
Immediate Release Formulation of Inhaled Beclomethasone Dipropionate-Hydroxypropyl-Beta-Cyclodextrin Composite Particles Produced Using Supercritical Assisted Atomization


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Phase Solubility Studies
2.3. HPLC Analysis of BDP
2.4. Production of Drug–HP-β-CD Composite Particles
2.5. Solid-State Characterization
2.6. In Vitro Aerosol Performance Determined Using an ACI
2.7. Drug Content Determination and In Vitro Dissolution Tests
3. Results and Discussion
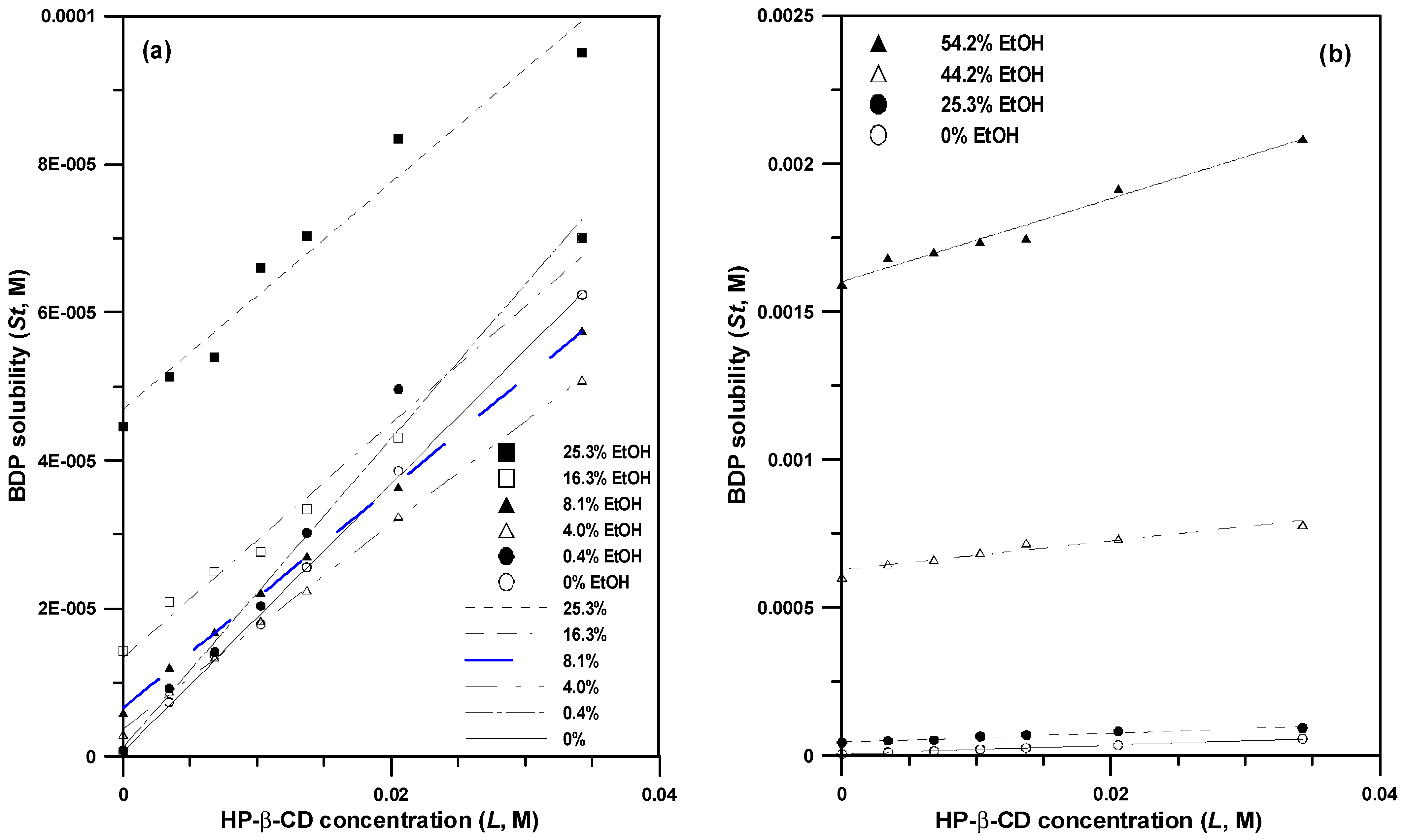
3.1. Phase Solubility Study
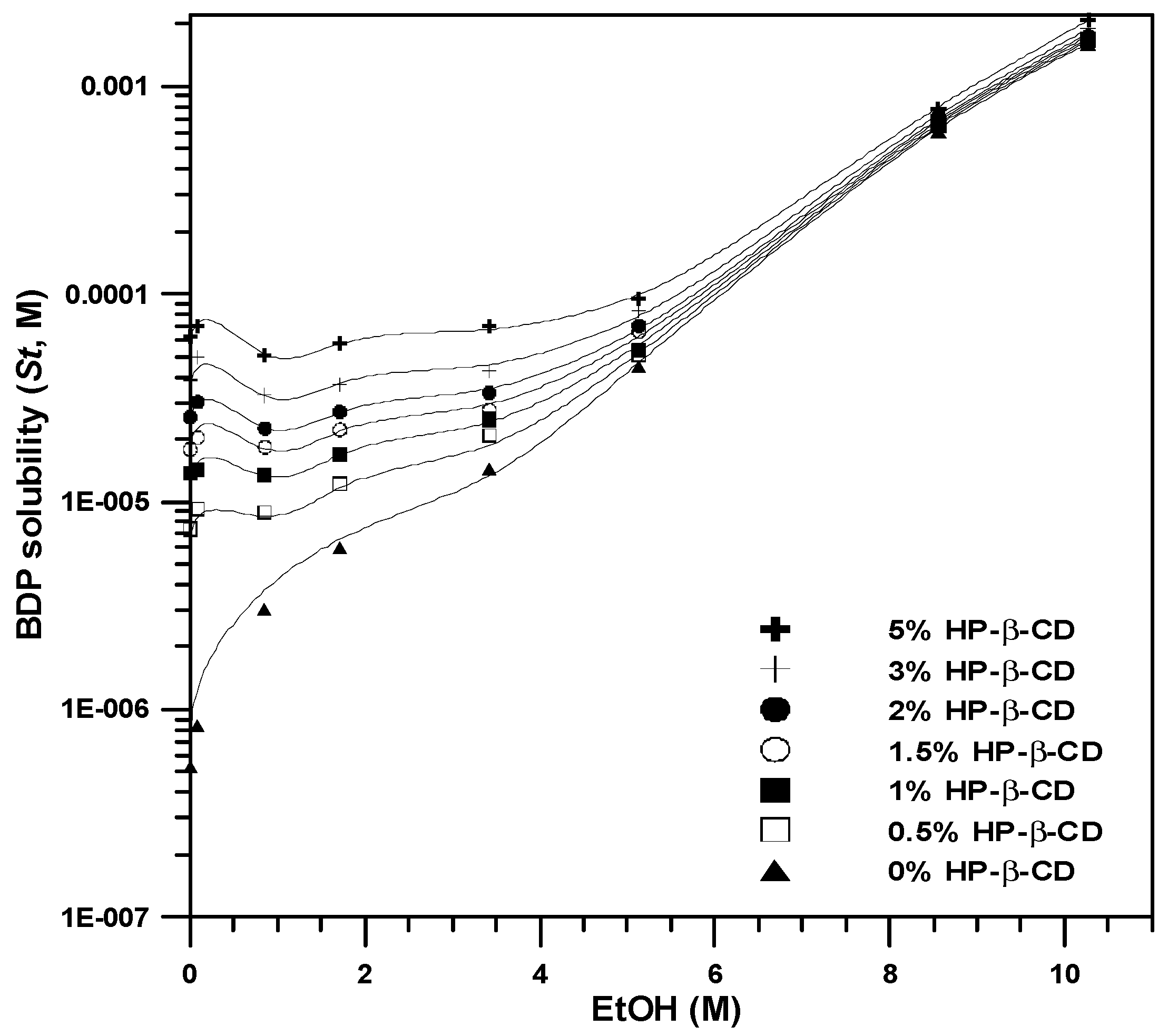
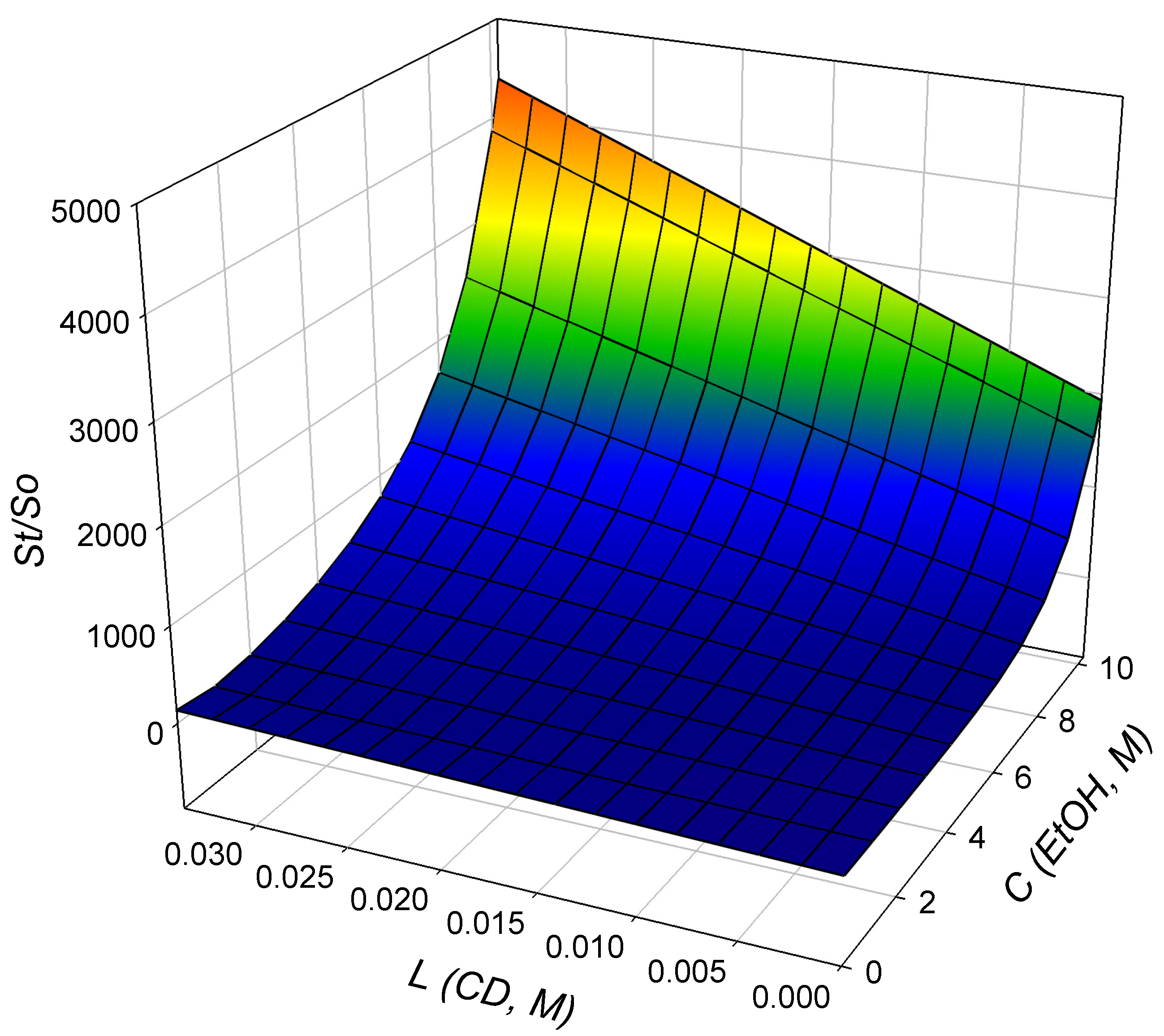
3.2. Total Drug Solubility of BDP
3.3. In Vitro Aerosolization Performance of the Drug–HP-β-CD Formulation
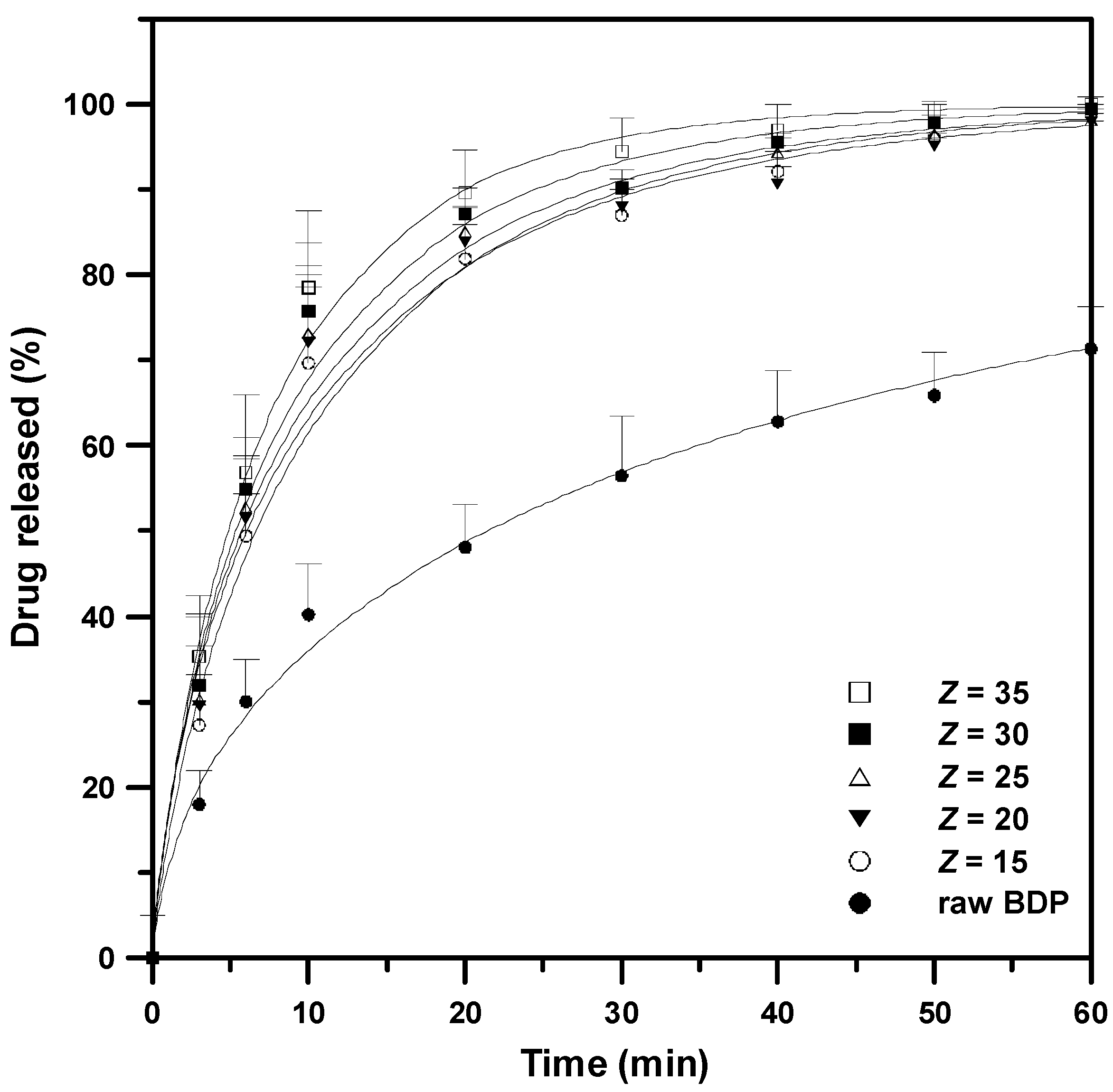
3.4. In Vitro Dissolution Performance of the Drug–HP-β-CD Formulation
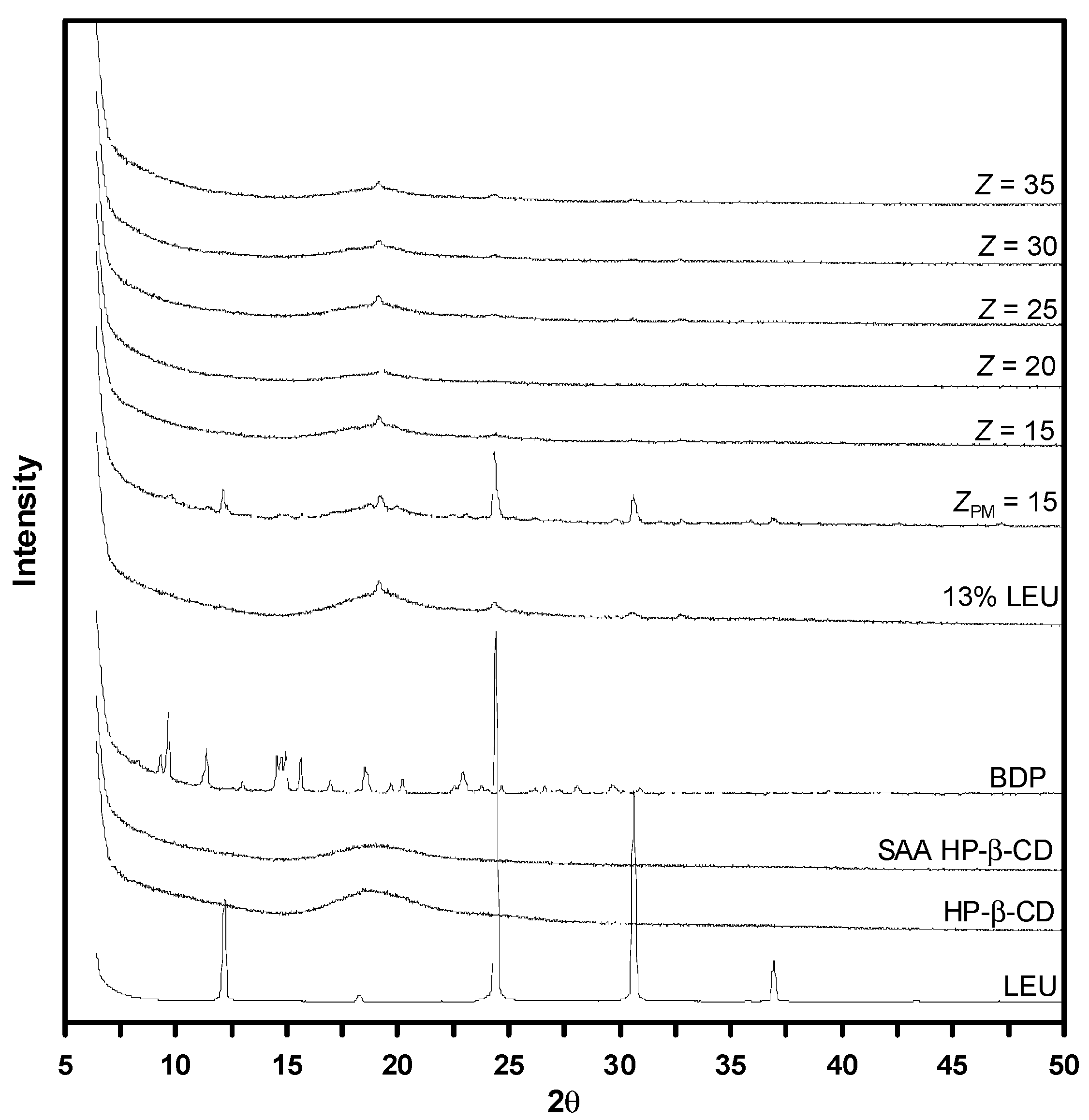
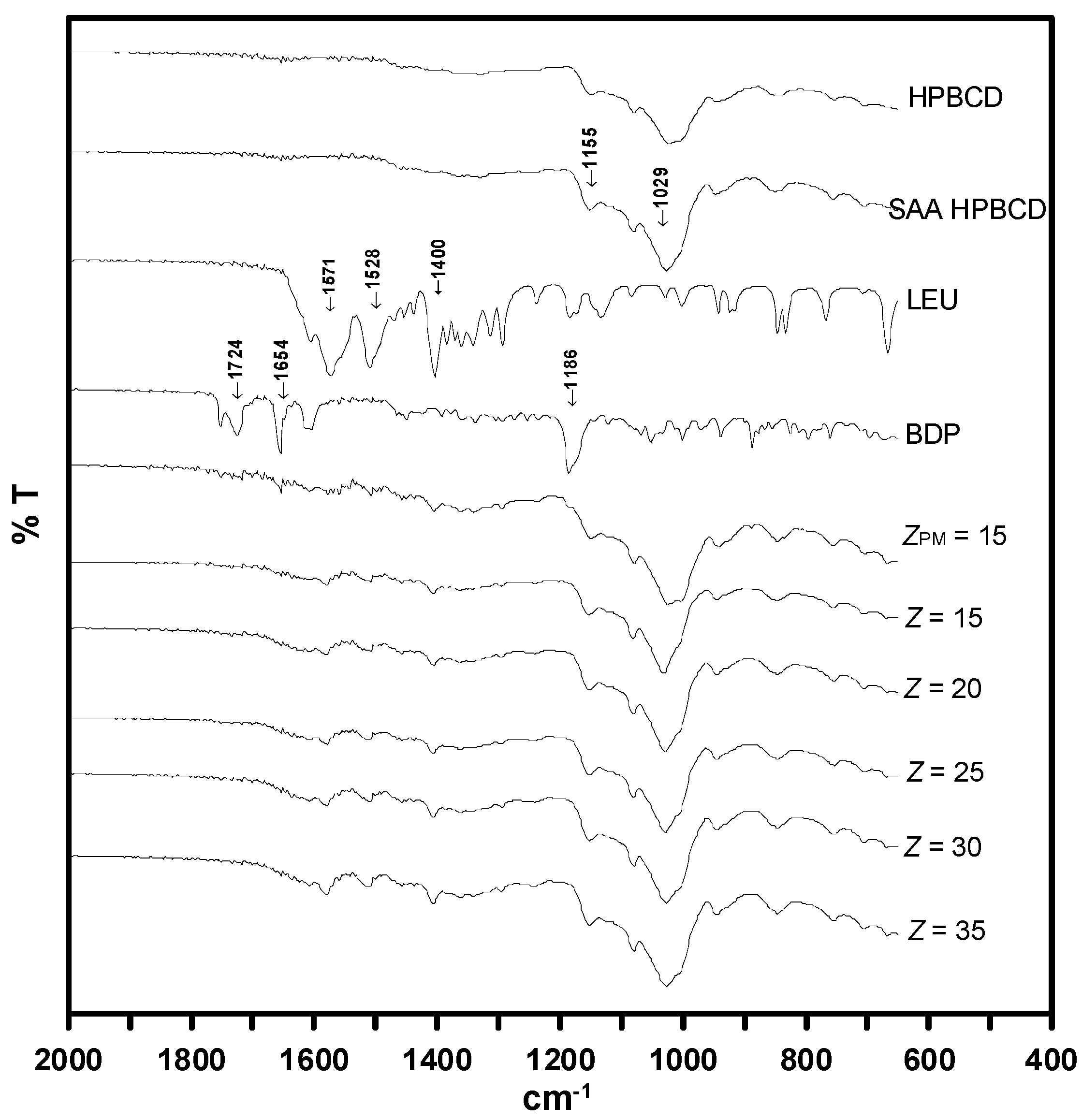
3.5. Solid Characterization
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Medicines Agency. Cyclodextrins Used as Excipients. 2017. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/questions-answers-cyclodextrins-used-excipients-medicinal-products-human-use_en.pdf (accessed on 9 October 2017).
- Vartiainen, V.; Bimbo, L.M.; Hirvonen, J.; Kauppinen, E.I.; Raula, J. Aerosolization, drug permeation and cellular interaction of dry powder pulmonary formulations of corticosteroids with hydroxypropyl-β-cyclodextrin as a solubilizer. Pharm. Res. 2017, 34, 25–35. [Google Scholar] [CrossRef]
- George, S.J.; Vasudevan, D.T. Studies on the preparation, characterization, and solubility of 2-HP-β-cyclodextrin-Meclizine HCl inclusion complexes. J. Young Pharm. 2012, 4, 220–227. [Google Scholar] [CrossRef] [Green Version]
- Bandi, N.; Wei, W.; Roberts, C.B.; Kotra, L.P.; Kompella, U.B. Preparation of budesonide– and indomethacin–hydroxypropyl-β-cyclodextrin (HPBCD) complexes using a single-step, organic-solvent-free supercritical fluid process. Eur. J. Pharm. Sci. 2004, 23, 159–168. [Google Scholar] [CrossRef]
- Matilainen, L.; Toropainen, T.; Vihola, H.; Hirvonen, J.; Järvinen, T.; Jarho, P.; Järvinen, K. In vitro toxicity and permeation of cyclodextrins in Calu-3 cells. J. Contr. Release 2008, 126, 10–16. [Google Scholar] [CrossRef]
- Gao, S.; Jiang, J.; Li, X.; Ye, F.; Fu, Y.; Zhao, L. Electrospun polymer-free nanofibers incorporating hydroxypropyl-β-cyclodextrin/difenoconazole via supramolecular assembly for antifungal activity. J. Agric. Food Chem. 2021, 69, 5871–5881. [Google Scholar] [CrossRef]
- Gao, S.; Li, X.; Jiang, J.; Zhao, L.; Fu, Y.; Ye, F. Fabrication and characterization of thiophanate methyl/hydroxypropyl-β-cyclodextrin inclusion complex nanofibers by electrospinning. J. Mol. Liq. 2021, 335, 11622. [Google Scholar] [CrossRef]
- Gilani, K.; Najafabadi, A.R.; Barghi, M.; Rafiee-Tehrani, M. Aerosolisation of beclomethasone dipropionate using spray dried lactose/polyethylene glycol carriers. Eur. J. Pharm. Biopharm. 2004, 58, 595–606. [Google Scholar] [CrossRef]
- Cabral-Marques, H.; Almeida, R. Optimisation of spray-drying process variables for dry powder inhalation (DPI) formulations of corticosteroid/cyclodextrin inclusion complexes. Eur. J. Pharm. Biopharm. 2009, 73, 121–129. [Google Scholar] [CrossRef]
- Amaro, M.I.; Tajber, L.; Corrigan, O.I.; Healy, A.M. Co-spray dried carbohydrate microparticles: Crystallisation delay/inhibition and improved aerosolization characteristics through the incorporation of hydroxypropyl-β-cyclodextrin with amorphous raffinose or trehalose. Pharm. Res. 2015, 32, 180–195. [Google Scholar] [CrossRef]
- Mohtar, N.; Taylor, K.M.G.; Sheikh, K.; Somavarapu, S. Design and development of dry powder sulfobutylether-β-cyclodextrin complex for pulmonary delivery of fisetin. Eur. J. Pharm. Biopharm. 2017, 113, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, É.Y.; Amaro, M.I.; de Almeida, G.S.; Cabral, L.M.; Healy, A.M.; de Sousa, V.P. Development of a new formulation of roflumilast for pulmonary drug delivery to treat inflammatory lung conditions. Int. J. Pharm. 2018, 550, 89–99. [Google Scholar] [CrossRef]
- Wu, H.T.; Huang, S.C.; Yang, C.P.; Chien, L.J. Precipitation parameters and the cytotoxicity of chitosan hydrochloride microparticles production by supercritical assisted atomization. J. Supercrit. Fluids 2015, 102, 123–132. [Google Scholar] [CrossRef]
- Reverchon, E.; Adami, R.; Caputo, G. Supercritical assisted atomization: Performance comparison between laboratory and pilot scale. J. Supercrit. Fluids 2006, 37, 298–306. [Google Scholar] [CrossRef]
- Adami, R.; Liparoti, S.; Reverchon, E. A new supercritical assisted atomization configuration, for the micronization of thermolabile compounds. Chem. Eng. J. 2011, 173, 55–61. [Google Scholar] [CrossRef]
- Liparoti, S.; Adami, R.; Reverchon, E. PEG micronization by supercritical assisted atomization, operated under reduced pressure. J. Supercrit. Fluids 2012, 72, 46–51. [Google Scholar] [CrossRef]
- Wu, H.T.; Tsai, H.M.; Li, T.H. Formation of polyethylene glycol particles using a low-temperature supercritical assisted atomization process. Molecules 2019, 24, 2235. [Google Scholar] [CrossRef] [Green Version]
- De Marco, I.; Franco, P. Effect of the carrier on the coprecipitation of curcumin through supercritical-assisted atomization. ChemEngineering 2021, 5, 59. [Google Scholar] [CrossRef]
- Di Capua, A.; Bejarano, A.; Adami, R.; Reverchon, E. Preparation and characterization of Chilean propolis coprecipitates using supercritical assisted atomization. Chem. Eng. Res. Des. 2018, 136, 776–785. [Google Scholar] [CrossRef]
- Higuchi, T.; Connors, K.A. Phase-solubility techniques. Adv. Anal. Chem. Instrum. 1965, 4, 117–212. [Google Scholar]
- Sahib, M.N.; Darwis, Y.; Khiang, P.K.; Tan, Y.T.F. Aerodynamic characterization of beclomethasone dipropionate from Beclate-50 Inhaler® by HPLC-UV. J. Liq. Chromatogr. Relat. 2011, 34, 613–621. [Google Scholar] [CrossRef]
- Wu, H.T.; Li, T.H.; Tsai, H.M.; Chien, L.J.; Chuang, Y.H. Formulation of inhalable beclomethasone dipropionate-mannitol composite particles through low-temperature supercritical assisted atomization. J. Supercrit. Fluids 2021, 168, 105095. [Google Scholar] [CrossRef]
- Reverchon, E.; Antonacci, A. Cyclodextrins micrometric powders obtained by supercritical fluid processing. Biotechnol. Bioeng. 2006, 94, 753–761. [Google Scholar] [CrossRef]
- Day, C.P.F.; Miloserdov, A.; Wildish-Jones, K.; Pearson, E.; Carruthers, A.E. Quantifying the hygroscopic properties of cyclodextrin containing aerosol for drug delivery to the lungs. Phys. Chem. Chem. Phys. 2020, 22, 11327–11336. [Google Scholar] [CrossRef]
- O’Shaughnessy, P.T.; Raabe, O.G. A comparison of cascade impactor data reduction methods. Aerosol Sci. Technol. 2003, 37, 187–200. [Google Scholar] [CrossRef]
- Assem, M.; Khowessah, O.M.; Ghorab, D. Nano-crystallization as a tool for the enhancement of beclomethasone dipropionate dermal deposition: Formulation, in vitro characterization and ex vivo study. J. Drug Deliv. Sci. Technol. 2019, 54, 101318. [Google Scholar] [CrossRef]
- Terakosolphan, W.; Hassoun, M.; Kumar, A.; Forbes, B. Solubility of fluticasone propionate and beclomethasone dipropionate in simulated lung lining fluids. Drug Deliv. Lungs 2016, 27. Available online: https://aerosol-soc.com/assets/uploads/2016/11/59.Terakosolphan.pdf (accessed on 16 April 2022).
- Malaekeh-Nikouei, B.; Tabassi, S.A.S.; Gerayeli, G.; Salmani, M.A.; Gholamzadeh, A. The effect of cyclodextrin mixtures on aqueous solubility of beclomethasone dipropionate. J. Incl. Phenom. Macrocycl. Chem. 2012, 72, 383–387. [Google Scholar] [CrossRef]
- Sakagami, M.; Kinoshita, W.; Sakon, K.; Sato, J.-I.; Makino, Y. Mucoadhesive beclomethasone microspheres for powder inhalation: Their pharmacokinetics and pharmacodynamics evaluation. J. Control. Release 2002, 80, 207–218. [Google Scholar] [CrossRef]
- Junyaprasert, V.B.; Morakul, B. Nanocrystals for enhancement of oral bioavailability of poorly water-soluble drugs. Asian J. Pharm. Sci. 2015, 10, 13–23. [Google Scholar] [CrossRef] [Green Version]
- Loftsson, T.; Hreinsdottir, D.; Masson, M. Evaluation of cyclodextrin solubilization of drugs. Int. J. Pharm. 2005, 302, 18–28. [Google Scholar] [CrossRef]
- Charumanee, S.; Okonogi, S.; Sirithunyalug, J.; Wolschann, P. Effect of cyclodextrin types and co-solvent on solubility of a poorly water soluble drug. Sci. Pharm. 2016, 84, 694–704. [Google Scholar] [CrossRef] [Green Version]
- Nakhle, L.; Kfoury, M.; Greige-Gerges, H.; Fourmentin, S. Effect of dimethylsulfoxide, ethanol, α- and β-cyclodextrins and their association on the solubility of natural bioactive compounds. J. Mol. Liq. 2020, 310, 113156. [Google Scholar] [CrossRef]
- He, Y.; Li, P.; Yalkowsky, S.H. Solubilization of fluasterone in cosolvent/cyclodextrin combinations. Int. J. Pharm. 2003, 264, 25–34. [Google Scholar] [CrossRef]
- Li, R.; Quan, P.; Liu, D.F.; Wei, F.D.; Zhang, Q.; Xu, Q.W. The Influence of cosolvent on the complexation of HP-β-cyclodextrins with oleanolic acid and ursolic acid. AAPS PharmSciTech 2009, 10, 1137–1144. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.T.; Chuang, Y.H.; Lin, H.C.; Chien, L.J. Characterization and aerosolization performance of hydroxypropyl-β-xyclodextrin particles produced using supercritical assisted atomization. Polymers 2021, 13, 2260. [Google Scholar] [CrossRef]
- Chvatal, A.; Ambrus, R.; Party, P.; Katona, G.; Jójárt-Laczkovich, O.; Szabó-Révész, P.; Fattal, E.; Tsapis, N. Formulation and comparison of spray dried non-porous and large porous particles containing meloxicam for pulmonary drug delivery. Int. J. Pharm. 2019, 559, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Molina, C.; Kaialy, W.; Nokhodchi, A. The crucial role of leucine concentration on spray dried mannitol-leucine as a single carrier to enhance the aerosolization performance of albuterol sulfate. J. Drug Deliv. Sci. Technol. 2019, 49, 97–106. [Google Scholar] [CrossRef]
- Raula, J.; Kuivanen, A.; Lähde, A.; Jiang, H.; Antopolsky, M.; Kansikase, J.; Kauppinen, E.I. Synthesis of L-leucine nanoparticles via physical vapor deposition at varying saturation conditions. J. Aerosol Sci. 2007, 38, 1172–1184. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.F.; Le, Y. Preparation of ultrafine beclomethasone dipropionate drug powder by antisolvent precipitation. Ind. Eng. Chem. Res. 2007, 46, 4839–4845. [Google Scholar] [CrossRef]
- Casula, L.; Lai, F.; Pini, E.; Valenti, D.; Sinico, C.; Cardia, M.C.; Marceddu, S.; Ailuno, G.; Fadda, A.M. Pulmonary delivery of curcumin and beclomethasone dipropionate in a multicomponent nanosuspension for the treatment of bronchial asthma. Pharmaceutics 2021, 13, 1300. [Google Scholar] [CrossRef]
- Veiga, M.D.; Merino, M.; Fernández, D.; Lozano, R. Characterization of some cyclodextrin derivatives by thermal analysis. J. Therm. Anal. Calorim. 2002, 68, 511–516. [Google Scholar] [CrossRef]
- Cabral Marques, H.M.; Hadgraft, J.; Kellaway, I.W. Studies of cyclodextrin inclusion complexes. I. The salbutamol-cyclodextrin complex as studied by phase solubility and DSC. Int. J. Pharm. 1990, 63, 259–266. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EtOH (%, w/w) | Complexation Efficiency (CE) | R2 |
|---|---|---|
| 0 | 0.00181 | 0.99 |
| 0.4 | 0.00197 | 0.99 |
| 4.0 | 0.00139 | 0.99 |
| 8.1 | 0.00149 | 0.99 |
| 16.3 | 0.00158 | 0.99 |
| 25.3 | 0.00154 | 0.96 |
| 44.2 | 0.00488 | 0.95 |
| 54.2 | 0.0143 | 0.98 |
| Parameter Symbol | Calculated Parameters |
|---|---|
| σ (mM−1) | 0.3200 |
| Kb (mM−1) | 3481.6 |
| Kt (mM−2) | 220.61 |
| ρb (mM−1) | 0.4863 |
| ρt (mM−1) | 0.1704 |
| R2 | 0.93 |
| Run | Z | CBDP | CLEU | ED | FPF | MMAD | Drug Content | dno | d4,3 | ρtap | HR |
|---|---|---|---|---|---|---|---|---|---|---|---|
| mg/mL | mg/mL | % | % | µm | % | µm | µm | g/cm3 | - | ||
| B1 1 | - | 0 | 97.4 | 21.8 ± 2.0 | 3.95 | - | - | - | - | - | |
| B2 | 15 | 0.67 | 0.1 | 95.8 | 25.5 ± 1.0 | 3.43 | 6.20 ± 0.1 | - | - | 0.22 ± 0.01 | 1.44 ± 0.03 |
| B3 | 15 | 0.67 | 1.5 | 97.4 | 41.8 ± 2.3 | 2.70 | 5.53 ± 0.1 | 1.20 ± 0.20 | 1.41 ± 0.20 | 0.20 ± 0.02 | 1.39 ± 0.01 |
| B4 | 20 | 0.5 | 1.5 | 96.5 | 43.8 ± 5.0 | 3.13 | 4.13 ± 0.2 | 1.39 ± 0.15 | 1.46 ± 0.30 | 0.21 ± 0.02 | 1.38 ± 0.02 |
| B5 | 25 | 0.4 | 1.5 | 99.5 | 45.6 ± 4.0 | 2.52 | 3.34 ± 0.1 | 1.45 ± 0.20 | 1.53 ± 0.25 | 0.20 ± 0.01 | 1.33 ± 0.02 |
| B6 | 30 | 0.33 | 1.5 | 94.2 | 49.2 ± 5.0 | 2.22 | 2.79 ± 0.3 | 1.46 ± 0.22 | 1.55 ± 0.15 | 0.21 ± 0.02 | 1.36 ± 0.04 |
| B7 | 35 | 0.29 | 1.5 | 98.4 | 52.2 ± 3.0 | 2.48 | 2.37 ± 0.2 | 1.49 ± 0.20 | 1.71 ± 0.18 | 0.19 ± 0.01 | 1.30 ± 0.05 |
| Run | Z | Weibull 1 | |||
|---|---|---|---|---|---|
| a | b | r2 | Td2 | ||
| B1 3 | - | 8.385 | 0.574 | 0.98 | 40.5 |
| B3 | 15 | 6.473 | 0.791 | 0.98 | 10.6 |
| B4 | 20 | 5.316 | 0.726 | 0.97 | 10.0 |
| B5 | 25 | 5.328 | 0.750 | 0.98 | 9.3 |
| B6 | 30 | 5.407 | 0.788 | 0.98 | 8.5 |
| B7 | 35 | 5.445 | 0.843 | 0.98 | 7.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.-T.; Chuang, Y.-H.; Lin, H.-C.; Hu, T.-C.; Tu, Y.-J.; Chien, L.-J. Immediate Release Formulation of Inhaled Beclomethasone Dipropionate-Hydroxypropyl-Beta-Cyclodextrin Composite Particles Produced Using Supercritical Assisted Atomization. Polymers 2022, 14, 2114. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14102114
Wu H-T, Chuang Y-H, Lin H-C, Hu T-C, Tu Y-J, Chien L-J. Immediate Release Formulation of Inhaled Beclomethasone Dipropionate-Hydroxypropyl-Beta-Cyclodextrin Composite Particles Produced Using Supercritical Assisted Atomization. Polymers. 2022; 14(10):2114. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14102114
Chicago/Turabian StyleWu, Hsien-Tsung, Yao-Hsiang Chuang, Han-Cyuan Lin, Tzu-Chieh Hu, Yi-Jia Tu, and Liang-Jung Chien. 2022. "Immediate Release Formulation of Inhaled Beclomethasone Dipropionate-Hydroxypropyl-Beta-Cyclodextrin Composite Particles Produced Using Supercritical Assisted Atomization" Polymers 14, no. 10: 2114. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14102114