
Tablet Formulations of Polymeric Electrospun Fibers for the Controlled Release of Drugs with pH-Dependent Solubility



Abstract
:1. Introduction
2. Materials and Methods
2.1. Polymer Electrospun Nanofibers Preparation
2.2. Formulations and Tablets Preparation
2.3. Morphological Characterization: Scanning Electron Microscopy (SEM)
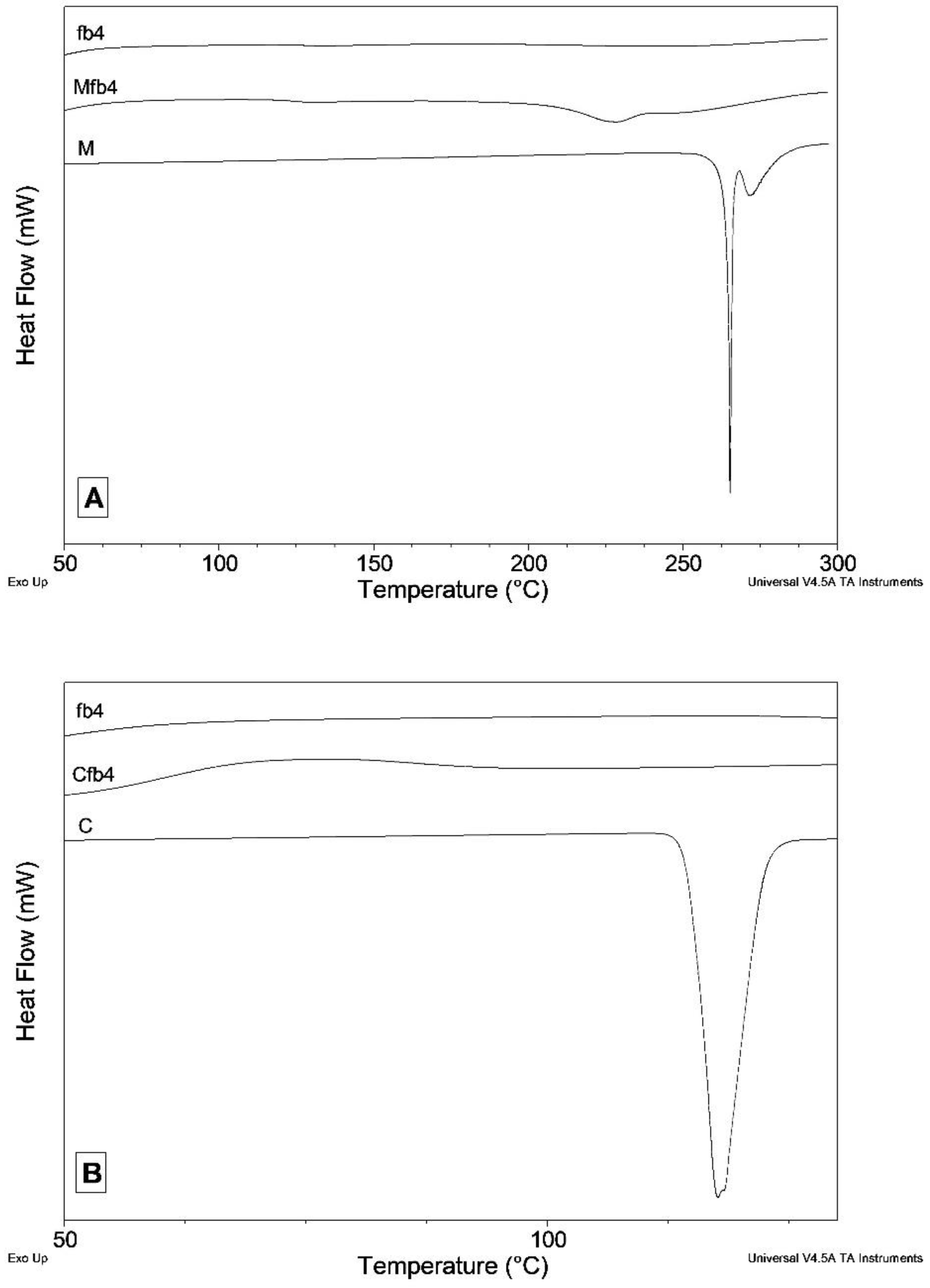
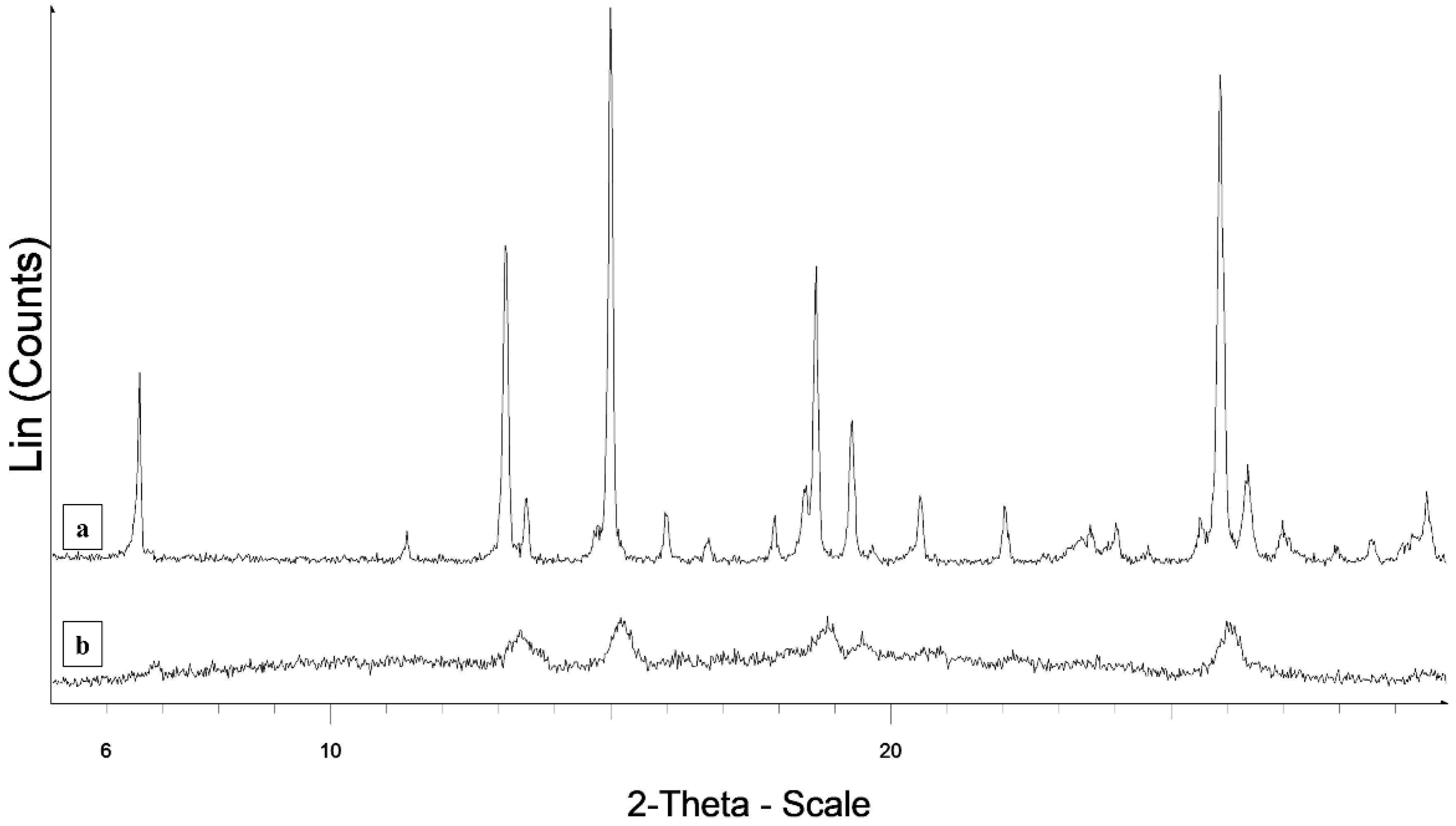
2.4. Differential Scanning Calorimetry (DSC) and X-ray Powder Diffraction (XRPD)
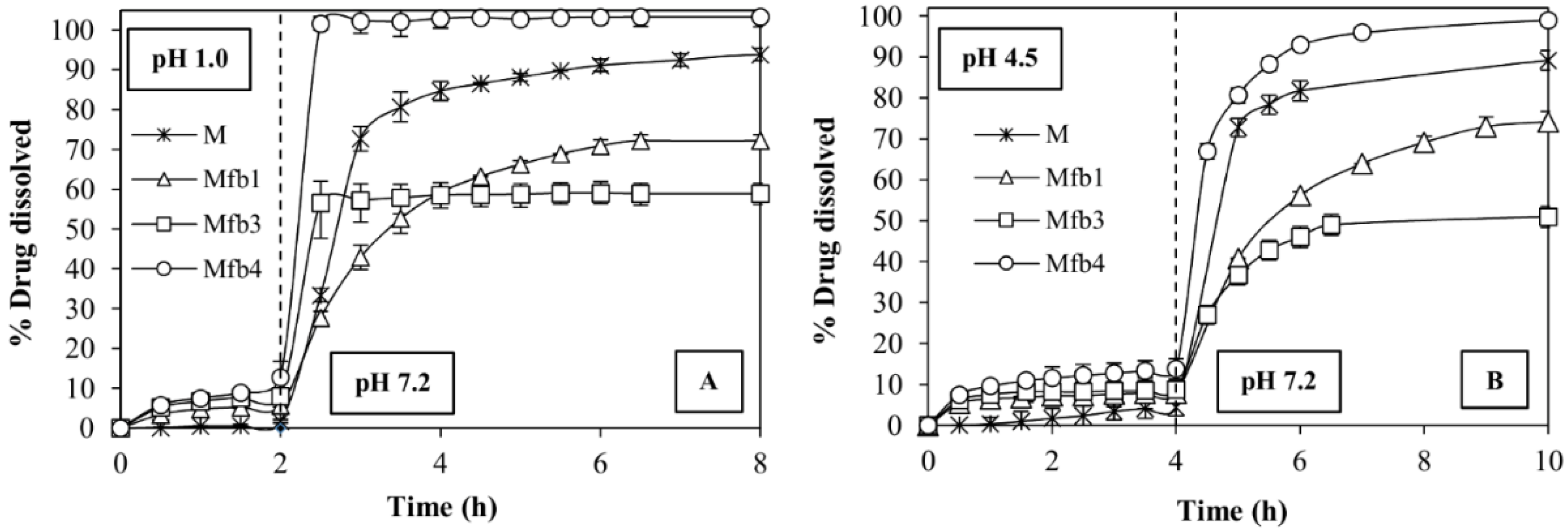
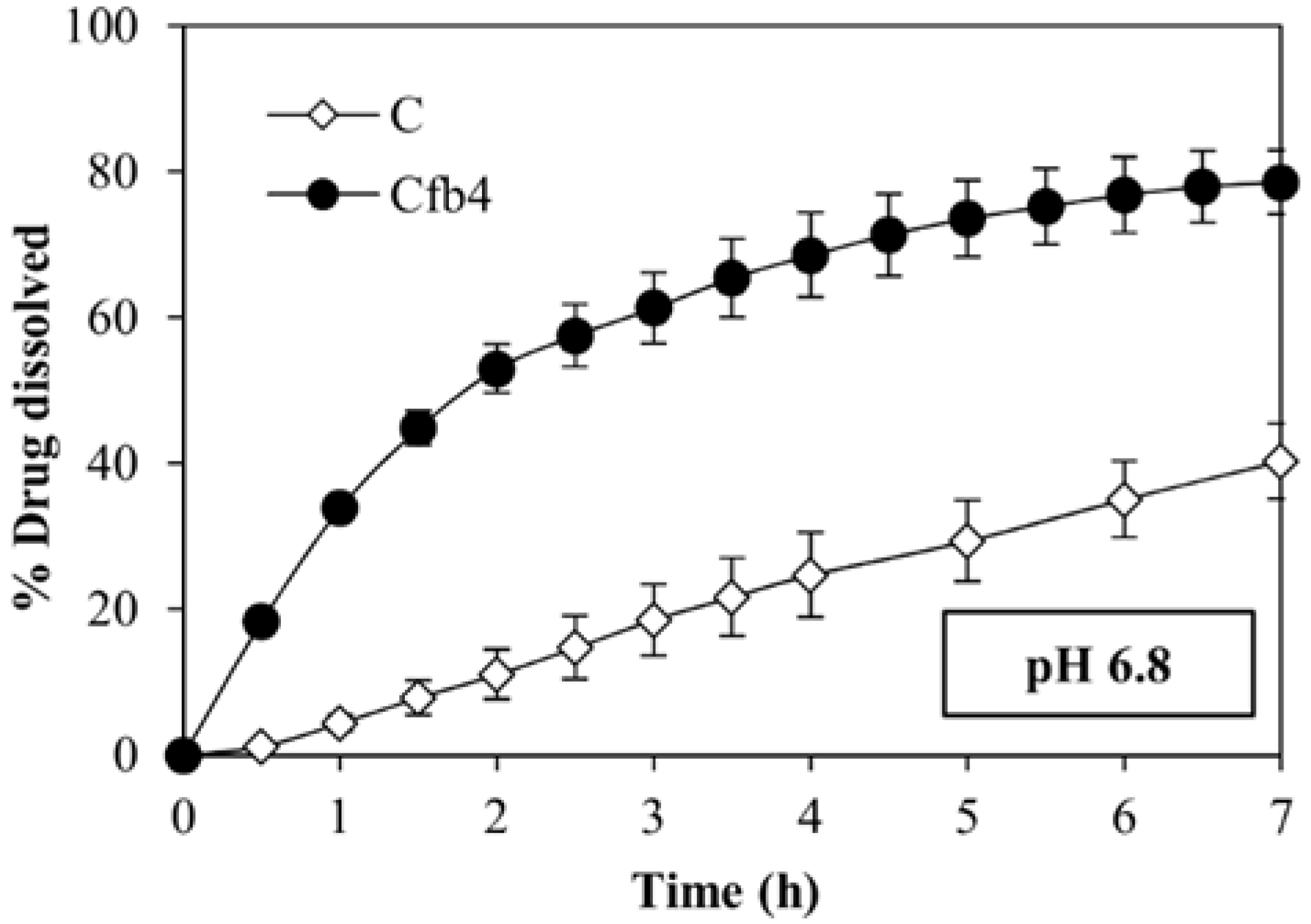
2.5. Dissolution Tests
2.6. Contact Angle
2.7. Release Kinetics
3. Results
3.1. Polymeric Electrospun Nanofibers Preparation
3.2. SEM Measurements
3.3. DSC and XRPD Measurements
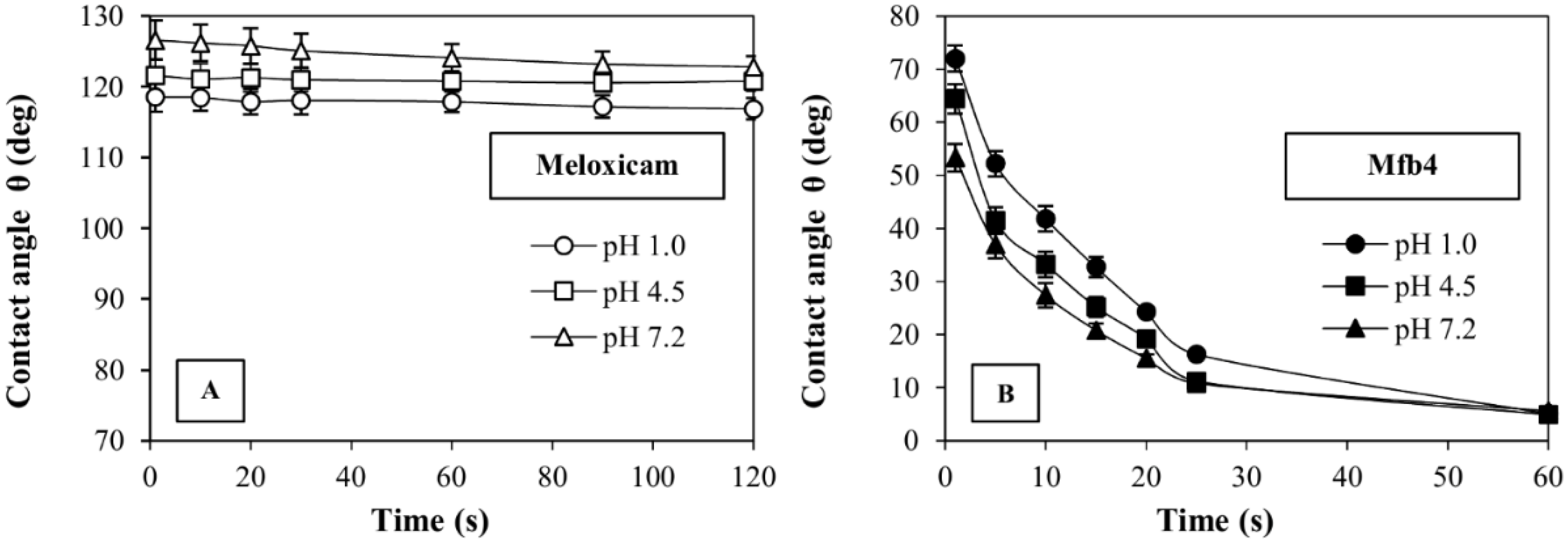
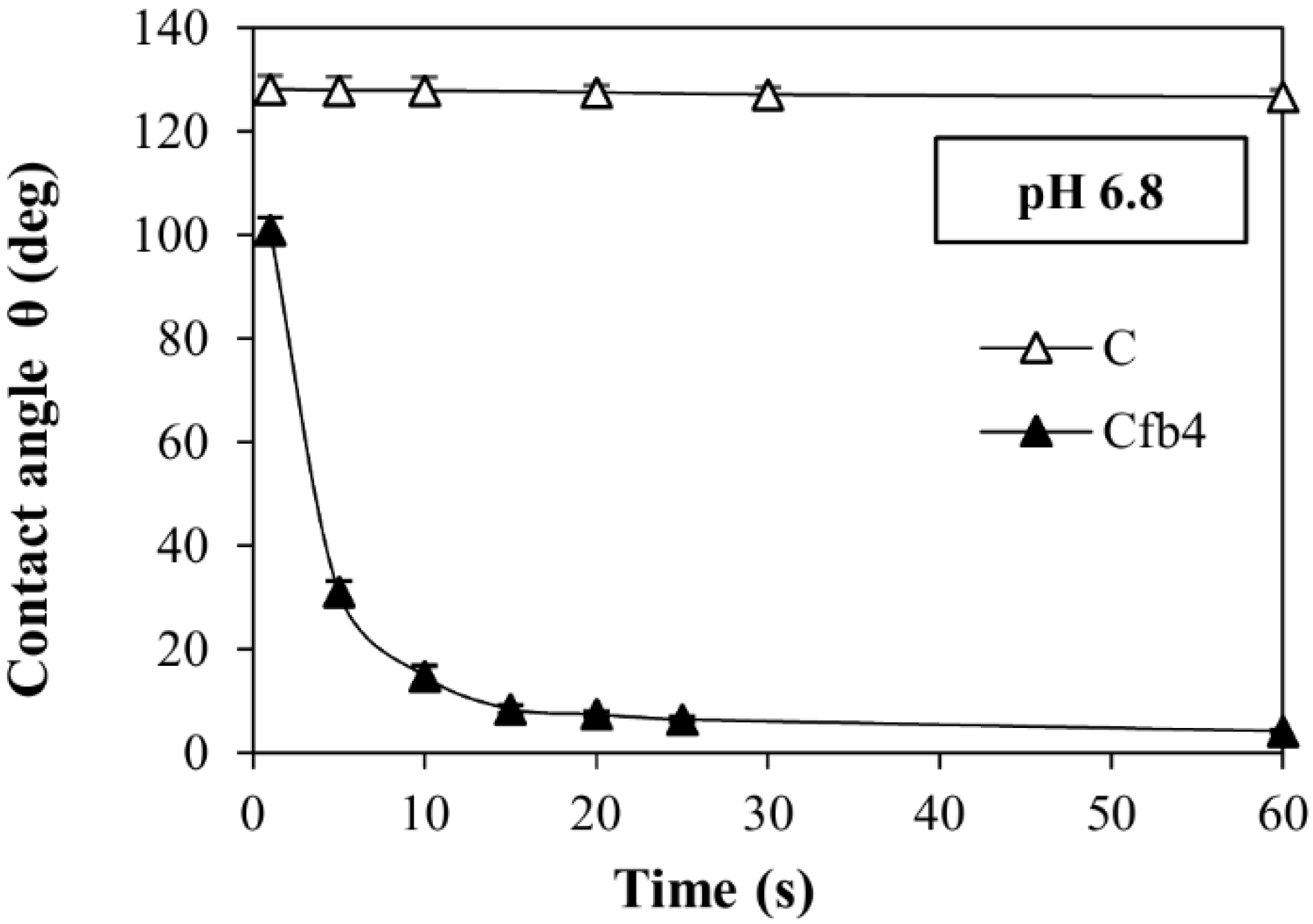
3.4. Dissolution Tests and Contact Angle
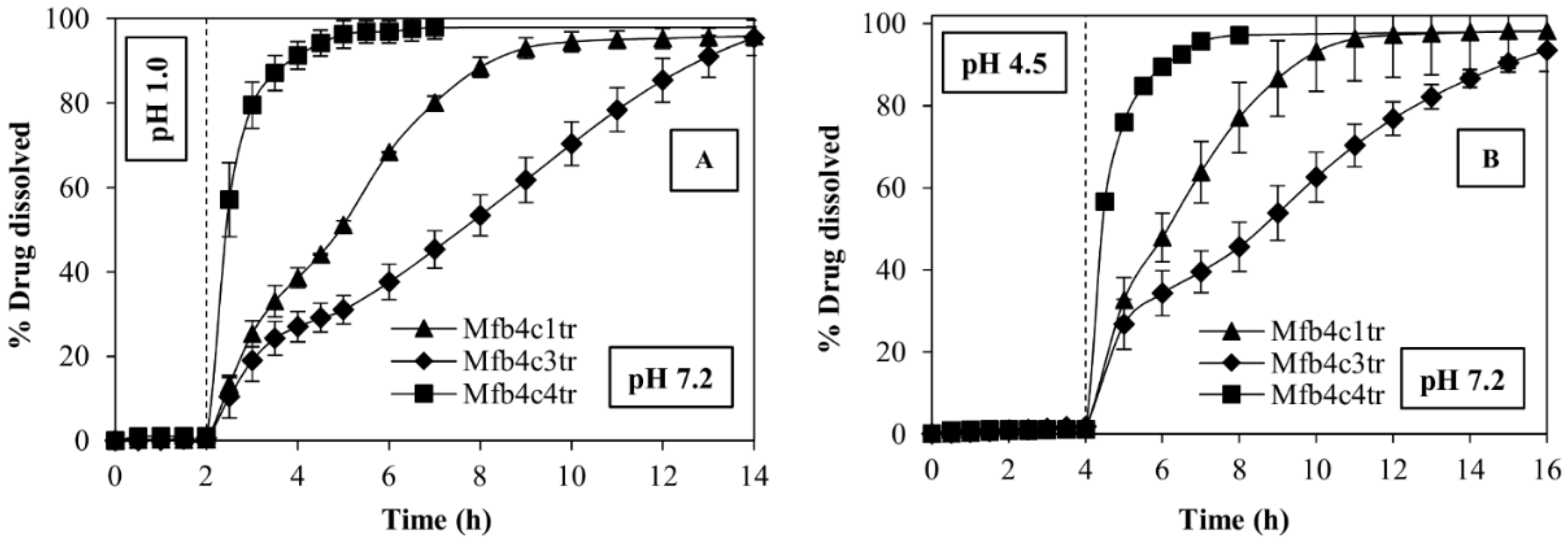
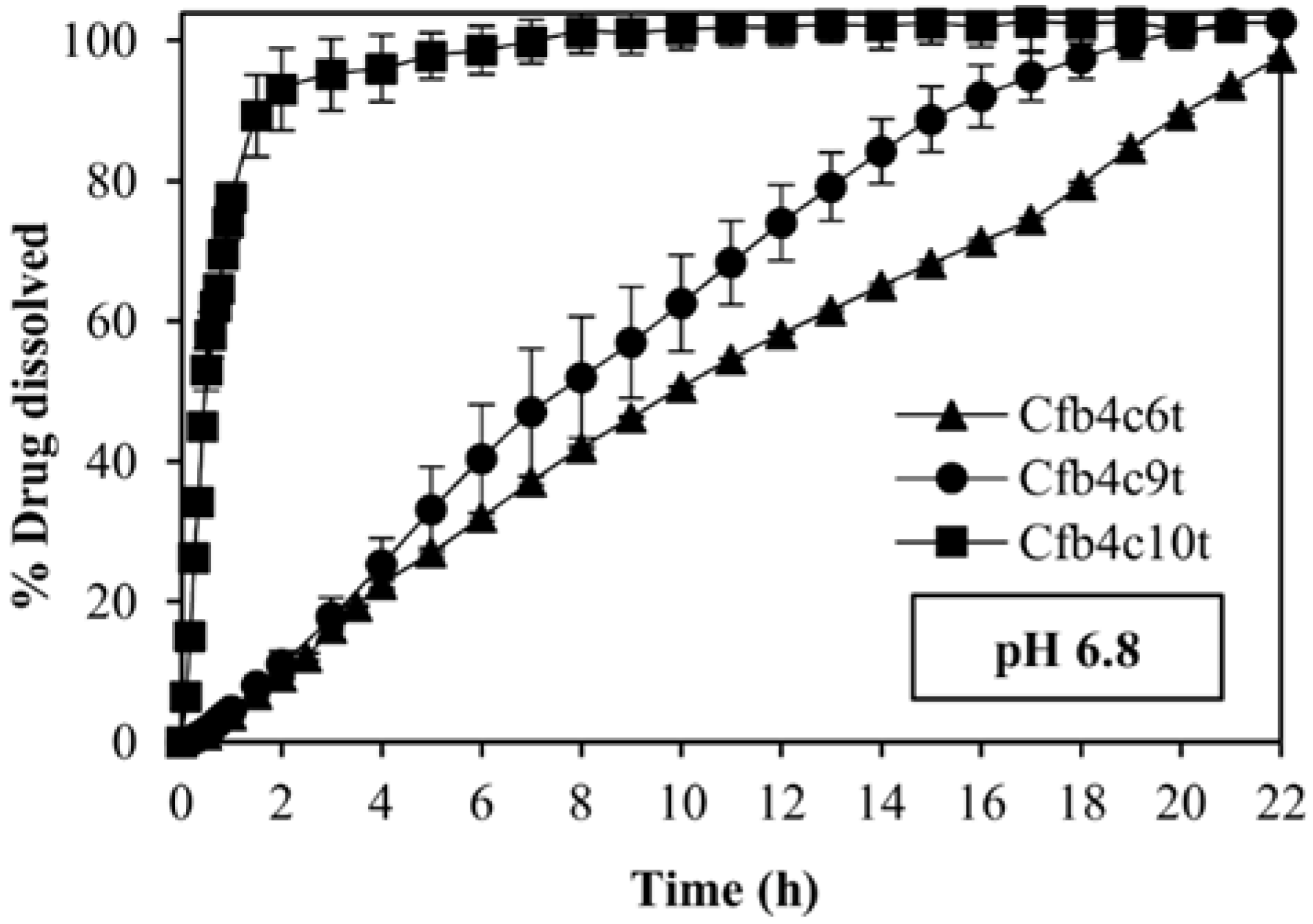
3.5. Release Kinetics
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Villegas, I.; La Casa, C.; de la Lastra, C.A.; Motilva, V.; Herrerías, J.M.; Martín, M.J. Mucosal damage induced by preferential COX-1 and COX-2 inhibitors: Role of prostaglandins and inflammatory response. Life Sci. 2004, 74, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Pandit, J.K.; Singh, S.; Muthu, M.S. Controlled release formulations in neurology practice. Ann. Indian Acad. Neurol. 2006, 9, 207. [Google Scholar] [CrossRef]
- Conti, S.; Maggi, L.; Segale, L.; Ochoa Machiste, E.; Conte, U.; Grenier, P.; Vergnault, G. Matrices containing NaCMC and HPMC 1. Dissolution performance characterization. Int. J. Pharm. 2007, 2, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wen, H.; Park, K. Challenges and new technologies of oral controlled release. In Oral Controlled Release Formulation Design and Drug Delivery: Theory to Practice; Wiley: Hoboken, NJ, USA, 2010; pp. 257–277. [Google Scholar]
- Ghorpade, V.S.; Yadav, A.V.; Dias, R.J. Citric acid crosslinked β-cyclodextrin/carboxymethylcellulose hydrogel films for controlled delivery of poorly soluble drugs. Carbohydr. Polym. 2017, 164, 339–348. [Google Scholar] [CrossRef]
- Kaur, G.; Grewal, J.; Jyoti, K.; Jain, U.K.; Chandra, R.; Madan, J. Chapter 15—Oral controlled and sustained drug delivery systems: Concepts, advances, preclinical, and clinical status, in Drug Targeting and Stimuli Sensitive Drug Delivery Systems; Grumezescu, A.M., Ed.; William Andrew Publishing: Norwich, UK, 2018; pp. 567–626. [Google Scholar]
- Bruni, G.; Maggi, L.; Mustarelli, P.; Sakaj, M.; Friuli, V.; Ferrara, C.; Berbenni, V.; Girella, A.; Milanese, C.; Marini, A. Enhancing the pharmaceutical behavior of nateglinide by cocrystallization: Physicochemical assessment of cocrystal formation and informed use of differential scanning calorimetry for its quantitative characterization. J. Pharm. Sci. 2019, 108, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- Bruni, G.; Monteforte, F.; Maggi Friuli, F.; Ferrara, C.; Mustarelli, P.; Girella, A.; Berbenni, V.; Capsoni, D.; Milanese, C.; Marini, A. Probenecid and benzamide: Cocrystal prepared by a green method and its physic-chemical and pharmaceutical characterization. J. Therm. Anal. Calorim. 2020, 140, 1859–1869. [Google Scholar] [CrossRef]
- Monteforte, F.; Bruni, G.; Quinzeni, I.; Friuli, V.; Maggi, L.; Capsoni, D.; Bini, M. Meloxicam-LDH hybrid compound: A successful strategy to improve solubility. J. Inorg. Organomet. Polym. Mater. 2020, 30, 637–648. [Google Scholar] [CrossRef]
- Guagliano, M.; Monteforte, F.; Bruni, G.; Friuli, V.; Maggi, L.; Quinzeni, I.; Bini, M. The peculiar dissolution behaviour of Piretanide hosted in layered double hydroxides. Appl. Clay Sci. 2020, 198, 105826. [Google Scholar] [CrossRef]
- La Rocca, M.; Rinaldi, A.; Bruni, G.; Friuli, V.; Maggi, L.; Bini, M. New Emerging Inorganic–Organic Systems for Drug-Delivery: Hydroxyapatite@ Furosemide Hybrids. J. Inorg. Organomet. Polym. Mater. 2022, 1, 1–11. [Google Scholar] [CrossRef]
- Van den Mooter, G. The use of amorphous solid dispersions: A formulation strategy to overcome poor solubility and dissolution rate. Drug Discov. Today Technol. 2012, 9, e79–e85. [Google Scholar] [CrossRef]
- Priya, A.S.; Sivakamavalli, J.; Vaseeharan, B.; Stalin, T. Improvement on dissolution rate of inclusion complex of Rifabutin drug with β-cyclodextrin. Int. J. Biol. Macromol. 2013, 62, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Prosapio, V.; De Marco, I.; Scognamiglio, M.; Reverchon, E. Folic acid–PVP nanostructured composite microparticles by supercritical antisolvent precipitation. Chem. Eng. J. 2015, 277, 286–294. [Google Scholar] [CrossRef]
- Chiou, A.H.J.; Yeh, M.K.; Chen, C.Y.; Wang, D.P. Micronization of meloxicam using a supercritical fluids process. J. Supercrit. Fluids 2007, 42, 120–128. [Google Scholar] [CrossRef]
- Bruni, G.; Maggi, L.; Tammaro, L.; Di Lorenzo, R.; Friuli, V.; Maietta, M.; Berbenni, V.; Milanese, C.; Girella, A.; Marini, A. Electrospun fibers as potential carrier systems for enhanced drug release of perphenazine. Int. J. Pharm. 2016, 511, 190–197. [Google Scholar] [CrossRef]
- Maggi, L.; Friuli, V.; Chiesa, E.; Pisani, S.; Sakaj, M.; Celestini, P.; Bruni, G. Improvement of the firocoxib dissolution performance using electrospun fibers obtained from different polymer/surfactant associations. Int. J. Mol. Sci. 2019, 20, 3084. [Google Scholar] [CrossRef] [Green Version]
- Torres-Martinez, E.J.; Cornejo Bravo, J.M.; Serrano Medina, A.; Pérez González, G.L.; Villarreal, L.J. A Summary of Electrospun Nanofibers as Drug Delivery System: Drugs Loaded and Biopolymers Used as Matrices. Curr. Drug Deliv. 2018, 15, 1360–1374. [Google Scholar] [CrossRef]
- Pisani, S.; Friuli, V.; Conti, B.; Bruni, G.; Maggi, L. Tableted hydrophilic electrospun nanofibers to promote meloxicam dissolution rate. J. Drug Deliv. Sci. Technol. 2021, 66, 102878. [Google Scholar] [CrossRef]
- The Fluid Dynamics of Taylor Cones, Annual Review of Fluid Mechanics. Available online: https://0-www-annualreviews-org.brum.beds.ac.uk/doi/abs/10.1146/annurev.fluid.39.050905.110159 (accessed on 21 April 2022).
- Mansour, H.M.; Sohn, M.; Al-Ghananeem, A.; DeLuca, P.P. Materials for pharmaceutical dosage forms: Molecular pharmaceutics and controlled release drug delivery aspects. Int. J. Mol. Sci. 2010, 11, 3298–3322. [Google Scholar] [CrossRef] [Green Version]
- Van der Merwe, J.; Steenekamp, J.; Steyn, D.; Hamman, J. The role of functional excipients in solid oral dosage forms to overcome poor drug dissolution and bioavailability. Pharmaceutics 2020, 12, 393. [Google Scholar] [CrossRef]
- Mamidi, N.; Delgadillo RM, V.; González-Ortiz, A. Engineering of carbon nano-onion bioconjugates for biomedical applications. Mater. Sci. Eng. C 2021, 120, 111698. [Google Scholar] [CrossRef]
- Foox, M.; Zilberman, M. Drug delivery from gelatin-based systems. Expert Opin. Drug Discov. 2015, 12, 1547–1563. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, N.; Velasco Delgadillo, R.M.; Barrera, E.V. Covalently functionalized carbon nano-onions integrated gelatin methacryloyl nanocomposite hydrogel containing γ-cyclodextrin as drug carrier for high-performance pH-triggered drug release. Pharmaceuticals 2021, 14, 291. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, N.; Castrejón, J.V.; González-Ortiz, A. Rational design and engineering of carbon nano-onions reinforced natural protein nanocomposite hydrogels for biomedical applications. J. Mech. Behav. Biomed. Mater. 2020, 104, 103696. [Google Scholar] [CrossRef] [PubMed]
- Labib, G. Overview on zein protein: A promising pharmaceutical excipient in drug delivery systems and tissue engineering. Expert Opin. Drug Discov. 2018, 15, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Mamidi, N.; González-Ortiz, A.; Lopez Romo, I.; VBarrera, E. Development of functionalized carbon nano-onions reinforced zein protein hydrogel interfaces for controlled drug release. Pharmaceutics 2019, 11, 621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mamidi, N.; Delgadillo RM, V.; Castrejón, J.V. Unconventional and facile production of a stimuli-responsive multifunctional system for simultaneous drug delivery and environmental remediation. Environ. Sci. Nano 2021, 8, 2081–2097. [Google Scholar] [CrossRef]
- Mamidi, N.; Zuníga, A.E.; Villela-Castrejón, J. Engineering and evaluation of forcespun functionalized carbon nano-onions reinforced poly (ε-caprolactone) composite nanofibers for pH-responsive drug release. Mater. Sci. Eng. C 2020, 112, 110928. [Google Scholar] [CrossRef]
- Pisani, S.; Dorati, R.; Chiesa, E.; Genta, I.; Modena, T.; Bruni, G.; Grisoli, P.; Conti, B. Release profile of gentamicin sulfate from polylactide-co-polycaprolactone electrospun nanofiber matrices. Pharmaceutics 2019, 11, 161. [Google Scholar] [CrossRef] [Green Version]
- Al-Jbour, N.D.; Beg, M.D.; Gimbun, J.; Alam, A.M. An overview of chitosan nanofibers and their applications in the drug delivery process. Curr. Drug Deliv. 2019, 16, 272–294. [Google Scholar] [CrossRef]
- Mamidi, N.; Delgadillo, R.M.V. Design, fabrication and drug release potential of dual stimuli-responsive composite hydrogel nanoparticle interfaces. Colloids Surf. B 2021, 204, 111819. [Google Scholar] [CrossRef]
- Li, C.L.; Martini, L.G.; Ford, J.L.; Roberts, M. The use of hypromellose in oral drug delivery. J. Pharm. Pharmacol. 2005, 57, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Timmins, P.; Pygall, S.R.; Melia, C.D. Hydrophilic Matrix Tablets for Oral Controlled Release. Springer: Berlin, Germany, 2014; pp. 1–326. [Google Scholar]
- Rane, M.; Parmar, J.; Rajabi-Siahboomi, A. Hydrophilic matrices for oral extended release: Influence of fillers on drug release from HPMC matrices. Pharma Times 2010, 42, 41–45. [Google Scholar]
- Generally Recognized as Safe (GRAS), F.D.A. Available online: www.fda.gov/food/generally-recognized-safe-gras/gras-substances-scogs-database (accessed on 14 April 2022).
- Tanno, F.; Nishiyama, Y.; Kokubo, H.; Obara, S. Evaluation of hypromellose acetate succinate (HPMCAS) as a carrier in solid dispersions. Drug Dev. Ind. Pharm. 2004, 30, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Yuvaraja, K.; Khanam, J. Enhancement of carvedilol solubility by solid dispersion technique using cyclodextrins, water soluble polymers and hydroxyl-acid. J. Pharm. Biomed. Anal. 2014, 96, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Hotaling, N.A.; Bharti, K.; Kriel, H.; Simon, C.G., Jr. Diameter J: A validated open source nanofiber diameter measurement tool. Biomaterials 2015, 61, 327–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- <711> Dissolution: Extended release dosage forms. In The United States Pharmacopeia (USP40-NF35); United States Pharmacopeial Convention, Inc.: Rockville, MD, USA, 2017; pp. 138–140. Available online: https://www.usp.org/sites/default/files/usp/document/harmonization/gen-method/stage_6_monograph.pdf (accessed on 1 April 2022).
- Yang, Q.; Wang, S.; Fan, P.; Wang, L.; Di, Y.; Lin, K.; Xiao, F.S. pH-responsive carrier system based on carboxylic acid modified mesoporous silica and polyelectrolyte for drug delivery. Chem. Mater. 2005, 17, 5999–6003. [Google Scholar] [CrossRef]
- Reagents: Solutions/Buffer Solutions. In The United States Pharmacopeia (USP40-NF35); United States Pharmacopeial Convention, Inc.: Rockville, MD, USA, 2017; pp. 2409–2411.
- Gouda, R.; Baishya, H.; Qing, Z. Application of mathematical models in drug release kinetics of carbidopa and levodopa ER tablets. J. Dev. Drugs 2017, 6, 1–8. [Google Scholar]
- Laracuente, M.L.; Marina, H.Y.; McHugh, K.J. Zero-order drug delivery: State of the art and future prospects. J. Control Release 2020, 327, 834–856. [Google Scholar] [CrossRef]
- Paarakh, M.P.; Jose, P.A.; Setty, C.M.; Christoper, G.P. Release kinetics–concepts and applications. Int. J. Pharm. Res. Technol. 2018, 8, 12–20. [Google Scholar]
- Siepmann, J.; Peppas, N.A. Higuchi equation: Derivation, applications, use and misuse. Int. J. Pharm. 2011, 418, 6–12. [Google Scholar] [CrossRef]
- Korsmeyer, R.W.; Peppas, N.A. Effect of the morphology of hydrophilic polymeric matrices on the diffusion and release of water soluble drugs. J. Membr. Sci. 1981, 9, 211–227. [Google Scholar] [CrossRef]
- Singh, J.P. Martin’s Physical Pharmacy and Pharmaceutical Sciences; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2006; pp. 589–609. [Google Scholar]
- Bruschi, M.L. Strategies to Modify the Drug Release from Pharmaceutical Systems, 1st ed.; Elsevier Woodhead Publishing: Sawston, CA, USA, 2015. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Composition of Electrospun Polymer Solutions | Electrospinning Process Parameters | ||||||
|---|---|---|---|---|---|---|---|
| Fibers | HPMC-AS716 (%w/v) | HPMC-AS912 (%w/v) | HPMC K100LV (%w/v) | M (%w/v) | C (%w/v) | Voltage (kV) | Flow Rate (mL/h) |
| fb1 | 20.0 | - | - | - | - | 28 | 0.6 |
| fb3 | - | 15.0 | - | - | - | 30 | 0.6 |
| fb4 | - | 15.0 | 2.0 | - | - | 28 | 0.8 |
| Mfb1 | 20.0 | - | - | 5.0 | - | 30 | 0.5 |
| Mfb3 | - | 15.0 | - | 5.0 | - | 30 | 0.5 |
| Mfb4 | - | 15.0 | 2.0 | 5.0 | - | 30 | 0.5 |
| Cfb4 | 15.0 | 2.0 | - | 5.0 | 30 | 0.4 | |
| Ingredients | Fast Release Formulations | Sustained Release Formulations | ||||
|---|---|---|---|---|---|---|
| Mfb4c6 | Cfb4c10 | Mfb4c1 | Mfb4c3 | Cfb4c6 | Cfb4c9 | |
| Mfb4 (corresponding 7.5 mg of meloxicam) | 33.0 mg | - | 33.0 mg | 33.0 mg | - | - |
| Cfb4 (corresponding 6.25 mg of carvedilol) | - | 27.5 mg | - | - | 27.5 mg | 27.5 mg |
| Sodium starch glycolate | 50.0 mg | - | - | - | - | |
| Microcrystalline cellulose | 75.0 mg | 100.0 mg | - | - | - | |
| Cross-linked polyvinylpyrrolidone | - | 50.0 mg | - | - | - | |
| HPMC K4M | - | - | 10.0 mg | 30.0 mg | 45.0 mg | 33.0 mg |
| Mannitol | - | - | - | - | 70.0 mg | 70.0 mg |
| Magnesium stearate | 1.0 mg | 1.0 mg | 1.0 mg | 1.0 mg | 1.0 mg | 1.0 mg |
| Total weight | 159.0 mg | 178.5 mg | 44.0 mg | 64.0 mg | 143.5 mg | 131.5 mg |
| Diameter of tablet | 8 mm | 8 mm | 5 mm | 6 mm | 8 mm | 8 mm |
| Sample | td 90% After 2 h at pH 1.0 | td 90% After 4 h at pH 4.5 |
|---|---|---|
| Fast release formulation (Mfb4c4tr) | 2 h | 2 h |
| Sustained release formulation 1 (Mfb4c1tr) | 6.5 h | 5.5 h |
| Sustained release formulation 2 (Mfb4c3tr) | 11 h | 11 h |
| Sustained Release Formulation | Zero-Order | First-Order | Higuchi | Korsmeyer–Peppas | Hixson–Crowell | ||
|---|---|---|---|---|---|---|---|
| R2 | R2 | K1 | R2 | R2 | n | R2 | |
| MFb4c1tr after 2 h in pH 1.0 | 0.9902 | 0.9628 | −0.1547 | 0.9945 | 0.9505 | 1.7155 | 0.9866 |
| Mfb4c3tr after 2 h in pH 1.0 | 0.9873 | 0.8986 | −0.0966 | 0.9889 | 0.9454 | 1.1917 | 0.9593 |
| MFb4c1tr after 4 h in pH 4.5 | 0.9860 | 0.9789 | −0.1797 | 0.9943 | 0.9481 | 2.5252 | 0.9921 |
| Mfb4c3tr after 4 h in pH 4.5 | 0.9879 | 0.9643 | −0.0862 | 0.9934 | 0.9609 | 1.3467 | 0.9885 |
| Cfb4c6t | 0.9929 | 0.8764 | −0.0334 | 0.9680 | 0.9351 | 2.4742 | 0.9586 |
| Cfb4c9t | 0.9896 | 0.9338 | −0.0434 | 0.9664 | 0.9226 | 1.3374 | 0.9835 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Friuli, V.; Pisani, S.; Conti, B.; Bruni, G.; Maggi, L. Tablet Formulations of Polymeric Electrospun Fibers for the Controlled Release of Drugs with pH-Dependent Solubility. Polymers 2022, 14, 2127. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14102127
Friuli V, Pisani S, Conti B, Bruni G, Maggi L. Tablet Formulations of Polymeric Electrospun Fibers for the Controlled Release of Drugs with pH-Dependent Solubility. Polymers. 2022; 14(10):2127. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14102127
Chicago/Turabian StyleFriuli, Valeria, Silvia Pisani, Bice Conti, Giovanna Bruni, and Lauretta Maggi. 2022. "Tablet Formulations of Polymeric Electrospun Fibers for the Controlled Release of Drugs with pH-Dependent Solubility" Polymers 14, no. 10: 2127. https://0-doi-org.brum.beds.ac.uk/10.3390/polym14102127