
Determination of Dental Adhesive Composition throughout Solvent Drying and Polymerization Using ATR–FTIR Spectroscopy


Abstract
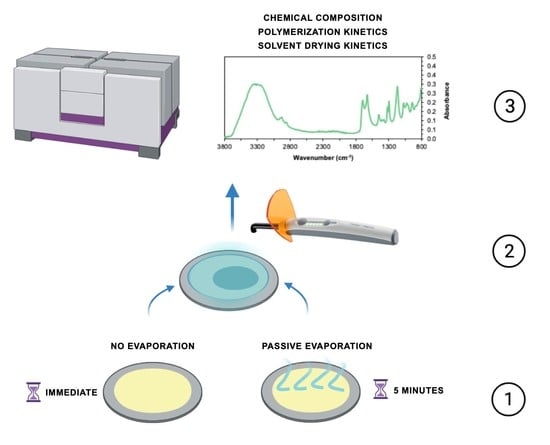
:
1. Introduction
2. Materials and Methods
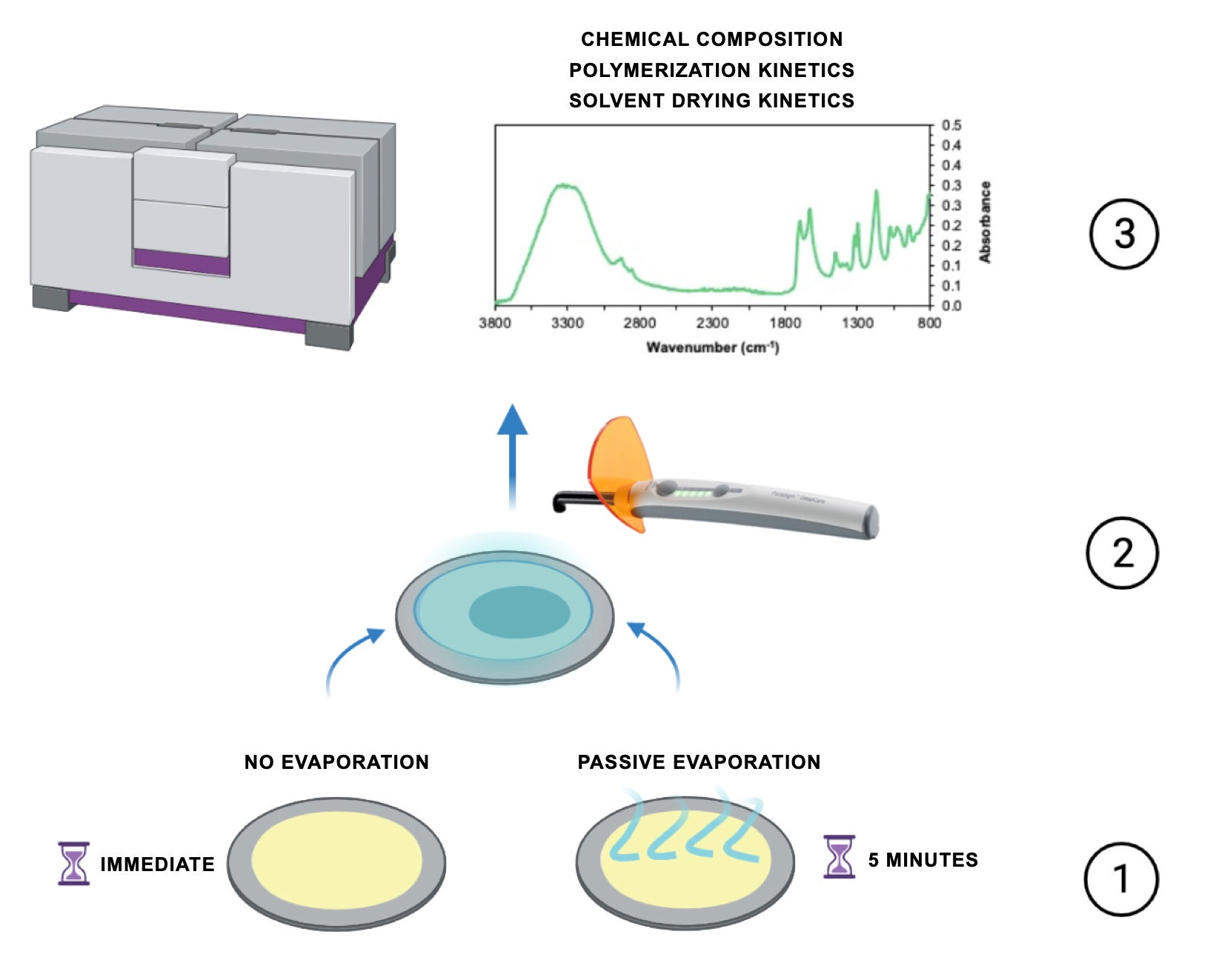
2.1. Material
2.2. Filler Isolation
2.3. FTIR Spectra of Components and Adhesives
2.4. Solvent Evaporation and Polymerization
2.5. Statistical Analysis
3. Results
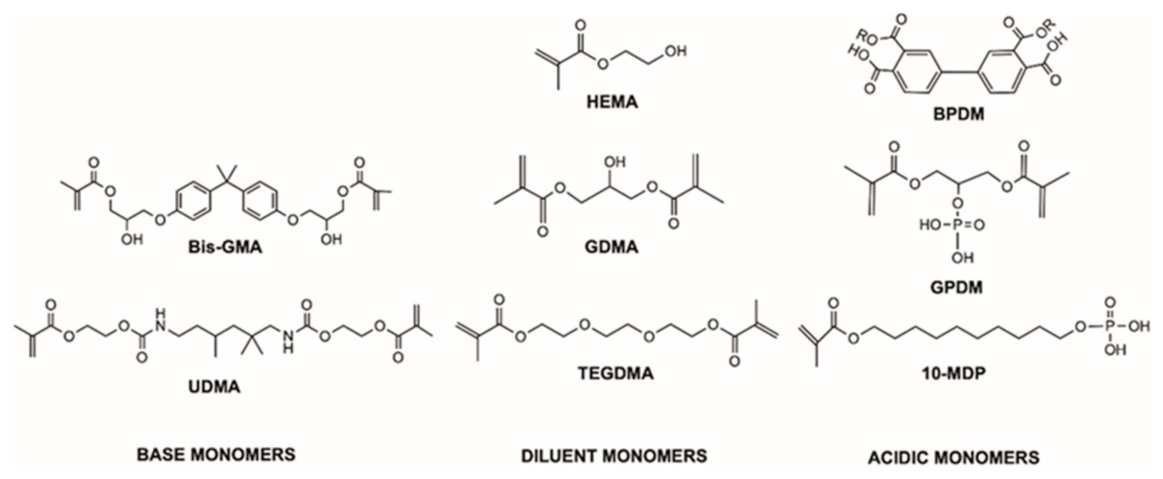
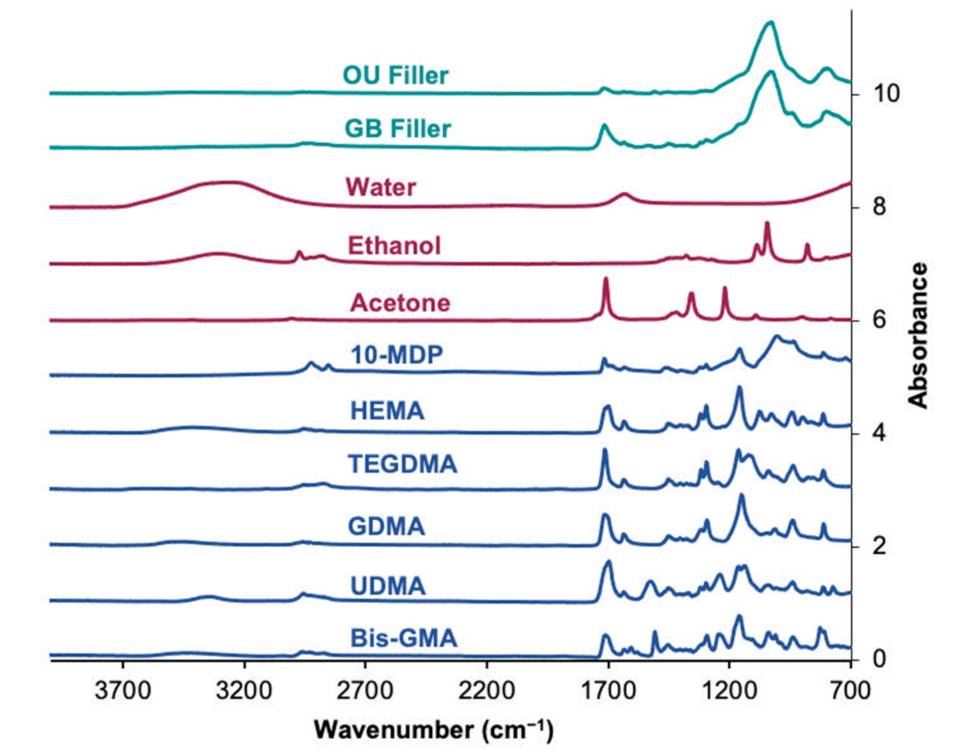
3.1. FTIR Spectra of Pure Monomers, Solvents, Fillers, and Filler Fractions
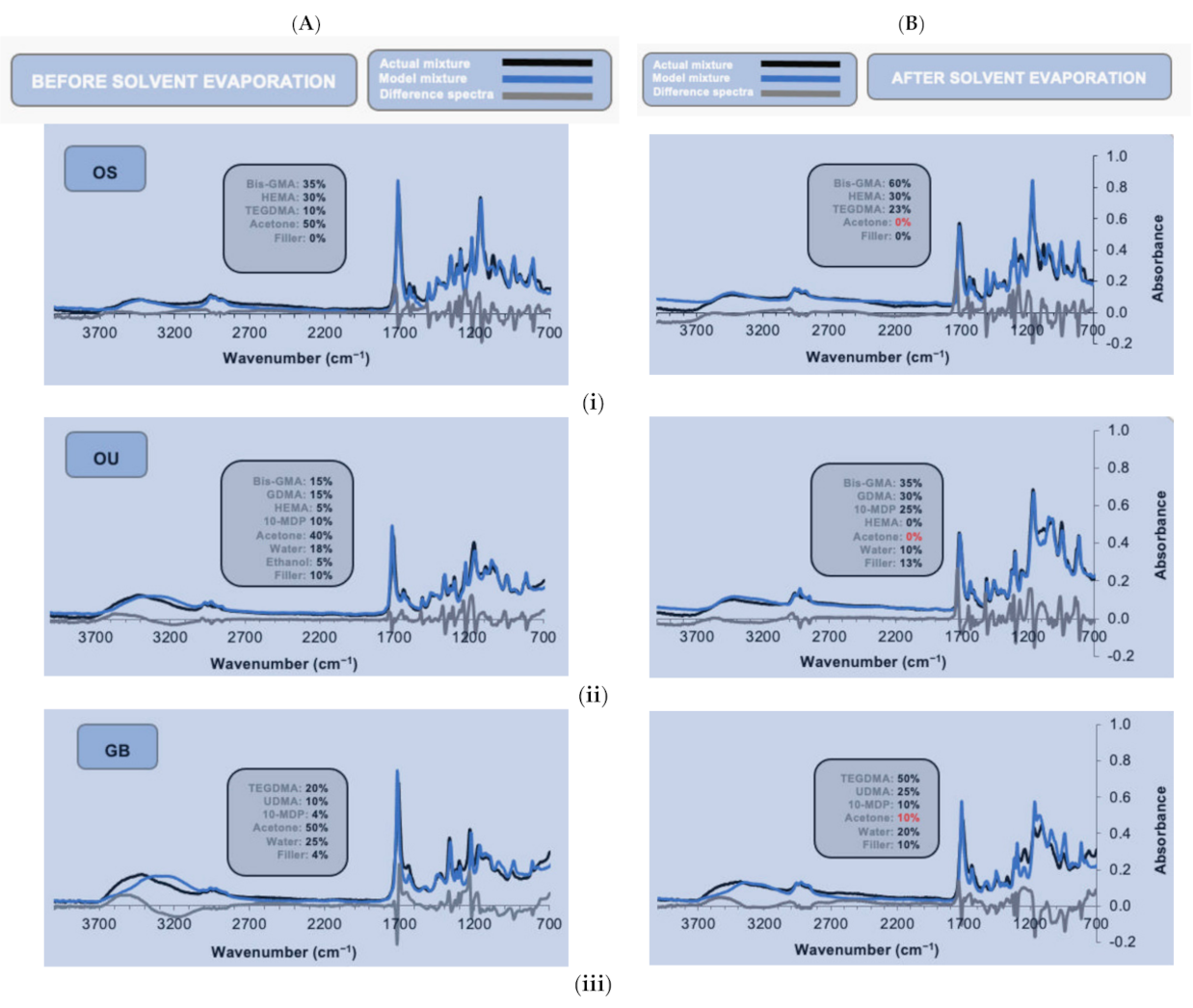
3.2. FTIR Compositional Modelling of Adhesives
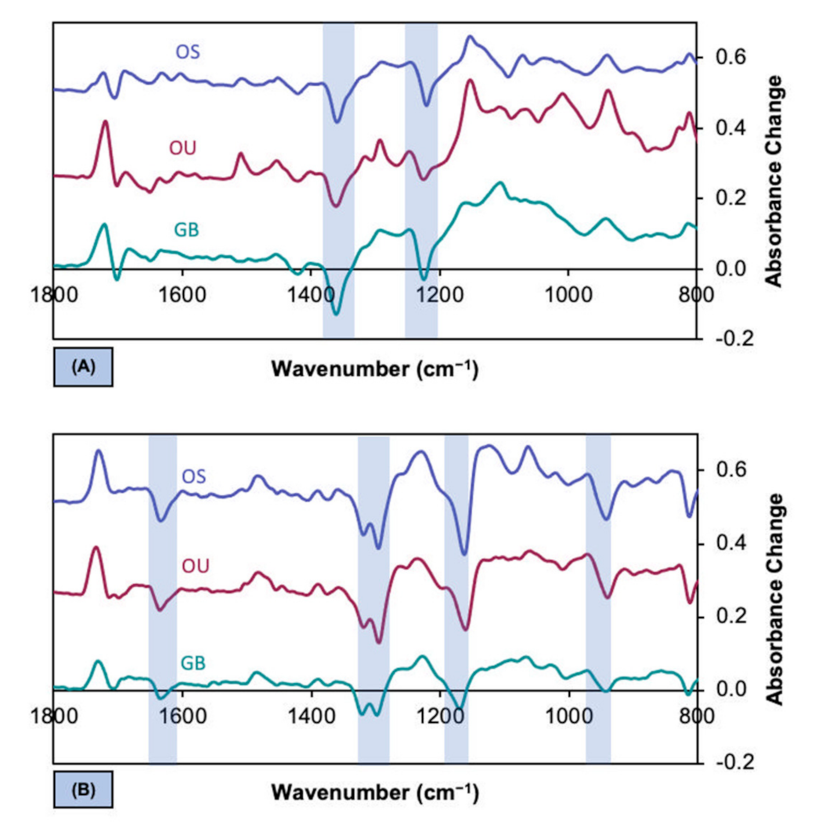
3.3. Difference Spectra for Evaporation Versus Polymerisation
3.4. Compositional Changes upon Evaporation
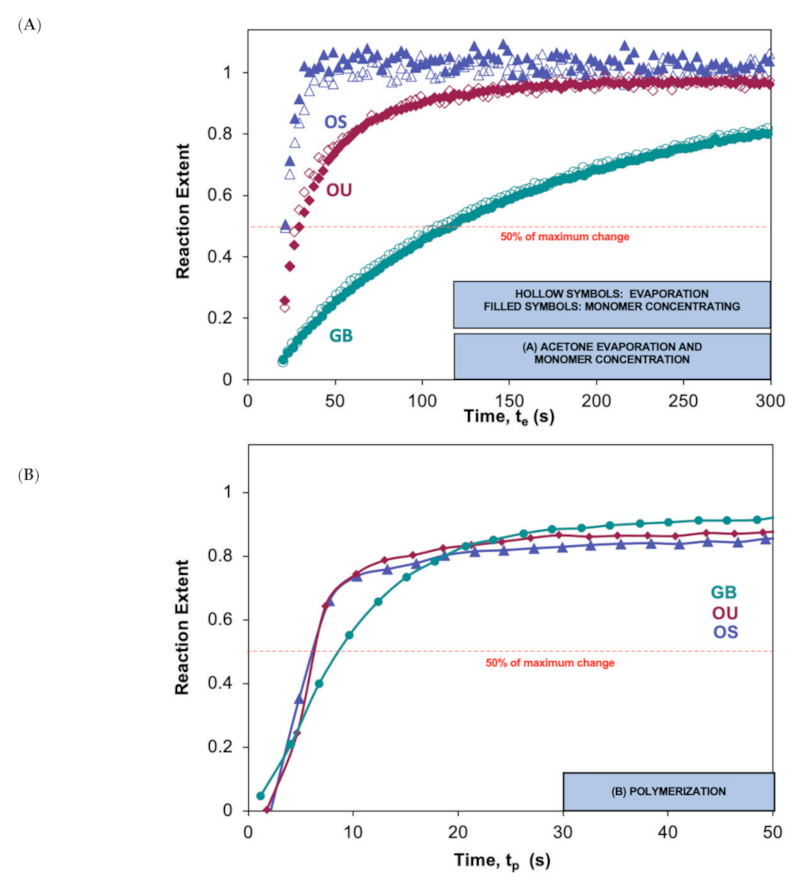
3.5. Polymerization Extents and Final Conversions
4. Discussion
4.1. FTIR Method
4.2. Material Selection
4.3. Initial FTIR Spectra of Adhesives
4.4. Spectral Changes upon Drying and Polymerization
4.5. Evaporation Kinetics
4.6. Polymerisation Kinetics
4.7. Study Limitations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yoshida, Y.; Inoue, S. Chemical analyses in dental adhesive technology. Jpn. Dent. Sci. Rev. 2012, 48, 141–152. [Google Scholar] [CrossRef] [Green Version]
- Perdigão, J. Current perspectives on dental adhesion: (1) Dentin adhesion—Not there yet. Jpn. Dent. Sci. Rev. 2020, 56, 190–207. [Google Scholar] [CrossRef] [PubMed]
- Van Meerbeek, B.; Yoshihara, K.; Van Landuyt, K.; Yoshida, Y.; Peumans, M. From Buonocore’s Pioneering Acid-Etch Technique to Self-Adhering Restoratives. A Status Perspective of Rapidly Advancing Dental Adhesive Technology. J. Adhes. Dent. 2020, 22, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Spencer, P.; Ye, Q.; Park, J.; Topp, E.M.; Misra, A.; Marangos, O.; Wang, Y.; Bohaty, B.S.; Singh, V.; Sene, F.; et al. Adhesive/Dentin interface: The weak link in the composite restoration. Ann. Biomed. Eng. 2010, 38, 1989–2003. [Google Scholar] [CrossRef] [Green Version]
- Ye, Q.; Park, J.; Laurence, J.S.; Parthasarathy, R.; Misra, A.; Spencer, P. Ternary phase diagram of model dentin adhesive exposed to over-wet environments. J. Dent. Res. 2011, 90, 1434–1438. [Google Scholar] [CrossRef]
- Yoshihara, K.; Hayakawa, S.; Nagaoka, N.; Okihara, T.; Yoshida, Y.; Van Meerbeek, B. Etching Efficacy of Self-Etching Functional Monomers. J. Dent. Res. 2018, 97, 1010–1016. [Google Scholar] [CrossRef]
- Bae, J.H.; Cho, B.H.; Kim, J.S.; Kim, M.S.; Lee, I.B.; Son, H.H.; Um, C.M.; Kim, C.K.; Kim, O.Y. Adhesive layer properties as a determinant of dentin bond strength. J. Biomed. Mater. Res. Part B Appl. Biomater. 2005, 74, 822–828. [Google Scholar] [CrossRef]
- Leal, F.B.; Madruga, F.C.; Prochnow, E.P.; Lima, G.S.; Ogliari, F.A.; Piva, E.; Moraes, R.R. Effect of acidic monomer concentration on the dentin bond stability of self-etch adhesives. Int. J. Adhes. Adhes. 2011, 31, 571–574. [Google Scholar] [CrossRef]
- Delgado, A.H.; Young, A.M. Modelling ATR-FTIR Spectra of Dental Bonding Systems to Investigate Composition and Polymerisation Kinetics. Materials 2021, 14, 760. [Google Scholar] [CrossRef]
- Ekambaram, M.; Yiu, C.K.Y.; Matinlinna, J.P. An overview of solvents in resin-dentin bonding. Int. J. Adhes. Adhes. 2015, 57, 22–33. [Google Scholar] [CrossRef]
- Van Landuyt, K.L.; Snauwaert, J.; De Munck, J.; Peumans, M.; Yoshida, Y.; Poitevin, A.; Coutinho, E.; Suzuki, K.; Lambrechts, P.; Van Meerbeek, B. Systematic review of the chemical composition of contemporary dental adhesives. Biomaterials 2007, 28, 3757–3785. [Google Scholar] [CrossRef]
- Cho, B.H.; Dickens, S.H. Effects of the acetone content of single solution dentin bonding agents on the adhesive layer thickness and the microtensile bond strength. Dent. Mater. 2004, 20, 107–115. [Google Scholar] [CrossRef]
- Ikeda, T.; De Munck, J.; Shirai, K.; Hikita, K.; Inoue, S.; Sano, H.; Lambrechts, P.; Van Meerbeek, B. Effect of evaporation of primer components on ultimate tensile strengths of primer-adhesive mixture. Dent. Mater. 2005, 21, 1051–1058. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.N.; Lanza, M.D.S.; Dourado, L.G.; Carvalho, E.M.; Bauer, J. Impact of Solvent Evaporation and Curing Protocol on Degree of Conversion of Etch-and-Rinse and Multimode Adhesives Systems. Int. J. Dent. 2019, 2019, 5496784. [Google Scholar] [CrossRef] [Green Version]
- Yiu, C.K.Y.; Pashley, E.L.; Hiraishi, N.; King, N.M.; Goracci, C.; Ferrari, M.; Carvalho, R.M.; Pashley, D.H.; Tay, F.R. Solvent and water retention in dental adhesive blends after evaporation. Biomaterials 2005, 26, 6863–6872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pashley, D.H.; Tay, F.R.; Breschi, L.; Tjäderhane, L.; Carvalho, R.M.; Carrilho, M.; Tezvergil-Mutluay, A. State of the art etch-and-rinse adhesives. Dent. Mater. 2011, 27, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Giannini, M.; Arrais, C.A.G.; Vermelho, P.M.; Reis, R.S.A.; Dos Santos, L.P.S.; Leite, E.R. Effects of the solvent evaporation technique on the degree of conversion of one-bottle adhesive systems. Oper. Dent. 2008, 33, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Klein-Júnior, C.A.; Zander-Grande, C.; Amaral, R.; Stanislawczuk, R.; Garcia, E.J.; Baumhardt-Neto, R.; Meier, M.M.; Loguercio, A.D.; Reis, A. Evaporating solvents with a warm air-stream: Effects on adhesive layer properties and resin-dentin bond strengths. J. Dent. 2008, 36, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Bail, M.; Malacarne-Zanon, J.; Silva, S.M.A.; Anauate-Netto, A.; Nascimento, F.D.; Amore, R.; Lewgoy, H.; Pashley, D.H.; Carrilho, M.R. Effect of air-drying on the solvent evaporation, degree of conversion and water sorption/solubility of dental adhesive models. J. Mater. Sci. Mater. Med. 2012, 23, 629–638. [Google Scholar] [CrossRef]
- Ferracane, J.L.; Hilton, T.J.; Stansbury, J.W.; Watts, D.C.; Silikas, N.; Ilie, N.; Heintze, S.; Cadenaro, M.; Hickel, R. Academy of Dental Materials guidance—Resin composites: Part II—Technique sensitivity (handling, polymerization, dimensional changes). Dent. Mater. 2017, 33, 1171–1191. [Google Scholar] [CrossRef] [Green Version]
- Algamaiah, H.; Silikas, N.; Watts, D.C. Conversion kinetics of rapid photo-polymerized resin composites. Dent. Mater. 2020, 36, 1266–1274. [Google Scholar] [CrossRef]
- Atai, M.; Watts, D.C. A new kinetic model for the photopolymerization shrinkage-strain of dental composites and resin-monomers. Dent. Mater. 2006, 22, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Delgado, A.H.S.; Young, A.M. Methacrylate peak determination and selection recommendations using ATR-FTIR to investigate polymerisation of dental methacrylate mixtures. PLoS ONE 2021, 16, e0252999. [Google Scholar] [CrossRef] [PubMed]
- Germscheid, W.; de Gorre, L.G.; Sullivan, B.; O’Neill, C.; Price, R.B.; Labrie, D. Post-curing in dental resin-based composites. Dent. Mater. 2018, 34, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Aljabo, A.; Xia, W.; Liaqat, S.; Khan, M.A.; Knowles, J.C.; Ashley, P.; Young, A.M. Conversion, shrinkage, water sorption, flexural strength and modulus of re-mineralizing dental composites. Dent. Mater. 2015, 31, 1279–1289. [Google Scholar] [CrossRef] [Green Version]
- Dressano, D.; Salvador, M.V.; Oliveira, M.T.; Marchi, G.M.; Fronza, B.M.; Hadis, M.; Palin, W.M.; Lima, A.F. Chemistry of novel and contemporary resin-based dental adhesives. J. Mech. Behav. Biomed. Mater. 2020, 110, 103875. [Google Scholar] [CrossRef] [PubMed]
- Kameyama, A.; Haruyama, A.; Abo, H.; Kojima, M.; Nakazawa, Y.; Muramatsu, T. Influence of solvent evaporation on ultimate tensile strength of contemporary dental adhesives. Appl. Adhes. Sci. 2019, 7, 4. [Google Scholar] [CrossRef]
- Parthasarathy, R.; Misra, A.; Song, L.; Ye, Q.; Spencer, P. Structure–property relationships for wet dentin adhesive polymers. Biointerphases 2018, 13, 061004. [Google Scholar] [CrossRef] [Green Version]
- Delgado, A.H.S.; Jamal, H.; Young, A.; Ashley, P. Scoping review of trials evaluating adhesive strategies in pediatric dentistry: Where do simplified strategies lie? BMC Oral Health 2021, 21, 33. [Google Scholar] [CrossRef]
- Mayerhöfer, T.G.; Pahlow, S.; Popp, J. The Bouguer-Beer-Lambert Law: Shining Light on the Obscure. ChemPhysChem 2020, 21, 2029–2046. [Google Scholar] [CrossRef]
- Coates, J. Interpretation of Infrared Spectra, A Practical Approach. In Encyclopedia of Analytical Chemistry; John Wiley & Sons: Chichester, UK, 2006. [Google Scholar]
- Qian, X.; Suh, B.; Hamer, M.; Tobias, R.H. One-Component Primer/Bonding-Resin Syste. Google Patents US5749733A, 12 May 1998. [Google Scholar]
- Yoshihara, K.; Nagaoka, N.; Hayakawa, S.; Okihara, T.; Yoshida, Y.; Van Meerbeek, B. Chemical interaction of glycero-phosphate dimethacrylate (GPDM) with hydroxyapatite and dentin. Dent. Mater. 2018, 34, 1072–1081. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Qian, X. One-Component Dental Adhesive Compositions and Method of Use. U.S. Patent 7968618B2, 3 August 2010. [Google Scholar]
- Young, A.M. FTIR investigation of polymerisation and polyacid neutralisation kinetics in resin-modified glass-ionomer dental cements. Biomaterials 2002, 23, 3289–3295. [Google Scholar] [CrossRef]
- Amaral, C.M.; Diniz, A.M.; Arantes, E.B.R.; dos Santos, G.B.; Noronha-Filho, J.D.; da Silva, E.M. Resin-dentin bond stability of experimental 4-META-based etch-and-rinse adhesives solvated by ethanol or acetone. J. Adhes. Dent. 2016, 18, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tjäderhane, L.; Breschi, L.; Mazzoni, A.; Li, N.; Mao, J.; Pashley, D.H.; Tay, F.R. Limitations in bonding to dentin and experimental strategies to prevent bond degradation. J. Dent. Res. 2011, 90, 953–968. [Google Scholar] [CrossRef]
- Frassetto, A.; Breschi, L.; Turco, G.; Marchesi, G.; Di Lenarda, R.; Tay, F.R.; Pashley, D.H.; Cadenaro, M. Mechanisms of degradation of the hybrid layer in adhesive dentistry and therapeutic agents to improve bond durability—A literature review. Dent. Mater. 2016, 32, e41–e53. [Google Scholar] [CrossRef]
- Cadenaro, M.; Breschi, L.; Rueggeberg, F.A.; Suchko, M.; Grodin, E.; Agee, K.; Di Lenarda, R.; Tay, F.R.; Pashley, D.H. Effects of residual ethanol on the rate and degree of conversion of five experimental resins. Dent. Mater. 2009, 25, 621–628. [Google Scholar] [CrossRef] [Green Version]
- Abu-Elenain, D.A.; Lewis, S.H.; Stansbury, J.W. Property evolution during vitrification of dimethacrylate photopolymer networks. Dent. Mater. 2013, 29, 1173–1181. [Google Scholar] [CrossRef] [Green Version]
- Sideridou, I.; Tserki, V.; Papanastasiou, G. Effect of chemical structure on degree of conversion in light-cured dimethacrylate-based dental resins. Biomaterials 2002, 23, 1819–1829. [Google Scholar] [CrossRef]
- Cadenaro, M.; Maravic, T.; Comba, A.; Mazzoni, A.; Fanfoni, L.; Hilton, T.; Ferracane, J.; Breschi, L. The role of polymerization in adhesive dentistry. Dent. Mater. 2018, 35, e1–e22. [Google Scholar] [CrossRef]
- Barszczewska-Rybarek, I.M. A guide through the dental dimethacrylate polymer network structural characterization and interpretation of physico-mechanical properties. Materials 2019, 12, 4057. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adhesive | Type/Batch | Composition |
|---|---|---|
| One-Step® (Bisco Inc., Anaheim, CA, USA) OS | Two-step etch-and-rinse1900003092 | Bis-GMA (10–30%), HEMA (10–30%), BPDM (10–30%) Solvent: Acetone (50–75%) Fillers: does not contain fillers |
| Optibond™ Universal (Kerr, Brea, CA, USA) OU | Universal/ Multimode 7020184 | HEMA (1–10%), GDMA (1–10%), GPDM (1–10%), Solvent: Acetone (30–60%), water (N/A), ethanol (1–10%) Fillers: N/A |
| G-Bond (GC, Tokyo, Japan) GB | One-step self-etch 2010091 | UDMA (5–10%), TEGDMA (5–10%), 10-MDP (2.5–5%) Solvent: Acetone (25–50%), water (15–25%) Fillers: Colloidal silica |
| Wavenumber (cm−1) | Methacrylate Peak Assignment | Wavenumber (cm−1) | Peak Assignment | Compound |
|---|---|---|---|---|
| 2950–2850 | C–H stretch | 3400 | O–H stretch | HEMA, Bis-GMA |
| 1700–1715 | C=O stretch | 3460 | O–H stretch | GDMA |
| 1635–1640 | C=C stretch | 3300 | O–H stretch/ N–H stretch | Water, Ethanol/ UDMA |
| 1390, 1365 | O–C–H bend | 1610 | Aromatic C=C stretch | Bis-GMA |
| 1320, 1300 | C–O stretch | 1510 | Aromatic C=C stretch | Bis-GMA |
| 1520 | N–H bend | UDMA | ||
| 1358 | CH3 bend | Acetone | ||
| 1235 | C–O stretch | Bis-GMA, UDMA | ||
| 1220 | C–C–C stretch | Acetone | ||
| 1145–1160 | C–O stretch | 10-MDP, MA | ||
| 1115 1075 | C–O–C stretch C–OH stretch | TEGDMA HEMA | ||
| 1100/1050 | C–OH stretch | Ethanol | ||
| 960 | P–O stretch | 10-MDP | ||
| 890 | C–C–O stretch | HEMA | ||
| 880 | C–C–O stretch | Ethanol |
| Components | Model Adhesive Chemical and Filler Fractions | |||||
|---|---|---|---|---|---|---|
| Monomers, Solvents or Fillers | OS Initial | OS 5 min | OU Initial | OU 5 min | GB Initial | GB 5 min |
| Bis-GMA | 0.35 | 0.60 | 0.15 | 0.35 | - | - |
| HEMA | 0.30 | 0.30 | 0.05 | - | - | - |
| GDMA | - | - | 0.15 | 0.30 | - | - |
| UDMA | - | - | - | - | 0.10 | 0.20 |
| TEGDMA (BPDM) | (0.10) | (0.23) | - | - | 0.20 | 0.35 |
| 10-MDP (GPDM) | - | - | (0.10) | (0.25) | 0.04 | 0.10 |
| Total monomers | 0.75 | 1.13 | 0.45 | 0.90 | 0.34 | 0.65 |
| Acetone | 0.50 | 0.00 | 0.40 | 0.00 | 0.50 | 0.10 |
| Water | - | - | 0.18 | 0.10 | 0.25 | 0.20 |
| Ethanol | - | - | 0.05 | - | - | - |
| Total solvent | 0.50 | 0.00 | 0.63 | 0.10 | 0.75 | 0.30 |
| Filler | - | - | 0.10 | 0.33 | 0.04 | 0.10 |
| Sum of fractions | 1.26 | 1.13 | 1.18 | 1.33 | 1.13 | 1.05 |
| Sum of Modulus of difference | 21 | 39 | 22 | 23 | 31 | 35 |
| Background absorbance | 0.01 | −0.004 | 0.00 | −0.004 | 0.003 | 0.03 |
| Adhesive | t0.5 (s) for Solvent Evaporation Determined Using Peaks for Acetone [1360 cm−1] Methacrylate [1320 cm−1] | DC (%) | |
|---|---|---|---|
| OS | 23 [1.3] A | 23 [1.3] A | 67.5 [0.01] A |
| OU | 25 [1.7] A | 26 [2.7] A | 68.8 [0.03] A |
| GB | 112 [15.6] B | 114 [13.3] B | 88.3 [0.02] B |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almusa, A.; Delgado, A.H.S.; Ashley, P.; Young, A.M. Determination of Dental Adhesive Composition throughout Solvent Drying and Polymerization Using ATR–FTIR Spectroscopy. Polymers 2021, 13, 3886. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13223886
Almusa A, Delgado AHS, Ashley P, Young AM. Determination of Dental Adhesive Composition throughout Solvent Drying and Polymerization Using ATR–FTIR Spectroscopy. Polymers. 2021; 13(22):3886. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13223886
Chicago/Turabian StyleAlmusa, Arwa, António H. S. Delgado, Paul Ashley, and Anne M. Young. 2021. "Determination of Dental Adhesive Composition throughout Solvent Drying and Polymerization Using ATR–FTIR Spectroscopy" Polymers 13, no. 22: 3886. https://0-doi-org.brum.beds.ac.uk/10.3390/polym13223886