
Unravelling Main- and Side-Chain Motions in Polymers with NMR Spectroscopy and Relaxometry: The Case of Polyvinyl Butyral

 , , , ,
, , , , 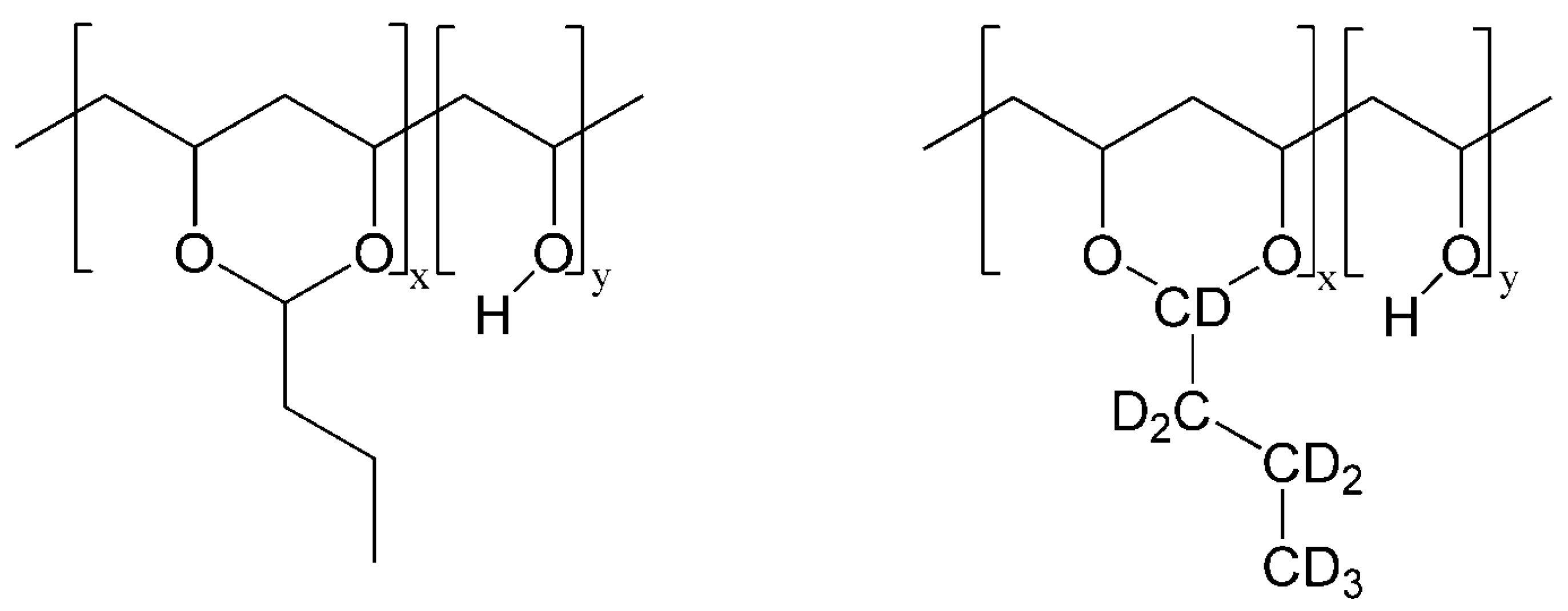{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
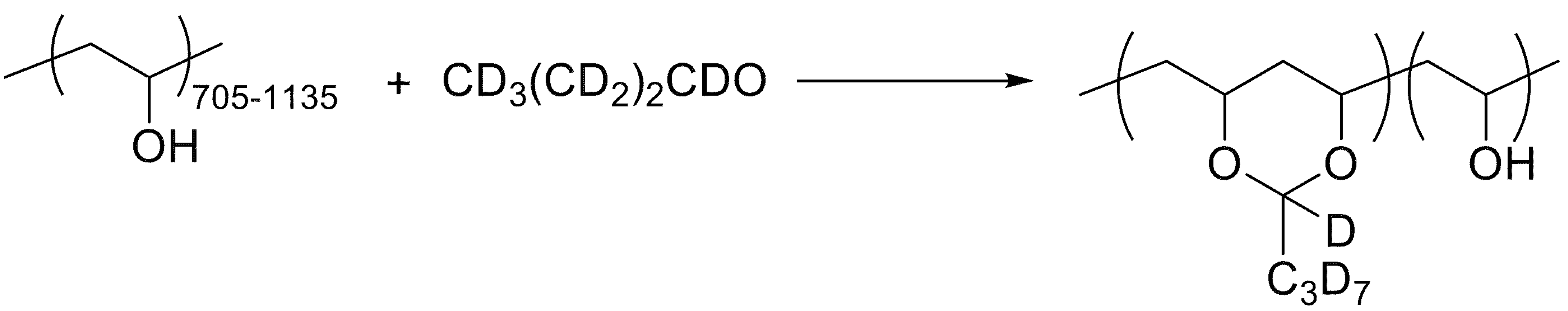{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Differential Scanning Calorimetry (DSC) and Size Exclusion Chromatography (SEC) Measurements
2.3. NMR Measurements
3. Results and Discussion
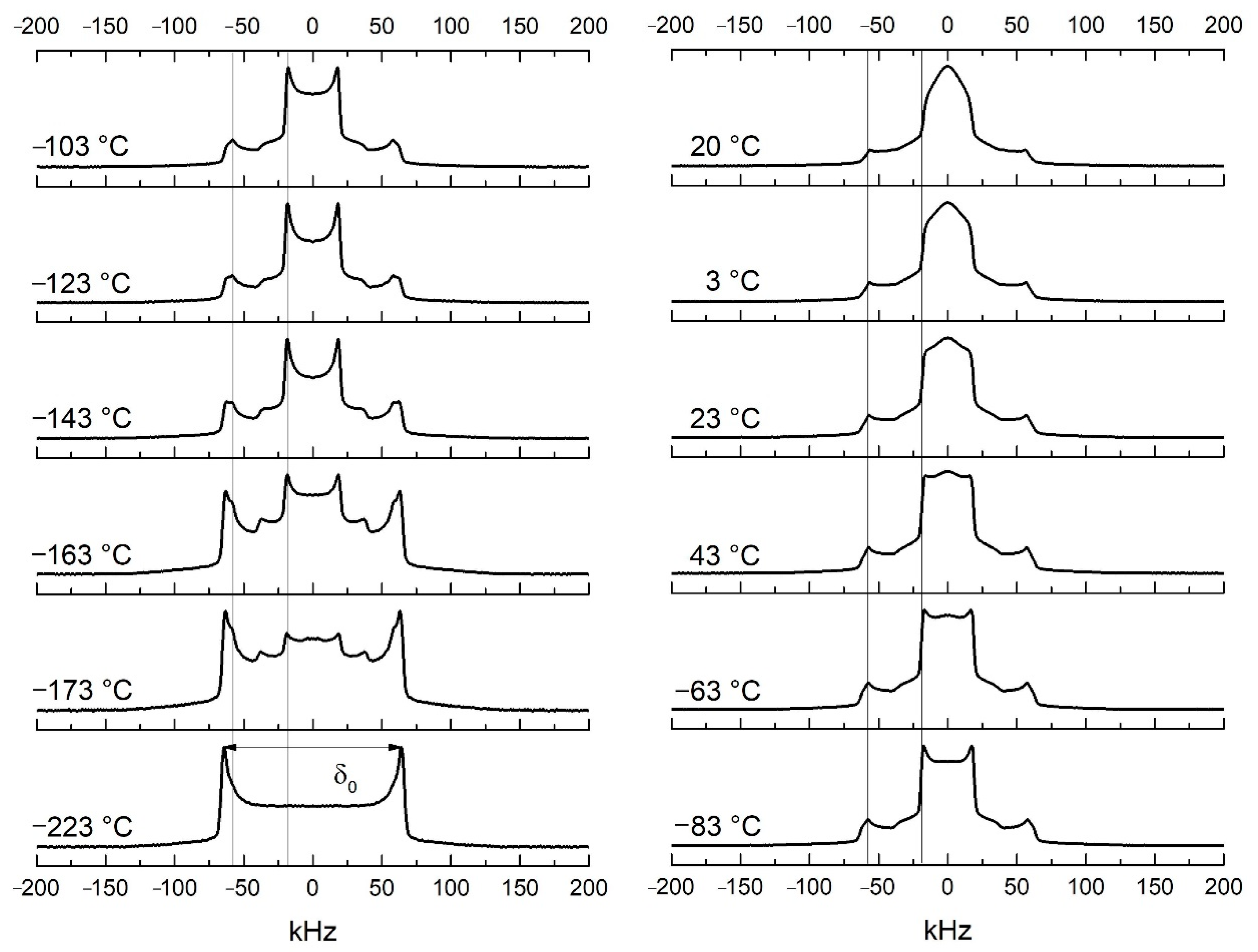
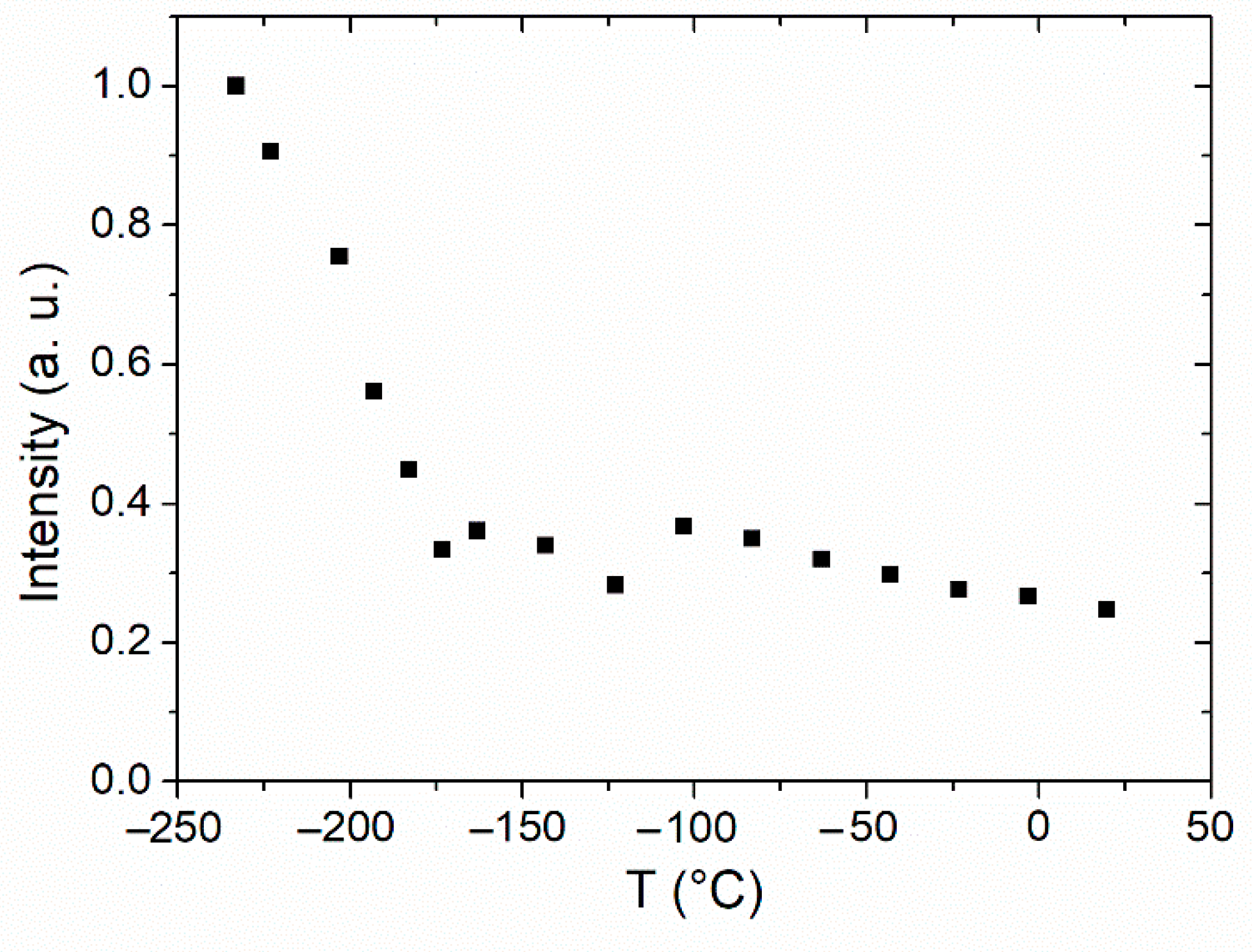
3.1. Dynamics of Side-Chains below Tg by 2H NMR Spectroscopy
3.2. Dynamics of Main- and Side-Chains across Tg from 1H FID Analysis
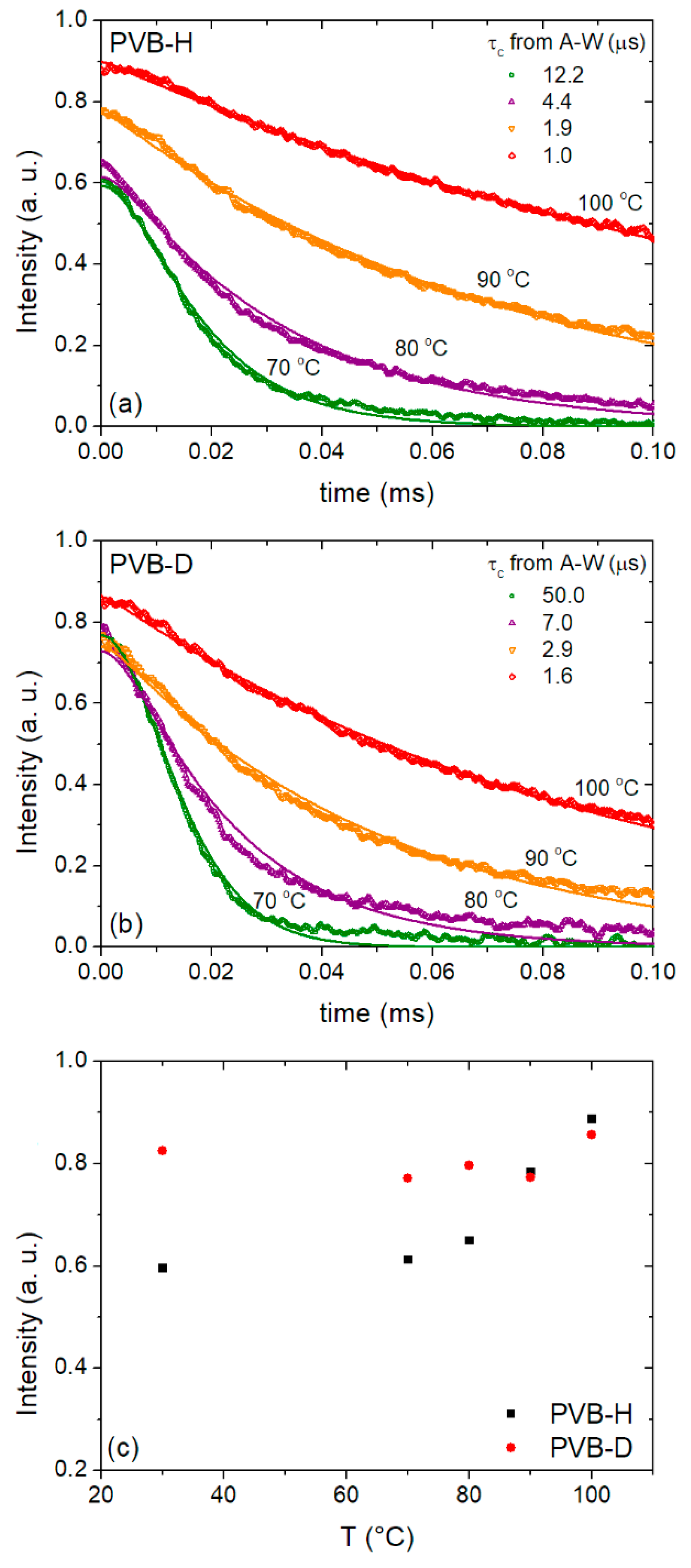
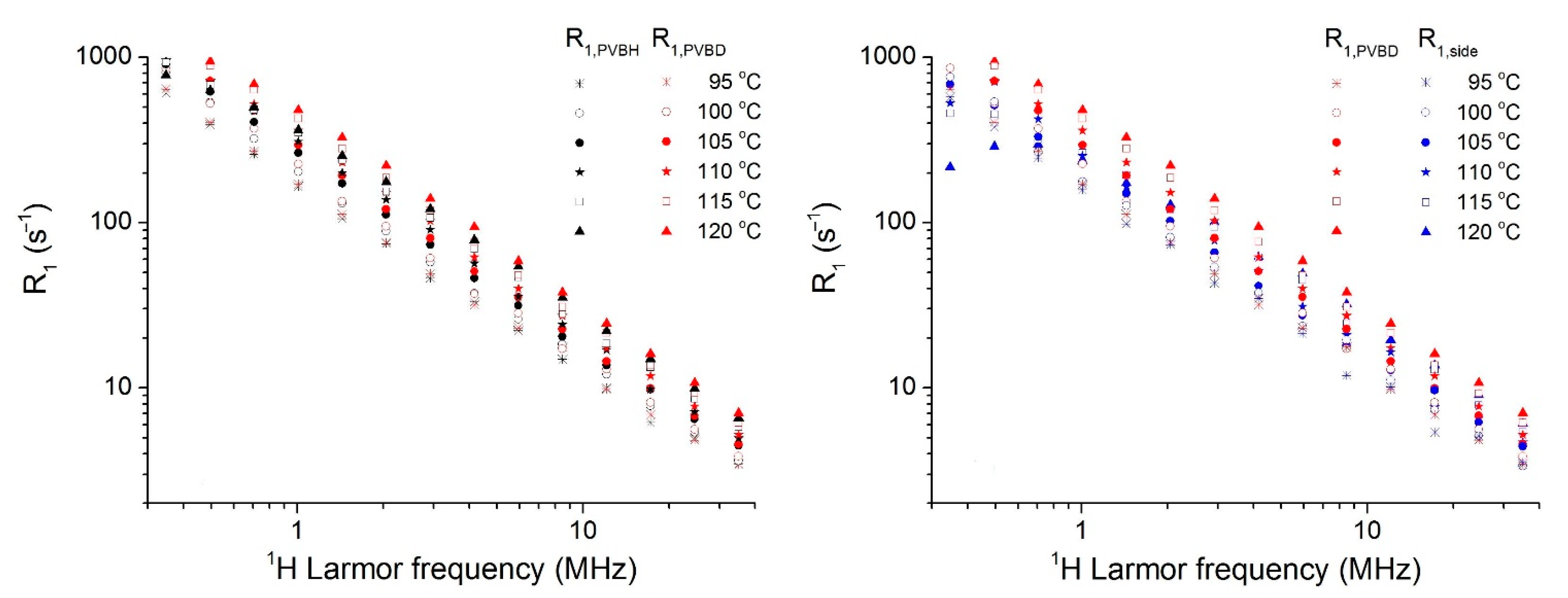
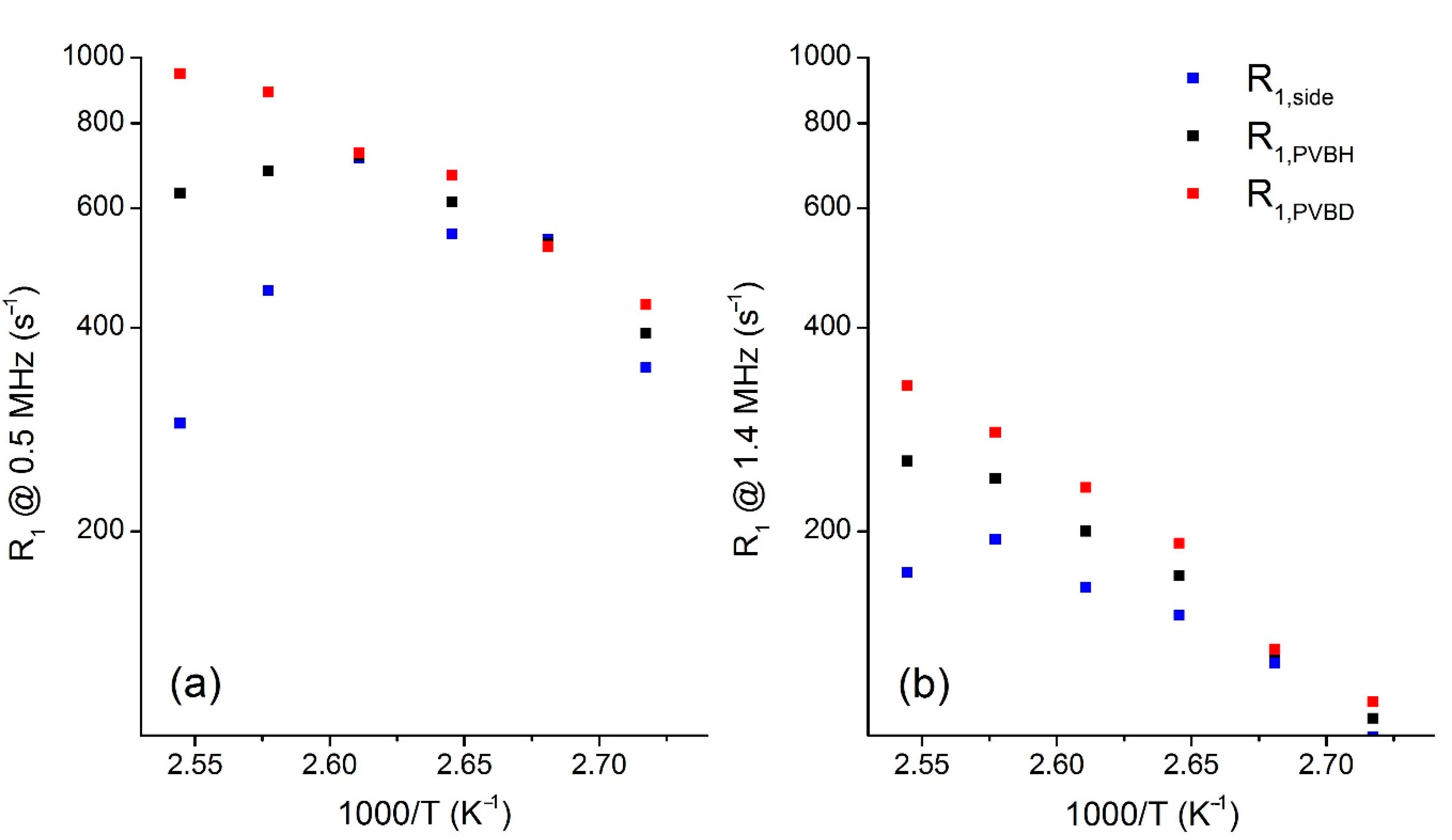
3.3. Dynamics of Main- and Side-Chains above Tg from 1H FC NMR Relaxometry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schönhals, A.; Kremer, F. Amorphous Polymers. In Polymer Science: A Comprehensive Reference; Matyjaszewski, K., Möller, M., Eds.; Elsevier BV: Amsterdam, The Netherlands, 2012; Volume 1, pp. 201–226. [Google Scholar]
- Kimmich, R.; Fatkullin, N. Polymer Chain Dynamics and NMR. Adv. Polym. Sci. 2004, 170, 1–113. [Google Scholar]
- Johari, G.P.; Goldstein, M. Viscous Liquids and the Glass Transition. II. Secondary Relaxations in Glasses of Rigid Molecules. J. Chem. Phys. 1970, 53, 2372–2388. [Google Scholar] [CrossRef]
- Schmidt-Rohr, K.; Kulik, A.S.; Beckham, H.W.; Ohlemacher, A.; Pawelzik, U.; Boeffel, C.; Spiess, H.W. Molecular Nature of the β Relaxation in Poly(methyl methacrylate) Investigated by Multidimensional NMR. Macromolecules 1994, 27, 4733–4745. [Google Scholar] [CrossRef]
- Kulik, A.S.; Beckham, H.W.; Schmidt-Rohr, K.; Radloff, D.; Pawelzik, U.; Boeffel, C.; Spiess, H.W. Coupling of the α and β Processes in Poly(ethyl methacrylate) Investigated by Multidimensional NMR. Macromolecules 1994, 27, 4746–4754. [Google Scholar] [CrossRef]
- Genix, A.-C.; Lauprêtre, F. Subglass and Glass Transitions of Poly(di-n-alkylitaconate)s with Various Side-Chain Lengths: Dielectric Relaxation Investigation. Macromolecules 2005, 38, 2786–2794. [Google Scholar] [CrossRef]
- Gaborieau, M.; Graf, R.; Kahle, S.; Pakula, T.; Spiess, H.W. Chain Dynamics in Poly(n-alkyl acrylates) by Solid-State NMR, Dielectric, and Mechanical Spectroscopies. Macromolecules 2007, 40, 6249–6256. [Google Scholar] [CrossRef]
- Mpoukouvalas, K.; Floudas, G.; Williams, G. Origin of the α, β, (βα), and “Slow” Dielectric Processes in Poly(ethyl methacrylate). Macromolecules 2009, 42, 4690–4700. [Google Scholar] [CrossRef]
- Liau, L.C.K.; Viswanath, D.S. Thermal Degradation of Poly(vinylbutyral)/Ceramic Composites: A Kinetic Approach. Ind. Eng. Chem. Res. 1998, 37, 49–57. [Google Scholar] [CrossRef]
- Boddu, V.M.; Viswanath, D.S.; Natarajan, G.; Knickerbocker, J.V. Activity and Reactivity of Carbon Formed by the Thermal Degradation of Poly(vinylbutyral) in Alumina Pellets. J. Am. Ceram. Soc. 1990, 73, 1620–1625. [Google Scholar] [CrossRef]
- Carrot, C.; Bendaoud, A.; Pillon, C. Handbook of Thermoplastics, 2nd ed.; Olabis, O., Adewale, K., Eds.; Taylor & Francis Group: Abingdon, UK; CRC Press: Boca Raton, FL, USA, 2016; Chapter 3. [Google Scholar]
- Martín, M.; Centelles, X.; Solé, A.; Barreneche, C.; Fernández, I.; Cabeza, L.F. Polymeric Interlayer Materials for Laminated Glass: A Review. Constr. Build. Mater. 2020, 230, 116897. [Google Scholar] [CrossRef]
- Hooper, P.A.; Blackman, B.R.K.; Dear, J.P. The Mechanical Behaviour of Poly(vinyl butyral) at Different Strain Magnitudes and Strain Rates. J. Mater. Sci. 2012, 47, 3564–3576. [Google Scholar] [CrossRef] [Green Version]
- Andreozzi, L.; Briccoli Bati, S.; Fagone, M.; Ranocchiai, G.; Zulli, F. Dynamic Torsion Tests to Characterize the Thermo-viscoelastic Properties of Polymeric Interlayers for Laminated Glass. Constr. Build. Mater. 2014, 65, 1–13. [Google Scholar] [CrossRef]
- Arayachukiat, S.; Siriprumpoonthum, M.; Nobukawa, S.; Yamaguchi, M. Viscoelastic Properties and Extrusion Processability of Poly(vinyl butyral). J. Appl. Polym. Sci. 2014, 131, 40337–40344. [Google Scholar] [CrossRef]
- Takahashi, Y. Dielectric α- and β-Dispersions in Polyvinyl Butyral (PVB). J. Phys. Soc. Jpn. 1961, 16, 1024. [Google Scholar] [CrossRef]
- Funt, B.L. Dielectric Dispersion in Solid Polyvinyl Butyral. Can. J. Chem. 1952, 30, 84–91. [Google Scholar] [CrossRef]
- Mehendru, P.C.; Kumar, N.; Arora, V.P.; Gupta, N.P. Dielectric Relaxation Studies in Polyvinyl butyral. J. Chem. Phys. 1982, 77, 4232–4235. [Google Scholar] [CrossRef]
- Saad, G.R.; El-Shafee, E.; Sabaa, M.W. Dielectric and Mechanical Properties in the Photodegradation of Poly(vinyl butyral) Films. Polym. Degrad. Stab. 1995, 47, 209–215. [Google Scholar] [CrossRef]
- Handge, U.A.; Wolff, M.F.H.; Abetz, V.; Heinrich, S. Viscoelastic and Dielectric Properties of Composites of Poly(vinyl butyral) and Alumina Particles with a High Filling Degree. Polymer 2016, 82, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Pizzanelli, S.; Prevosto, D.; Labardi, M.; Guazzini, T.; Bronco, S.; Forte, C.; Calucci, L. Dynamics of Poly(vinyl butyral) Studied Using Dielectric Spectroscopy and 1H NMR Relaxometry. Phys. Chem. Chem. Phys. 2017, 19, 31804–31812. [Google Scholar] [CrossRef] [Green Version]
- Pizzanelli, S.; Forte, C.; Bronco, S.; Guazzini, T.; Serraglini, C.; Calucci, L. PVB/ATO Nanocomposites for Glass Coating Applications: Effects of Nanoparticles on the PVB Matrix. Coatings 2019, 9, 247. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Rohr, K.; Spiess, H.W. Multidimensional Solid-State NMR and Polymers; Academic Press: London, UK, 1994. [Google Scholar]
- Macho, V.; Brombacher, L.; Spiess, H.W. The NMR-WEBLAB: An Internet Approach to NMR Lineshape Analysis. Appl. Magn. Reson. 2001, 20, 405–432. [Google Scholar] [CrossRef]
- Schäler, K.; Roos, M.; Micke, P.; Golitsyn, Y.; Seidlitz, A.; Thurn-Albrecht, T.; Schneider, H.; Hempel, G.; Saalwächter, K. Basic Principles of Static Proton Low-Resolution Spin Diffusion NMR in Nanophase-Separated Materials with Mobility Contrast. Solid State Nucl. Magn. Reson. 2015, 72, 50–63. [Google Scholar] [CrossRef]
- Hansen, E.W.; Kristiansen, P.E.; Pedersen, B. Crystallinity of Polyethylene Derived from Solid-State Proton NMR Free Induction Decay. J. Phys. Chem. B 1998, 102, 5444–5450. [Google Scholar] [CrossRef]
- Borsacchi, S.; Sudhakaran, U.P.; Calucci, L.; Martini, F.; Carignani, E.; Messori, M.; Geppi, M. Rubber-Filler Interactions in Polyisoprene Filled with In Situ Generated Silica: A Solid State NMR Study. Polymers 2018, 10, 822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimmich, R. Field-Cycling NMR Relaxometry: Instrumentation, Model Theories and Applications; Royal Society of Chemistry: London, UK, 2019. [Google Scholar]
- Fernández, M.D.; Fernández, M.J.; Hoces, P. Synthesis of Poly(vinyl butyral)s in Homogeneous Phase and Their Thermal Properties. J. Appl. Polym. Sci. 2006, 102, 5007–5017. [Google Scholar] [CrossRef]
- Dess, D.B.; Martin, J.C. A Useful 12-I-5 Triacetoxyperiodinane (The Dess-Martin Periodinane) for the Selective Oxidation of Primary or Secondary Alcohols and a Variety of Related 12-I-5 Species. J. Am. Chem. Soc. 1991, 113, 7277–7287. [Google Scholar] [CrossRef]
- Vega, A.J.; Luz, Z. Quadrupole Echo Distortion as a Tool for Dynamic NMR: Application to Molecular Reorientation in Solid Trimethylamine. J. Chem. Phys. 1987, 86, 1803–1813. [Google Scholar] [CrossRef]
- Powles, J.G.; Strange, J.H. Zero Time Resolution Nuclear Magnetic Resonance Transient in Solids. Proc. Phys. Soc. London 1963, 82, 6–15. [Google Scholar] [CrossRef]
- Anoardo, E.; Galli, G.; Ferrante, G. Fast-Field-Cycling NMR: Applications and Instrumentation. Appl. Magn. Reson. 2001, 20, 365–404. [Google Scholar] [CrossRef]
- Kimmich, R.; Anoardo, E. Field-Cycling NMR Relaxometry. Prog. Nucl. Magn. Reson. Spectrosc. 2004, 44, 257–320. [Google Scholar] [CrossRef]
- Mittermaier, A.; Kay, L.E. Measurement of Methyl 2H Quadrupolar Couplings in Oriented Proteins. How Uniform Is the Quadrupolar Coupling Constant? J. Am. Chem. Soc. 1999, 121, 10608–10613. [Google Scholar] [CrossRef]
- Burnett, L.H.; Muller, B.H. Deuteron Quadrupole Coupling Constants in Three Solid Deuterated Paraffin Hydrocarbons: C2D6, C4D10, C6D14. J. Chem. Phys. 1971, 55, 5829–5831. [Google Scholar] [CrossRef]
- Bloom, M.; Davis, J.H.; Valic, M.I. Spectral Distortion Effects Due to Finite Pulse Widths in Deuterium Nuclear Magnetic Resonance Spectroscopy. Can. J. Phys. 1980, 58, 1510–1517. [Google Scholar] [CrossRef]
- Kuebler, S.C.; Schaefer, D.J.; Boeffel, C.; Pawelzik, U.; Spiess, H.W. 2D Exchange NMR Investigation of the α-Relaxation in Poly(ethyl methacrylate) as Compared to Poly(methyl methacrylate). Macromolecules 1997, 30, 6597–6609. [Google Scholar] [CrossRef]
- Körber, T.; Mohamed, F.; Hofmann, M.; Lichtinger, A.; Willner, L.; Rössler, E.A. The Nature of Secondary Relaxations: The Case of Poly(ethylene-alt-propylene) Studied by Dielectric and Deuteron NMR Spectroscopy. Macromolecules 2017, 50, 1554–1568. [Google Scholar] [CrossRef]
- Litvinov, V.M.; Macho, V.; Spiess, H.W. Molecular Motions in Crystalline and Mesomorphic Forms of Poly(diethylsiloxane). Acta Polymer. 1997, 48, 471–477. [Google Scholar] [CrossRef]
- Lin, W.-Y.; Blum, F.D. Segmental Dynamics of Bulk Adsorbed Poly(methyl acrylate)-d3 by Deuterium NMR: Effect of Adsorbed Amount. Macromolecules 1997, 30, 5331–5338. [Google Scholar] [CrossRef]
- Maddumaarachchi, M.; Mathota Arachchige, Y.L.N.; Zhang, T.; Blum, F.D. Dynamics of Cetyltrimethylammonium Bromide Head Groups in Bulk by Solid-State Deuterium NMR Spectroscopy. Langmuir 2018, 34, 11058–11065. [Google Scholar] [CrossRef] [PubMed]
- Meirovitch, E.; Liang, Z.; Freed, J.H. Protein Dynamics in the Solid State from 2H NMR Line Shape Analysis: A Consistent Perspective. J. Phys. Chem. B 2015, 119, 2857–2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metin, B.; Blum, F.D. Molecular Mass and Dynamics of Poly(methyl acrylate) in the Glass-Transition Region. J. Chem. Phys. 2006, 124, 054908. [Google Scholar] [CrossRef]
- Nambiar, R.R.; Blum, F.D. Segmental Dynamics of Bulky Poly(vinyl acetate)-d3 by Solid State 2H NMR: Effect of Small Molecule Plasticizer. Macromolecules 2008, 41, 9837–9845. [Google Scholar] [CrossRef]
- Vogel, M.; Rössler, E.A. Slow β Process in Simple Organic Glass Formers Studied by One and Two Dimensional 2H Nuclear Magnetic Resonance. II. Discussion of Motional Models. J. Chem. Phys. 2001, 115, 10883–10891. [Google Scholar] [CrossRef]
- Bock, D.; Kalhau, R.; Micko, B.; Pötzschner, B.; Schneider, G.J.; Rössler, E.A. On the Cooperative Nature of the β-Process in Neat and Binary Glasses: A Dielectric and Nuclear Magnetic Resonance Spectroscopy Study. J. Chem. Phys. 2013, 139, 064508. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.; Rössler, E.A. Effects of Various Types of Molecular Dynamics on 1D and 2D 2H NMR Studied by Random Walk Simulations. J. Magn. Reson. 2000, 147, 43–58. [Google Scholar] [CrossRef]
- Papon, A.; Saalwächter, K.; Schäler, K.; Guy, L.; Lequeux, F.; Montes, H. Low-Field NMR Investigations of Nanocomposites: Polymer Dynamics and Network Effects. Macromolecules 2011, 44, 913–922. [Google Scholar] [CrossRef]
- Litvinov, V.M.; Penning, J.P. Phase Composition and Molecular Mobility in Nylon 6 Fibers as Studied by Proton NMR Transverse Magnetization Relaxation. Macromol. Chem. Phys. 2004, 205, 1721–1734. [Google Scholar] [CrossRef]
- Abragam, A. Principles of Nuclear Magnetism; Oxford University Press: Oxford, UK, 1961. [Google Scholar]
- Anderson, P.W.; Weiss, P.R. Exchange Narrowing in Paramagnetic Resonance. Rev. Mod. Phys. 1953, 25, 269–276. [Google Scholar] [CrossRef]
- Kehr, M.; Fatkullin, N.; Kimmich, R. Molecular Diffusion on a Time Scale Between Nano- and Milliseconds probed by Field-Cycling NMR Relaxometry of Intermolecular Dipolar interactions: Application to Polymer Melts. J. Chem. Phys. 2007, 126, 094903. [Google Scholar] [CrossRef]
- Kehr, M.; Fatkullin, N.; Kimmich, R. Deuteron and Proton Spin-Lattice Relaxation Dispersion of Polymer Melts: Intrasegment, Intrachain, and Interchain Contributions. J. Chem. Phys. 2007, 127, 084911. [Google Scholar] [CrossRef]
- Kariyo, S.; Stapf, S.; Blümich, B. Site Specific Proton and Deuteron NMR Relaxation Dispersion in Selectively Deuterated Polyisoprene Melts. Macromol. Chem. Phys. 2005, 206, 1292–1299. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Calucci, L.; Pizzanelli, S.; Mandoli, A.; Birczyński, A.; Lalowicz, Z.T.; De Monte, C.; Ricci, L.; Bronco, S. Unravelling Main- and Side-Chain Motions in Polymers with NMR Spectroscopy and Relaxometry: The Case of Polyvinyl Butyral. Polymers 2021, 13, 2686. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13162686
Calucci L, Pizzanelli S, Mandoli A, Birczyński A, Lalowicz ZT, De Monte C, Ricci L, Bronco S. Unravelling Main- and Side-Chain Motions in Polymers with NMR Spectroscopy and Relaxometry: The Case of Polyvinyl Butyral. Polymers. 2021; 13(16):2686. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13162686
Chicago/Turabian StyleCalucci, Lucia, Silvia Pizzanelli, Alessandro Mandoli, Artur Birczyński, Zdzisław T. Lalowicz, Cristina De Monte, Lucia Ricci, and Simona Bronco. 2021. "Unravelling Main- and Side-Chain Motions in Polymers with NMR Spectroscopy and Relaxometry: The Case of Polyvinyl Butyral" Polymers 13, no. 16: 2686. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13162686