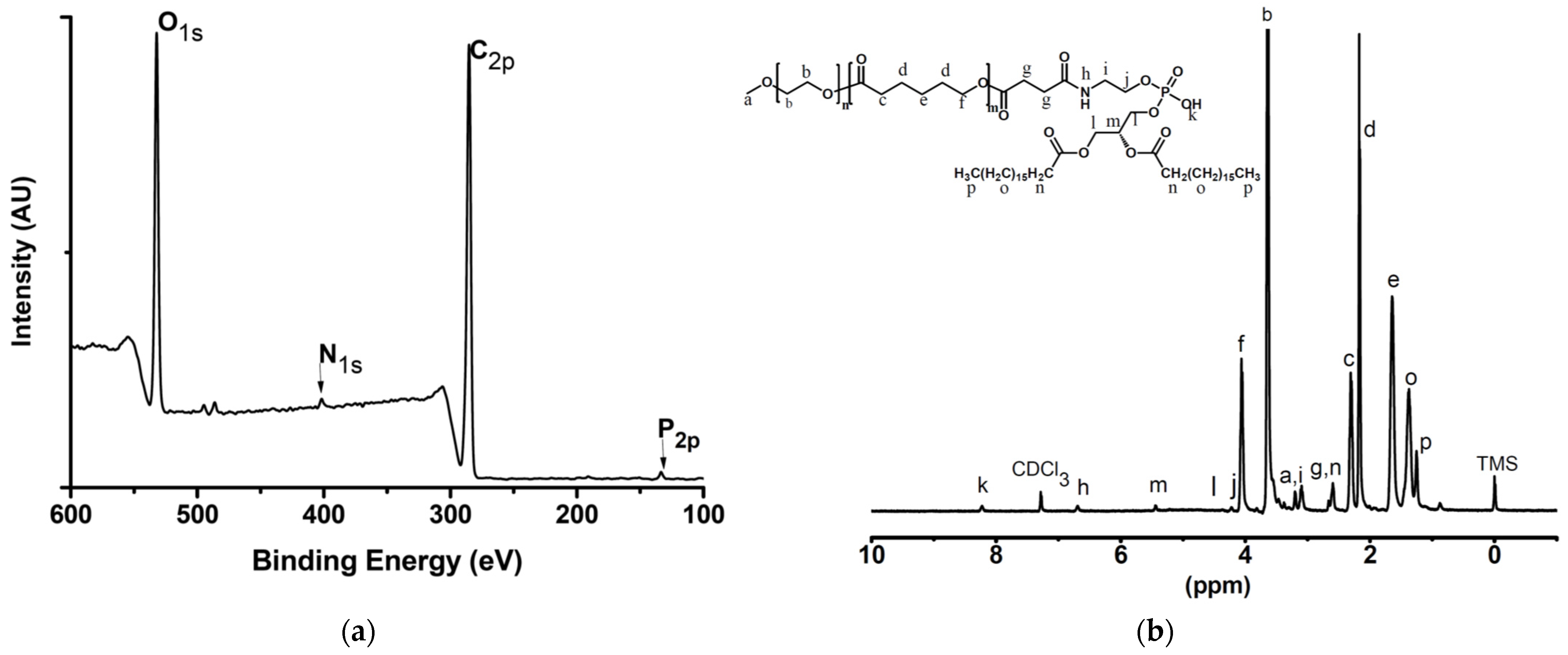
2.4. Characterisation Techniques
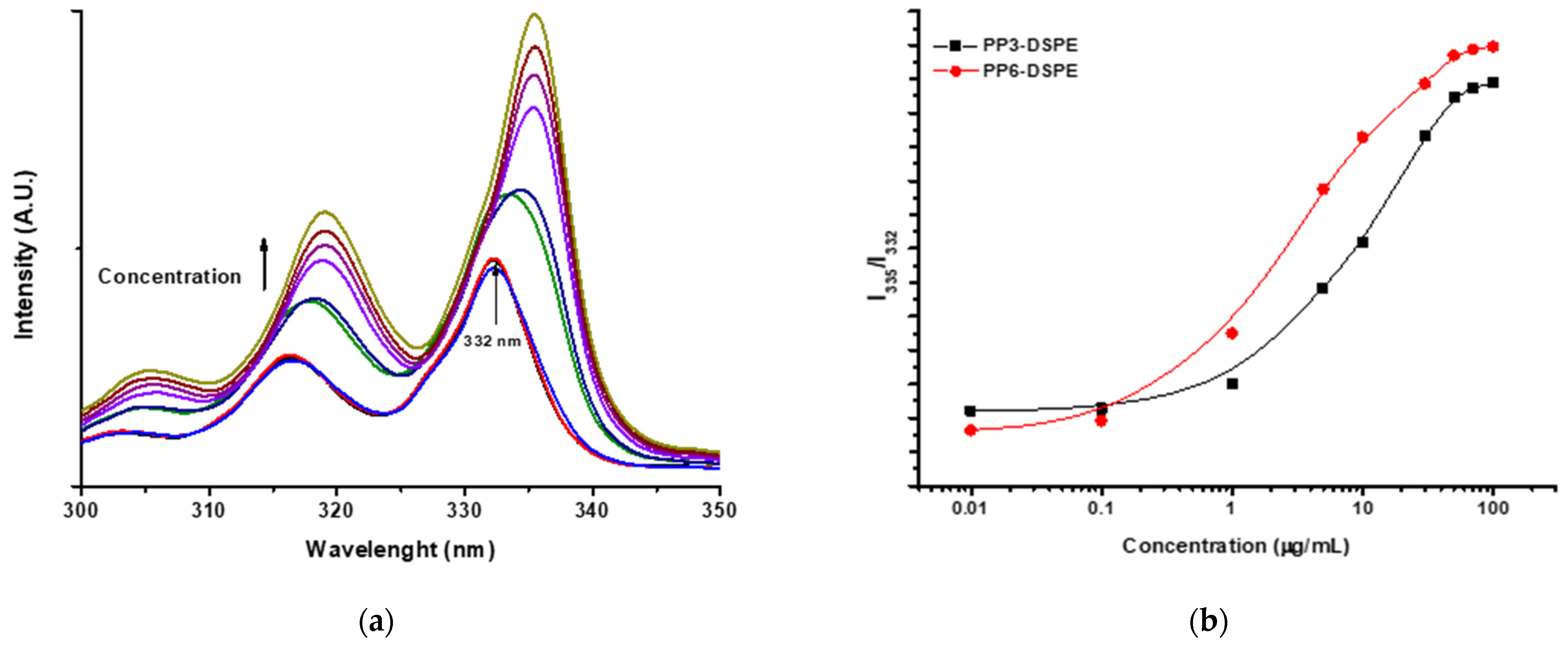
2.5. Critical Micelle Concentration Measurement
2.6. AmB Encapsulation
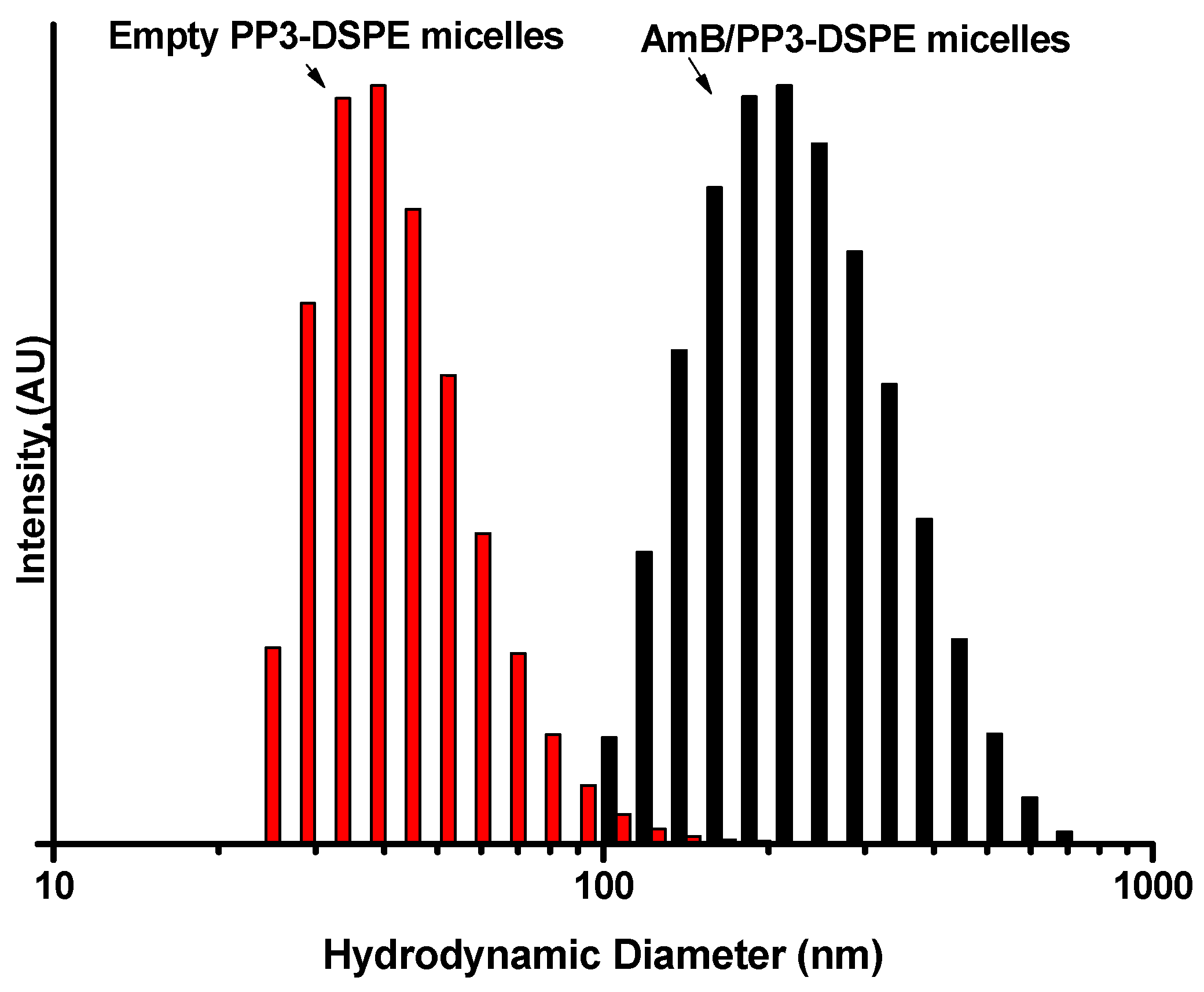
2.7. Hydrodynamic Diameter and ζ-Potential Measurements
2.8. Characterisation of AmB/PMs through DSC
2.9. X-ray Diffraction
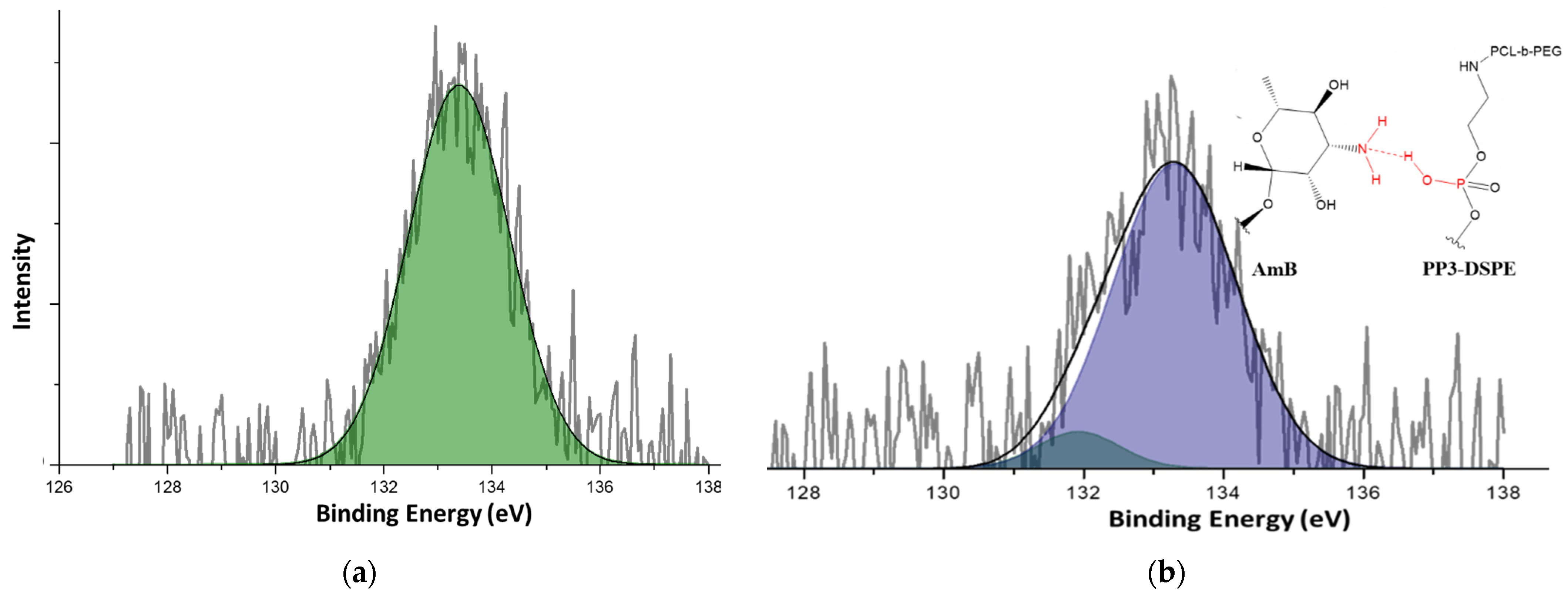
2.10. X-ray-Induced Photoelectron Spectrometry
2.11. AmB Release Assessment
2.12. Aggregation State
2.13. Haemolysis
2.14. Minimum Inhibitory Concentration
3. Results and Discussion
3.1. CMC
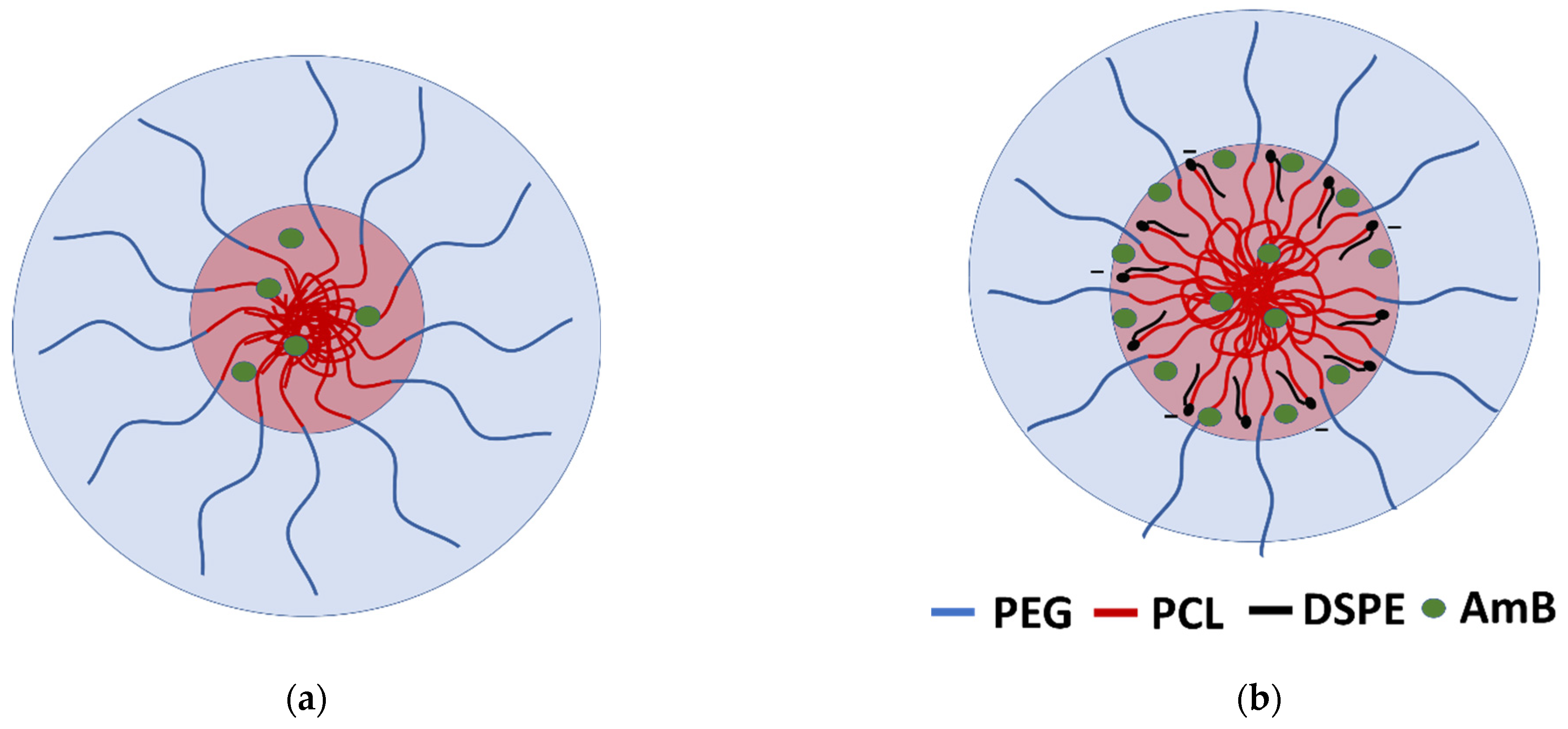
3.2. Encapsulation Capacity
3.3. Particle Size and ζ-Potential Measurement
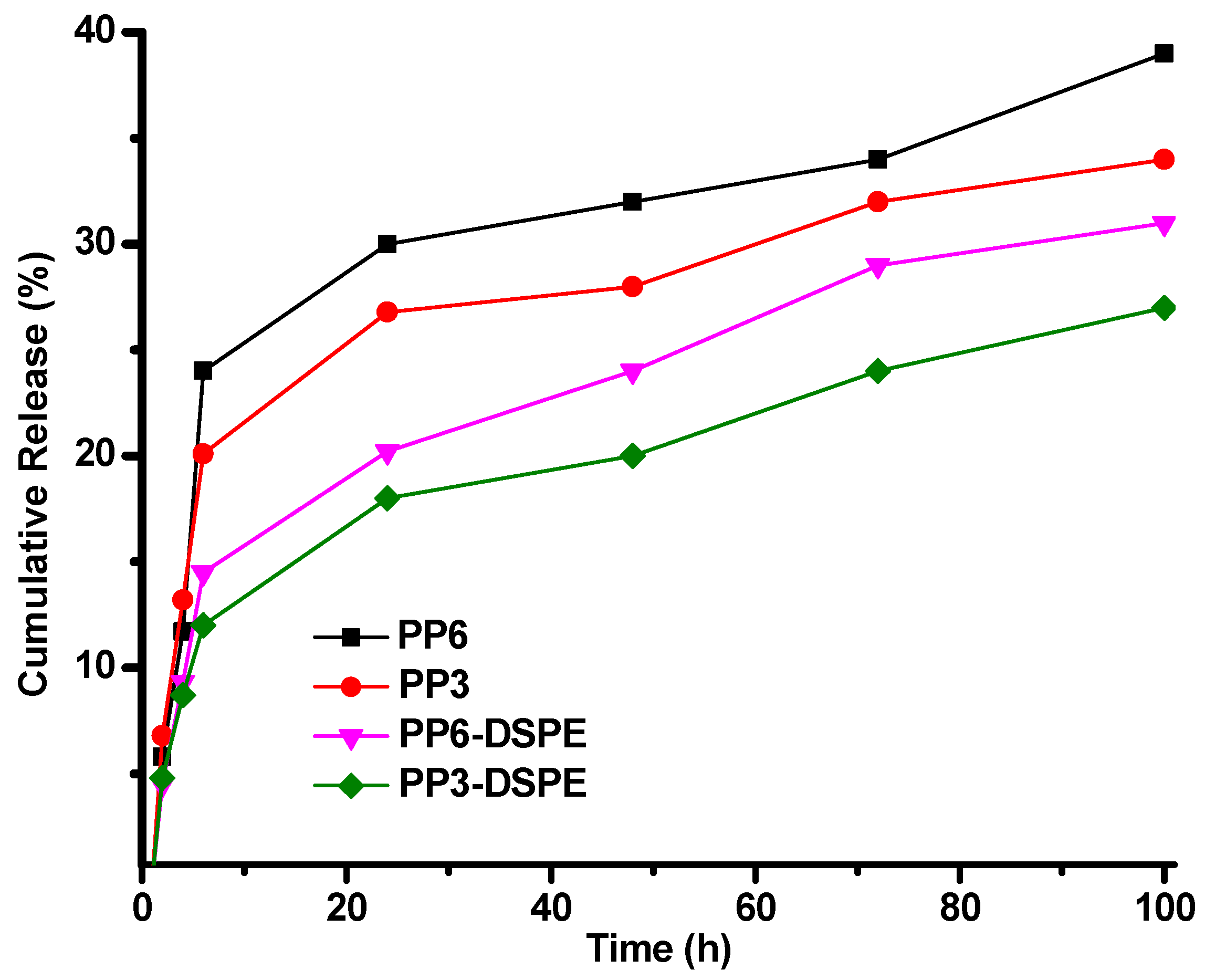
3.4. Release Study
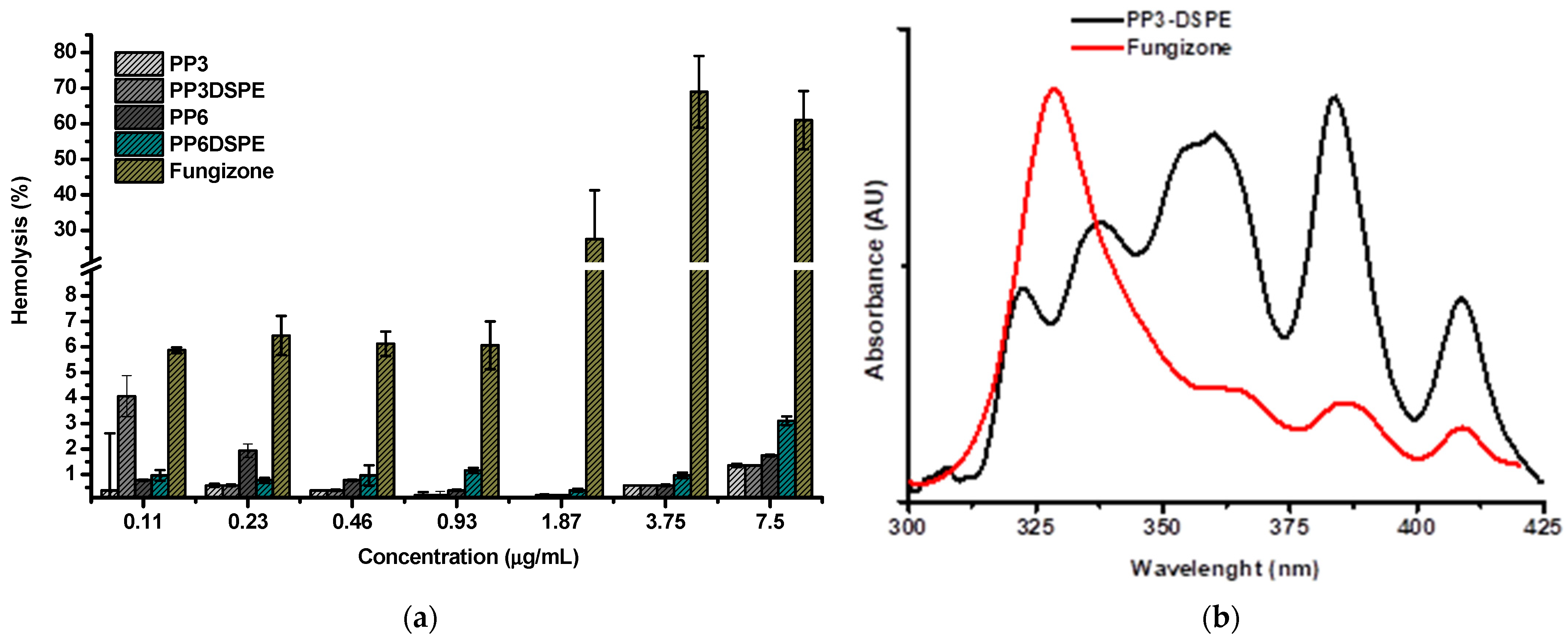
3.5. Haemolysis Behaviour
3.6. MIC
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, P.K.; Gorain, B.; Choudhury, H.; Singh, S.K.; Whadwa, P.; Sahu, S.; Gulati, M.; Kesharwani, P. Macro-phage targeted amphotericin B nanodelivery systems against visceral leishmaniasis. Mater. Sci. Eng. B 2020, 258, 114571. [Google Scholar] [CrossRef]
- Mosimann, V.; Neumayr, A.; Paris, D.H.; Blum, J. Liposomal amphotericin B treatment of Old World cutaneous and mucosal leishmaniasis: A literature review. Acta Trop. 2018, 182, 246–250. [Google Scholar] [CrossRef]
- Saravolatz, L.D.; Ostrosky-Zeichner, L.; Marr, K.A.; Rex, J.H.; Cohen, S.H. Amphotericin B: Time for a New “Gold Standard”. Clin. Infect. Dis. 2003, 37, 415–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarosi, G.A. Amphotericin B: Still the ’gold standard’ for antifungal therapy. Postgrad. Med. 1990, 88, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Calvo, B.; Melo, A.S.; Perozo-Mena, A.; Hernandez, M.; Francisco, E.C.; Hagen, F.; Meis, J.F.; Colombo, A.L. First report of Candida auris in America: Clinical and microbiological aspects of 18 episodes of candidemia. J. Infect. 2016, 73, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Vogelsinger, H.; Weiler, S.; Djanani, A.; Kountchev, J.; Bellmann-Weiler, R.; Wiedermann, C.J.; Bellmann, R. Amphotericin B tissue distribution in autopsy material after treatment with liposomal amphotericin B and amphotericin B colloidal dispersion. J. Antimicrob. Chemother. 2006, 57, 1153–1160. [Google Scholar] [CrossRef]
- Kamiński, D.M. Recent progress in the study of the interactions of amphotericin B with cholesterol and ergosterol in lipid environments. Eur. Biophys. J. 2014, 43, 453–467. [Google Scholar] [CrossRef] [Green Version]
- Torrado, J.J.; Espada, R.; Ballesteros, M.P.; Torrado-Santiago, S. Amphotericin B Formulations and Drug Targeting. J. Pharm. Sci. 2008, 97, 2405–2425. [Google Scholar] [CrossRef]
- Hamill, R.J. Amphotericin B Formulations: A Comparative Review of Efficacy and Toxicity. Drugs 2013, 73, 919–934. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, J.P.B.; Hamid, A.M.R. Amphotericin B deoxycholate versus liposomal amphotericin B: Effects on kidney function. Cochrane Database Syst. Rev. 2015, 11. [Google Scholar] [CrossRef] [Green Version]
- Barrett, J.P.; Vardulaki, K.A.; Conlon, C.; Cooke, J.; Daza-Ramirez, P.; Evans, E.G.V.; Hawkey, P.M.; Herbrecht, R.; Marks, D.I.; Moraleda, J.M.; et al. A systematic review of the antifungal effectiveness and tolerability of ampho-tericin B formulations. Clin. Ther. 2003, 25, 1295–1320. [Google Scholar] [CrossRef]
- Fanos, V.; Cataldi, L. Amphotericin B-Induced Nephrotoxicity: A Review. J. Chemother. 2000, 12, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Jensen, G.; Skenes, C.; Bunch, T.; Weissman, C.; Amirghahari, N.; Satorius, A.; Moynihan, K.; Eley, C. Determination of the rela-tive toxicity of amphotericin B formulations: A red blood cell potassium release assay. Drug Deliv. 1999, 6, 81–88. [Google Scholar] [CrossRef]
- Perlin, D.S.; Rautemaa-Richardson, R.; Alastruey-Izquierdo, A. The global problem of antifungal resistance: Prevalence, mechanisms, and management. Lancet Infect. Dis. 2017, 17, e383–e392. [Google Scholar] [CrossRef]
- Barwicz, J.; Christian, S.; Gruda, I. Effects of the aggregation state of amphotericin B on its toxicity to mice. Antimicrob. Agents Chemother. 1992, 36, 2310–2315. [Google Scholar] [CrossRef] [Green Version]
- Kaur, K.; Kumar, P.; Kush, P. Amphotericin B loaded ethyl cellulose nanoparticles with magnified oral bioavailability for safe and effective treatment of fungal infection. Biomed. Pharm. 2020, 128, 110297. [Google Scholar] [CrossRef]
- Yang, C.; Xue, B.; Song, W.; Kan, B.; Zhang, D.; Yu, H.; Shen, N.; Li, X.; Tang, Z.; Chen, X. Reducing the toxicity of amphotericin B by encapsulation using methoxy poly(ethylene glycol)-b-poly(l-glutamic acid-co-l-phenylalanine). Biomater. Sci. 2018, 6, 2189–2196. [Google Scholar] [CrossRef] [PubMed]
- Adler-Moore, J.P.; Gangneux, J.-P.; Pappas, P.G. Comparison between liposomal formulations of amphotericin B. Med. Mycol. 2016, 54, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhu, X.; Ke, F.; Ye, L.; Chen, E.-Q.; Zhang, A.-Y.; Feng, Z.-G. Preparation and self-assembly of amphiphilic triblock copolymers with polyrotaxane as a middle block and their application as carrier for the controlled release of Amphotericin B. Polymers 2009, 50, 4343–4351. [Google Scholar] [CrossRef]
- Vandermeulen, G.; Rouxhet, L.; Arien, A.; Brewster, M.; Préat, V. Encapsulation of amphotericin B in poly (ethylene gly-col)-block-poly (ɛ-caprolactone- co-trimethylenecarbonate) polymeric micelles. Int. J. Pharm. 2006, 309, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.L.; Kwon, G.S. Relative aggregation state and hemolytic activity of amphotericin B encapsulated by poly(ethylene oxide)-block–poly(N-hexyl-l-aspartamide)-acyl conjugate micelles: Effects of acyl chain length. J. Control. Release 2003, 87, 23–32. [Google Scholar] [CrossRef]
- Lavasanifar, A.; Samuel, J.; Sattari, S.; Kwon, G.S. Block copolymer micelles for the encapsulation and delivery of amphotericin B. Pharm. Res. 2002, 19, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Lavasanifar, A.; Samuel, J.; Kwon, G.S. Micelles self-assembled from poly(ethylene oxide)-block-poly(N-hexyl stearate l-aspartamide) by a solvent evaporation method: Effect on the solubilization and haemolytic activity of amphotericin B. J. Control. Release 2001, 77, 155–160. [Google Scholar] [CrossRef]
- Tang, X.; Dai, J.; Xie, J.; Zhu, Y.; Zhu, M.; Wang, Z.; Xie, C.; Yao, A.; Liu, T.; Wang, X.; et al. Enhanced Antifungal Activity by Ab-Modified Amphotericin B-Loaded Nanoparticles Using a pH-Responsive Block Copol-ymer. Nanoscale Res. Lett. 2015, 10, 256. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, C.; Shin, D.H.; Kwon, G.S. Reformulation of Fungizone by PEG-DSPE Micelles: Deaggregation and Detoxification of Amphotericin B. Pharm. Res. 2016, 33, 2098–2106. [Google Scholar] [CrossRef] [Green Version]
- Sheu, M.-T.; Chen, Y.-C.; Su, C.-Y.; Jhan, H.-J.; Ho, H.-O. Physical characterization and in vivo pharmacokinetic study of self-assembling amphotericin B-loaded lecithin-based mixed polymeric micelles. Int. J. Nanomed. 2015, 10, 7265–7274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villamil, J.C.; Parra-Giraldo, C.M.; Pérez, L.D. Enhancing the performance of PEG-b-PCL copolymers as precursors of micellar vehicles for amphotericin B through its conjugation with cholesterol. Colloids Surf. A Phys. Eng. Asp. 2019, 572, 79–87. [Google Scholar] [CrossRef]
- Rodriguez, Y.J.; Quejada, L.F.; Villamil, J.C.; Baena, Y.; Parra-Giraldo, C.M.; Perez, L.D. Development of amphotericin B micel-lar formulations based on copolymers of poly (ethylene glycol) and poly (ε-caprolactone) conjugated with retinol. Pharma-Ceutics 2020, 12, 196. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Zhu, W.; Liu, N.; Yang, F.; Feng, R. Linolenic acid-modified PEG-PCL micelles for curcumin delivery. Int. J. Pharm. 2014, 471, 312–321. [Google Scholar] [CrossRef]
- Liang, Y.; Deng, X.; Zhang, L.; Peng, X.; Gao, W.; Cao, J.; Gu, Z.; He, B. Terminal modification of polymeric micelles with π-conjugated moieties for efficient anticancer drug delivery. Biomaterials 2015, 71, 1–10. [Google Scholar] [CrossRef]
- Angelova, A.; Angelov, B. Dual and multi-drug delivery nanoparticles towards neuronal survival and synaptic repair. Neural Regen. Res. 2017, 12, 886–889. [Google Scholar] [CrossRef]
- Deng, H.; Liu, J.; Zhao, X.; Zhang, Y.; Liu, J.; Xu, S.; Deng, L.; Dong, A.; Zhang, J. PEG-b-PCL copolymer micelles with the ability of pH-controlled negative-to-positive charge reversal for intracellular delivery of doxorubicin. Biomacromolecules 2014, 15, 4281–4292. [Google Scholar] [CrossRef]
- Yoon, K.; Kang, H.C.; Li, L.; Cho, H.; Park, M.-K.; Lee, E.; Bae, Y.H.; Huh, K.M. Amphiphilic poly(ethylene gly-col)-poly(ε-caprolactone) AB2 miktoarm copolymers for self-assembled nanocarrier systems: Synthesis, characterization, and effects of morphology on antitumor activity. Polym. Chem. 2015, 6, 531–542. [Google Scholar] [CrossRef]
- Diaz, I.L.; Perez, L.D. Synthesis and micellization properties of triblock copolymers PDMAEMA-b-PCL-b-PDMAEMA and their applications in the fabrication of amphotericin B-loaded nanocontainers. Colloid Polym. Sci. 2014, 293, 913–923. [Google Scholar] [CrossRef]
- Rivas, C.J.M.; Tarhini, M.; Badri, W.; Miladi, K.; Greige-Gerges, H.; Nazari, Q.A.; Rodríguez, S.A.G.; Alvarez-Roman, R.; Fessi, H.; Elaissari, A. Nanoprecipitation process: From encapsulation to drug delivery. Int. J. Pharm. 2017, 532, 66–81. [Google Scholar] [CrossRef]
- Pfaller, M.; Diekema, D. Progress in antifungal susceptibility testing of Candida spp. by use of Clinical and Laboratory Stand-ards Institute broth microdilution methods, 2010 to 2012. J. Clin. Microbiol. 2021, 50, 2846–2856. [Google Scholar] [CrossRef] [Green Version]
- Sarker, S.D.; Nahar, L.; Kumarasamy, Y. Microtitre plate-based antibacterial assay incorporating resazurin as an indicator of cell growth, and its application in the in vitro antibacterial screening of phytochemicals. Methods 2007, 42, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Ohyashiki, T.; Mohri, T. Fluorometric analysis of the micelle formation process of surfactants in aqueous solution. I. Utility of pyrene in determination of the critical micelle concentration. Chem. Pharm. Bull. 1983, 31, 1296–1300. [Google Scholar] [CrossRef] [Green Version]
- Owen, S.C.; Chan, D.P.; Shoichet, M.S. Polymeric micelle stability. Nano Today 2012, 7, 53–65. [Google Scholar] [CrossRef]
- Xiao, R.; Wang, R.; Zeng, Z.; Xu, L.; Wang, J. Application of poly(ethylene glycol)—Distearoylphosphatidylethanolamine (PEG-DSPE) block copolymers and their derivatives as nanomaterials in drug delivery. Int. J. Nanomed. 2012, 7, 4185–4198. [Google Scholar] [CrossRef] [Green Version]
- Saha, U.; Banerjee, A.; Das, B. Drug-surfactant comicellization: Propranolol hydrochloride-surface active ionic liquid systems in aqueous medium. J. Mol. Liq. 2020, 309, 113164. [Google Scholar] [CrossRef]
- Fu, X.; Xiao, Y.; Hu, K.; Wang, J.; Lei, J.; Zhou, C. Thermosetting solid–solid phase change materials composed of poly(ethylene glycol)-based two components: Flexible application for thermal energy storage. Chem. Eng. J. 2016, 291, 138–148. [Google Scholar] [CrossRef]
- Peña, J.A.; Gutiérrez, S.J.; Villamil, J.C.; Agudelo, N.A.; Pérez, L.D. Polycaprolactone/polyvinylpyrrolidone/siloxane hybrid ma-terials: Synthesis and in vitro delivery of diclofenac and biocompatibility with periodontal ligament fibroblasts. Mater. Sci. Eng. C 2016, 58, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Barzegar-Jalali, M.; Adibkia, K.; Valizadeh, H.; Shadbad, M.R.S.; Nokhodchi, A.; Omidi, Y.; Mohammadi, G.; Nezhadi, S.H.; Hasan, M. Kinetic analysis of drug release from nanoparticles. J. Pharm. Pharm. Sci. 2008, 11, 167–177. [Google Scholar] [CrossRef] [Green Version]
- Son, G.-H.; Lee, B.-J.; Cho, C.-W. Mechanisms of drug release from advanced drug formulations such as polymeric-based drug-delivery systems and lipid nanoparticles. J. Pharm. Investig. 2017, 47, 287–296. [Google Scholar] [CrossRef]
- Bruschi, M. Mathematical models of drug reléase. In Strategies to Modify the Drug Release from Pharmaceutical Systems; Bruschi, M.L., Ed.; Woodhead Publishing: Cambridge, UK, 2015; pp. 63–86. [Google Scholar]
- Costa, P.; Lobo, J.M.S. Modeling and comparison of dissolution profiles. Eur. J. Pharm. Sci. 2001, 13, 123–133. [Google Scholar] [CrossRef]
- Fujii, G.; Chang, J.-E.; Coley, T.; Steere, B. The Formation of Amphotericin B Ion Channels in Lipid Bilayers. Biochemistry 1997, 36, 4959–4968. [Google Scholar] [CrossRef]
- Yoo, B.K.; Miah, A.J.; Lee, E.-S.; Han, K. Reduced renal toxicity of nanoparticular amphotericin B micelles prepared with partially benzylated poly-L-aspartic acid. Biol. Pharm. Bull. 2006, 29, 1700–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diezi, T.A.; Takemoto, J.K.; Davies, N.M.; Kwon, G.S. COMMUNICATION: Pharmacokinetics and Nephrotoxicity of Amphotericin B-Incorporated Poly(Ethylene Glycol)-Block-Poly(N-Hexyl Stearate l-aspartamide) Micelles. J. Pharm. Sci. 2011, 100, 2064–2070. [Google Scholar] [CrossRef]
- Brajtburg, J.; Elberg, S.; Bolard, J.; Kobayashi, G.S.; Levy, R.A.; Ostlund, R.E.; Schlessinger, D.; Medoff, G. Interaction of Plasma Proteins and Lipoproteins with Amphotericin B. J. Infect. Dis. 1984, 149, 986–997. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Average Composition | Mn (kDa) | Đ | CMC (μg/mL) |
|---|---|---|---|---|
| PEG | m(PEG)121 | 5.3 * | 1.1 | |
| PP6 | m(PEG)121-(PCL)75 | 13.8 | 1.2 | 0.66 |
| PP3 | m(PEG)121-(PCL)26 | 8.2 | 1.2 | 1.4 |
| PEG-DSPE | m(PEG)121-DSPE | 5.3 | 1.1 | 8.5 |
| PP6-DSPE | m(PEG)121-(PCL)75-DSPE | 14.5 | 1.2 | 0.30 |
| PP3-DSPE | m(PEG)121-(PCL)26-DSPE | 9.0 | 1.2 | 1.1 |
| Sample | AmB Loading (%wt.) | Empty PMs | AmB/PMs | ||
|---|---|---|---|---|---|
| Dh nm (PDI) | ζ-Potential (σ) mv | Dh nm (PDI) | ζ-Potential (σ) mv | ||
| PP6 | 8.37 ± 0.34 | 75 (0.41) | −5.8 (0.65) | 198 (0.27) | −4.2 (0.9) |
| PP3 | 10.62 ± 0.78 | 74 (0.43) | −5.4 (0.39) | 206 (0.33) | −3.0 (1.3) |
| PP6-DSPE | 15.29 ± 0.34 | 93 (0.40) | −18.0 (0.72) | 217 (0.26) | −7.5 (1.9) |
| PP3-DSPE | 16.40 ± 0.18 | 93 (0.44) | −13.3 (0.73) | 226 (0.25) | −8.8 (0.1) |
| PEG-DSPE | 84 (0.26) | −12.7 (0.67) | 149 (0.24) | −10.4 (1.6) | |
| Model | Parameter | PP6 | PP3 | PP6-DSPE | PP3-DSPE | PEG-DSPE |
|---|---|---|---|---|---|---|
| 0–6 h | ||||||
| Orden 0 | R2 | 0.978 | 0.979 | 0.987 | 0.952 | 0.988 |
| Ko | 4.570 | 3.833 | 2.789 | 2.265 | 5.227 | |
| Orden 1 | R2 | 0.664 | 0.621 | 0.632 | 0.603 | 0.635 |
| k | 1.026 | 0.957 | 0.976 | 0.897 | 0.966 | |
| Korsmeyer–Peppas | R2 | 0.843 | 0.814 | 0.823 | 0.799 | 0.825 |
| kKP | 1.895 | 1.858 | 1.922 | 1.888 | 1.818 | |
| N | 3.250 | 3.080 | 3.130 | 2.905 | 3.094 | |
| Higuchi | R2 | 0.952 | 0.994 | 0.996 | 0.986 | 0.995 |
| kH | 0.157 | 0.134 | 0.097 | 0.080 | 0.182 | |
| Hixson Crowell | R2 | 0.973 | 0.984 | 0.990 | 0.957 | 0.992 |
| kHC | 0.017 | 0.0138 | 0.0098 | 0.0079 | 0.0194 | |
| Baker–Lonsdale | R2 | 0.913 | 0.842 | 0.874 | 0.814 | 0.853 |
| kBL | 0.0601 | 0.0546 | 0.0476 | 0.0421 | 0.0625 | |
| Strain | Reference | Fungizone® | PP6 | PP3 | PP6-DSPE | PP3-DSPE |
|---|---|---|---|---|---|---|
| C. albicans | SC5314 | 0.46 | 0.23 | 0.11 | 0.23 | 0.11 |
| C. glabrata | ATCC 2001 | 0.93 | 0.46 | 0.11 | 0.23 | 0.11 |
| C. auris | 435-PUJ-HUSI | 0.93 | 0.93 | 0.23 | 0.23 | 0.23 |
| C. auris | 537-PUJ-HUSI | 3.75 | 0.93 | 0.23 | 0.93 | 0.23 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arias, E.R.; Angarita-Villamizar, V.; Baena, Y.; Parra-Giraldo, C.; Perez, L.D. Phospholipid-Conjugated PEG-b-PCL Copolymers as Precursors of Micellar Vehicles for Amphotericin B. Polymers 2021, 13, 1747. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13111747
Arias ER, Angarita-Villamizar V, Baena Y, Parra-Giraldo C, Perez LD. Phospholipid-Conjugated PEG-b-PCL Copolymers as Precursors of Micellar Vehicles for Amphotericin B. Polymers. 2021; 13(11):1747. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13111747
Chicago/Turabian StyleArias, Elsa R., Vivian Angarita-Villamizar, Yolima Baena, Claudia Parra-Giraldo, and Leon D. Perez. 2021. "Phospholipid-Conjugated PEG-b-PCL Copolymers as Precursors of Micellar Vehicles for Amphotericin B" Polymers 13, no. 11: 1747. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13111747