
Stimuli-Responsive Block Copolymer-Based Assemblies for Cargo Delivery and Theranostic Applications

Abstract
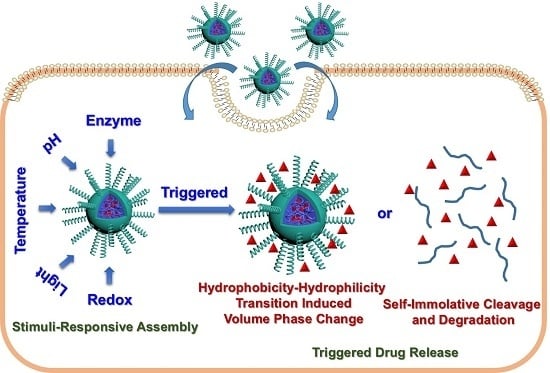
:
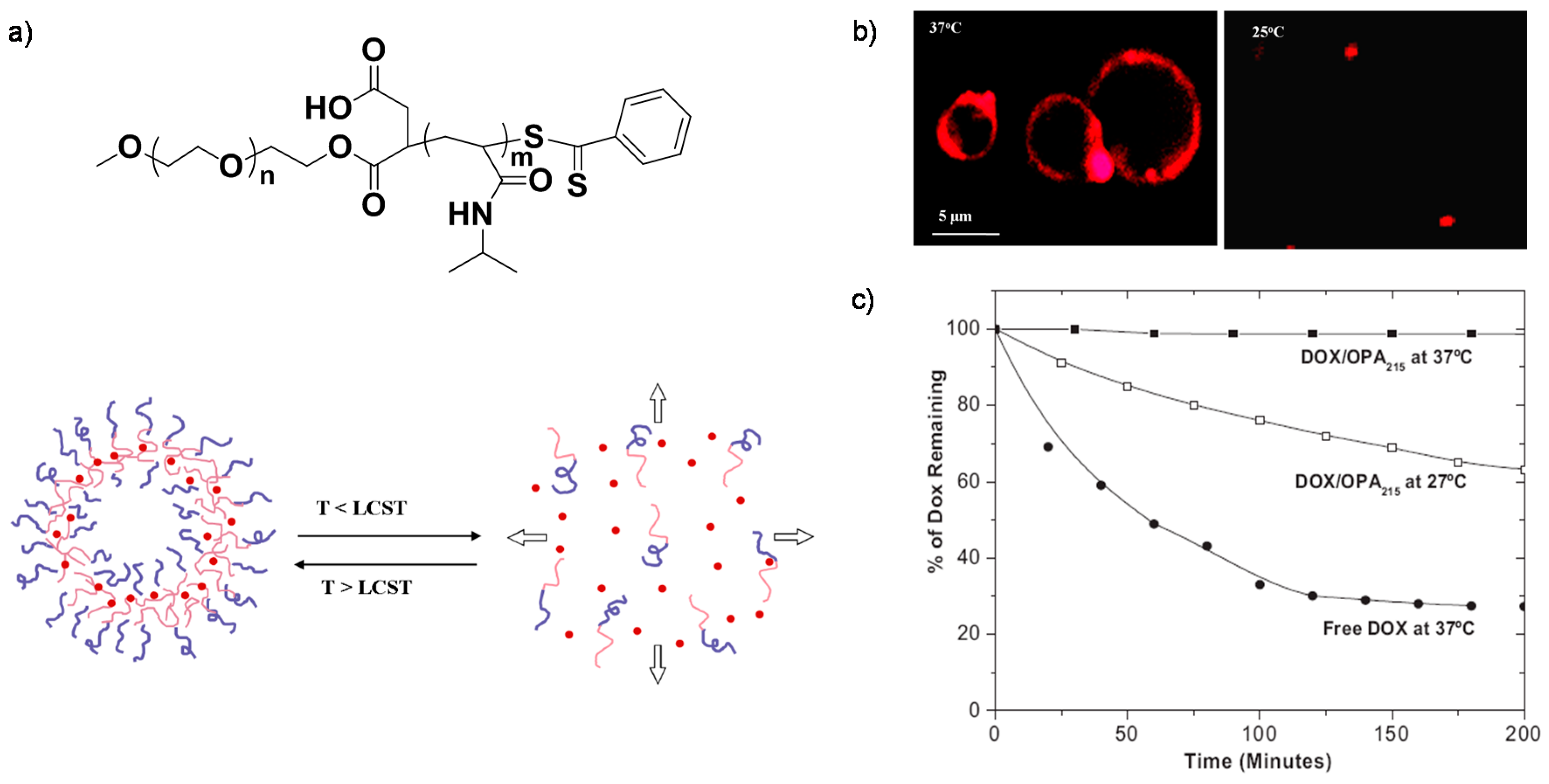
{kind=link}
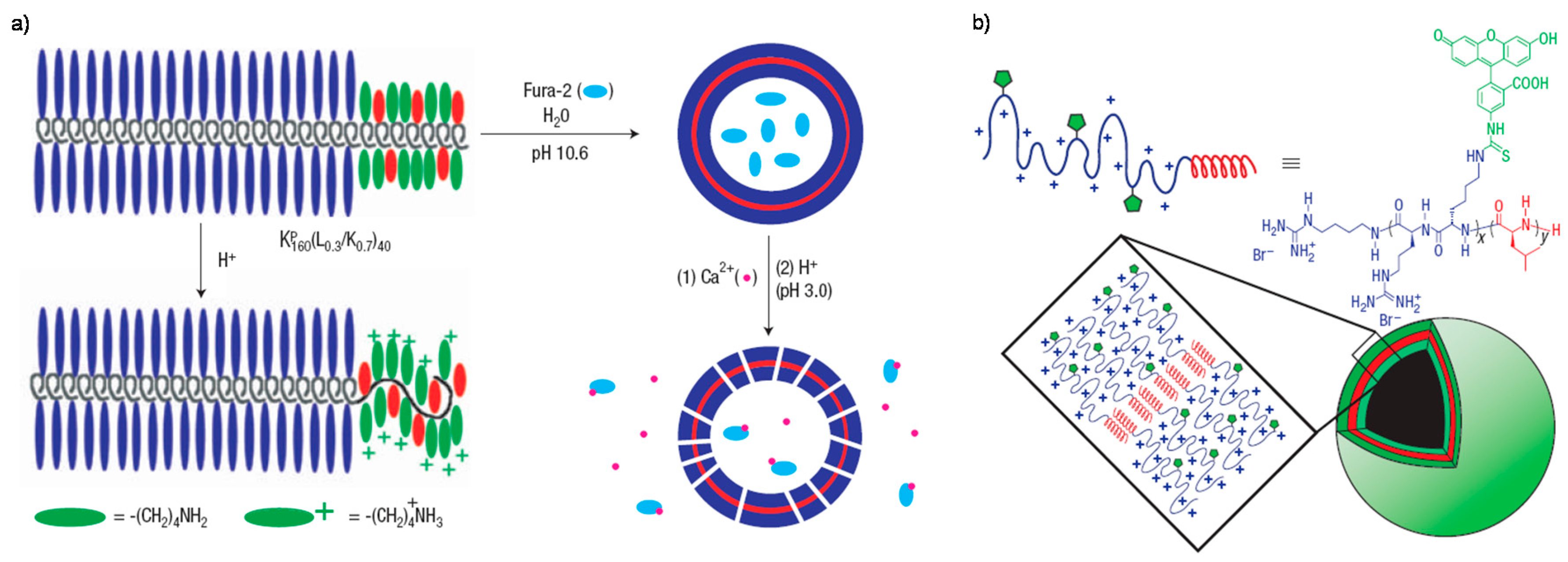
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Temperature-Sensitive Assemblies
3. pH-Sensitive Assemblies
4. Enzyme-Sensitive Assemblies
5. Redox-Sensitive Assemblies
6. Light-Sensitive Assemblies
7. Multi-Sensitive Assemblies
8. Conclusions and Perspectives
Acknowledgments
Conflicts of Interest
References
- Ross, J.S.; Schenkein, D.P.; Pietrusko, R.; Rolfe, M.; Linette, G.P.; Stec, J.; Stagliano, N.E.; Ginsburg, G.S.; Symmans, W.F.; Pusztai, L.; et al. Targeted therapies for cancer 2004. Am. J. Clin. Pathol. 2004, 122, 598–609. [Google Scholar] [CrossRef] [PubMed]
- Langer, R.; Tirrell, D.A. Designing materials for biology and medicine. Nature 2004, 428, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Harada, A.L.; Nagasaki, Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Deliv. Rev. 2001, 47, 113–131. [Google Scholar] [CrossRef]
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Panyam, J.; Labhasetwar, V. Biodegradable nanoparticles for drug and gene delivery to cells and tissue. Adv. Drug Deliv. Rev. 2003, 55, 329–347. [Google Scholar] [CrossRef]
- Sinha, V.R.; Trehan, A. Biodegradable microspheres for protein delivery. J. Control. Release 2003, 90, 261–280. [Google Scholar] [CrossRef]
- Ge, Z.S.; Liu, S.Y. Functional block copolymer assemblies responsive to tumor and intracellular microenvironments for site-specific drug delivery and enhanced imaging performance. Chem. Soc. Rev. 2013, 42, 7289–7325. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.M.; Zhang, G.Q.; Liu, S.Y. Enzyme-responsive polymeric assemblies, nanoparticles and hydrogels. Chem. Soc. Rev. 2012, 41, 5933–5949. [Google Scholar] [CrossRef] [PubMed]
- Ercole, F.; Davis, T.P.; Evans, R.A. Photo-responsive systems and biomaterials: Photochromic polymers, light-triggered self-assembly, surface modification, fluorescence modulation and beyond. Polym. Chem. 2010, 1, 37–54. [Google Scholar] [CrossRef]
- Hu, J.M.; Liu, S.Y. Engineering responsive polymer building blocks with host-guest molecular recognition for functional applications. Acc. Chem. Res. 2014, 47, 2084–2095. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.L.; Liu, S.Y. Recent advances towards the fabrication and biomedical applications of responsive polymeric assemblies and nanoparticle hybrid superstructures. Dalton Trans. 2015, 44, 3904–3922. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Liu, W.; Dong, C.M. UV- and NIR-responsive polymeric nanomedicines for on-demand drug delivery. Polym. Chem. 2013, 4, 3431–3443. [Google Scholar] [CrossRef]
- Choi, K.Y.; Liu, G.; Lee, S.; Chen, X.Y. Theranostic nanoplatforms for simultaneous cancer imaging and therapy: Current approaches and future perspectives. Nanoscale 2012, 4, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Samuel, A.M. PET/CT in pediatric oncology. Indian J. Cancer. 2010, 47, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.M.; Fang, R.H.; Luk, B.T.; Zhang, L. Polymeric nanotherapeutics: Clinical development and advances in stealth functionalization strategies. Nanoscale 2014, 6, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Ronak, M.; Kristi, L.K. Polymer-based therapeutics. Macromolecules 2009, 42, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Mai, Y.; Eisenberg, A. Self-assembly of block copolymers. Chem. Soc. Rev. 2012, 41, 5969–5985. [Google Scholar] [CrossRef] [PubMed]
- Brian, T.L.; Zhang, L.F. Current advances in polymer-based nanotheranostics for cancer treatment and diagnosis. ACS. Appl. Mater. Interfaces 2014, 6, 21859–21873. [Google Scholar]
- Matsumura, Y.; Maeda, H.A. New concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar] [PubMed]
- Iyer, A.K.; Khaled, G.; Fang, J.; Maeda, H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov. Today 2006, 11, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Xiao, Z.Y.; Valencia, P.M.; Radovic-Moreno, A.F.; Farokhzad, O.C. Targeted polymeric therapeutic nanoparticles: Design, development and clinical translation. Chem. Soc. Rev. 2012, 41, 2971–3010. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.D.; Betancourt, T.; Brannon-Peppas, L. Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv. Drug Delivery Rev. 2008, 60, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Stolnik, S.; Illum, L.; Davis, S.S. Long circulating microparticulate drug carriers. Adv. Drug Delivery Rev. 1995, 16, 195–214. [Google Scholar] [CrossRef]
- Simone, E.A.; Dziubla, T.D.; Muzykantov, V.R. Polymeric carriers: Role of geometry in drug delivery. Expert Opin. Drug Deliv. 2008, 5, 1283–1300. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, X.J.; Guo, M.; Yan, H.S.; Wang, C.H.; Liu, K.L. Cisplatin-loaded polymer/magnetite composite nanoparticles as multifunctional therapeutic nanomedicine. Chin. J. Polym. Sci. 2014, 32, 1329–1337. [Google Scholar] [CrossRef]
- Yuan, F.; Dellian, M.; Fukumura, D.; Leunig, M.; Berk, D.A.; Torchilin, V.P.; Jain, R.K. Vascular permeability in a human tumor xenograft: Molecular size dependence and cutoff size. Cancer Res. 1995, 55, 3752–3756. [Google Scholar] [PubMed]
- Cabral, H.; Matsumoto, Y.; Mizuno, K.; Chen, Q.; Murakami, M.; Kimura, M.; Terada, Y.; Kano, M.R.; Miyazono, K.; Uesaka, M.; et al. Accumulation of sub-100 nm polymeric micelles in poorly permeable tumours depends on size. Nat. Nanotechnol. 2011, 6, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, V.P.; Stylianopoulos, T.; Martin, J.D.; Popovic, Z.; Chen, O.; Kamoun, W.S.; Bawendi, M.G.; Fukumura, D.; Jain, R.K. Normalization of tumor blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat. Nanotechnol. 2012, 7, 383–388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Dalhaimer, P.; Cai, S.S.; Tsai, R.; Tewari, M.; Minko, T.; Discher, D.E. Shape effects of filaments versus spherical particles in flow and drug delivery. Nat. Nanotechnol. 2007, 2, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Christian, D.A.; Cai, S.S.; Garbuzenko, O.B.; Harada, T.; Zajac, A.L.; Minko, T.; Discher, D.E. Flexible filaments for in vivo imaging and delivery: Persistent circulation of filomicelles opens the dosage window for sustained tumor shrinkage. Mol. Pharm. 2009, 6, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, H.; Nagasaki, Y.; Kataoka, K. PEGylated nanoparticles for biological and pharmaceutical applications. Adv. Drug Deliv. Rev. 2003, 55, 403–419. [Google Scholar] [CrossRef]
- Talelli, M.; Rijcken, C.J.F.; Van, N.C.F.; Storm, G.; Hennink, W.E. Micelles based on HPMA copolymers. Adv. Drug Deliv. Rev. 2010, 62, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Rajora, A.K.; Ravishankar, D.; Osborn, H.M.I.; Greco, F. Impact of the enhanced permeability and retention (EPR) effect and cathepsins levels on the activity of polymer-drug conjugates. Polymers 2014, 6, 2186–2220. [Google Scholar] [CrossRef]
- Huang, M.M.; Zhao, K.J.; Wang, L.; Lin, S.Q.; Li, J.J.; Chen, J.B.; Zhao, C.G.; Ge, Z.S. Dual stimuli-responsive polymer prodrugs quantitatively loaded by nanoparticles for enhanced cellular internalization and triggered drug release. ACS Appl. Mater. Interfaces 2016, 8, 11226–11236. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ko, N.R.; Oh, J.K. Recent advances in stimuli-responsive degradable block copolymer micelles: Synthesis and controlled drug delivery applications. Chem. Commun. 2012, 48, 7542–7552. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.H.; Zhong, Z.Y.; Jan, F.J. Stimuli-responsive polymersomes for programmed drug delivery. Biomacromolecules 2009, 10, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Feng, A.C.; Yuan, J.Y. Smart nanocontainers: Progress on novel stimuli-responsive polymer vesicles. Macromol. Rapid Commun. 2014, 35, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Blum, A.P.; Kammeyer, J.K.; Rush, A.M.; Callmann, C.E.; Hahn, M.E.; Gianneschi, N.C. Stimuli-responsive nanomaterials for biomedical applications. J. Am. Chem. Soc. 2015, 137, 2140–2154. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Niu, Z.W. Temperature responsive 3D structure of rod-like bionanoparticles induced by depletion interaction. Chin. J. Polym. Sci. 2014, 32, 1271–1275. [Google Scholar] [CrossRef]
- Issels, R.D. Hyperthermia adds to chemotherapy. Eur. J. Cancer 2008, 44, 2546–2554. [Google Scholar] [CrossRef] [PubMed]
- Kizaka-Kondoh, S.; Inoue, M.; Harada, H.; Hiraoka, M. Tumor hypoxia: A target for selective cancer therapy. Cancer Sci. 2003, 94, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Seki, T.; Maeda, H. Therapeutic strategies by modulating oxygen stress in cancer and inflammation. Adv. Drug Deliv. Rev. 2009, 61, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, B.J.; Swindell, E.P.; MacRenaris, K.W.; Hankins, P.L.; Chipre, A.J.; Mastarone, D.; Ahn, R.W.; Meade, T.J.; O’Halloran, T.V.; Nguyen, S.T. pH-responsive theranostic polymer-caged nanobins: Enhanced cytotoxicity and T1 MRI contrast by Her2 targeting. Part. Part. Syst. Charact. 2013, 30, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Li, B.A. Novel upconversion nanotheranostic agent for multi-modality imaging-guided chemotherapy with on-demand drug release. Sci. China Chem. 2015, 58, 970–970. [Google Scholar] [CrossRef]
- Langer, A.A. Systematic review of PET and PET/CT in oncology: A way to personalize cancer treatment in a cost-effective manner. BMC Health Serv. Res. 2010, 10, 283. [Google Scholar] [CrossRef] [PubMed]
- Ke, C.Y.; Mathias, C.J.; Green, M.A. Folate-receptor-targeted radionuclide imaging agents. Adv. Drug Deliv. Rev. 2004, 56, 1143–1160. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.L.; Shih, Y.H.; Lee, P.C.; Hsieh, T.M.H.; Luo, T.Y.; Shieh, M.J. Multimodal image-guided photothermal therapy mediated by 188Re-labeled micelles containing a cyanine-type photosensitizer. ACS Nano 2011, 5, 5594–5607. [Google Scholar] [CrossRef] [PubMed]
- Yahara, T.; Koga, T.; Yoshida, S.; Nakagawa, S.; Deguchi, H.; Shirouzu, K. Relationship between microvessel density and thermographic hot areas in breast cancer. Surg. Today 2003, 33, 243–248. [Google Scholar] [PubMed]
- Tran, N.T.D.; Truong, N.P.; Gu, W.Y.; Jia, Z.F.; Cooper, M.A.; Monteiro, M.J. Timed-release polymer nanoparticles. Biomacromolecules 2013, 14, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.T.D.; Jia, Z.F.; Truong, N.P.; Cooper, M.A.; Monteiro, M.J. Fine tuning the disassembly time of thermoresponsive polymer nanoparticles. Biomacromolecules 2013, 14, 3463–3471. [Google Scholar] [CrossRef] [PubMed]
- Truong, N.P.; Whittaker, M.R.; Anastasaki, A.; Haddleton, D.M.; Quinn, J.F.; Davis, T.P. Facile production of nanoaggregates with tuneable morphologies from thermoresponsive P(DEGMA-co-HPMA). Polym. Chem. 2016, 7, 430–440. [Google Scholar] [CrossRef]
- Truong, N.P.; Quinn, J.F.; Anastasaki, A.; Haddleton, D.M.; Whittaker, M.R.; Davis, T.P. Facile access to thermoresponsive filomicelles with tuneable cores. Chem. Commun. 2016, 52, 4497–4500. [Google Scholar] [CrossRef] [PubMed]
- Schild, H.G. Poly(N-isopropylacrylamide): Experiment, theory and application. Prog. Polym. Sci. 1992, 17, 163–249. [Google Scholar] [CrossRef]
- Wei, H.; Cheng, S.X.; Zhang, X.Z.; Zhuo, R.X. Thermo-sensitive polymeric micelles based on poly(N-isopropylacrylamide) as drug carriers. Prog. Polym. Sci. 2009, 34, 893–910. [Google Scholar] [CrossRef]
- Hu, X.L.; Li, Y.; Liu, T.; Zhang, G.Y.; Liu, S.Y. Intracellular cascade FRET for temperature imaging of living cells with polymeric ratiometric fluorescent thermometers. ACS Appl. Mater. Interfaces 2015, 7, 15551–15560. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Geng, Y.; Discher, D.E.; Yang, S. Temperature-controlled assembly and release from polymer vesicles of poly(ethylene oxide)-block-poly(N-isopropylacrylamide). Adv. Mater. 2006, 18, 2905–2909. [Google Scholar] [CrossRef]
- Chung, J.E.; Yokoyama, M.; Okano, T. Inner core segment design for drug delivery control of thermo-responsive polymeric micelles. J. Control. Release 2000, 65, 93–103. [Google Scholar] [CrossRef]
- Nakayama, M.; Okano, T.; Miyazaki, T.; Kohori, F.; Sakai, K.; Yokoyama, M. Molecular design of biodegradable polymeric micelles for temperature-responsive drug release. J. Control. Release 2006, 115, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.J.; Liu, T.; Liu, S.Y. Synthesis of amphiphilic tadpole-shaped linear-cyclic diblock copolymers via ring-opening polymerization directly initiating from cyclic precursors and their application as drug nanocarriers. Biomacromolecules 2011, 12, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Wycisk, A.; Döring, A.; Schneider, M.; Schönhoff, M.; Kuckling, D. Synthesis of β-cyclodextrin-based star block copolymers with thermo-responsive behavior. Polymers 2015, 7, 921–938. [Google Scholar] [CrossRef]
- Cheng, C.; Wei, H.; Shi, B.X.; Cheng, H.; Li, C.; Gu, Z.W.; Cheng, S.X.; Zhang, X.Z.; Zhuo, R.X. Biotinylated thermoresponsive micelle self-assembled from double-hydrophilic block copolymer for drug delivery and tumor target. Biomaterials 2008, 29, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Kataoka, K. Precise control of lower critical solution temperature of thermosensitive poly(2-isopropyl-2-oxazoline) via gradient copolymerization with 2-ethyl-2-oxazoline as a hydrophilic comonomer. Macromolecules 2006, 39, 6622–6630. [Google Scholar] [CrossRef]
- Lutz, J.F. Polymerization of oligo(ethylene glycol) (meth)acrylates: Toward new generations of smart biocompatible materials. J. Polym. Sci. Part A 2008, 46, 3459–3470. [Google Scholar] [CrossRef]
- Soga, O.; Nostrum, C.F.V.; Hennink, W.E. Poly(N-(2-hydroxypropyl) methacrylamide mono/di lactate): A new class of biodegradable polymers with tunable thermosensitivity. Biomacromolecules 2004, 5, 818–821. [Google Scholar] [CrossRef] [PubMed]
- Herrero, V.R.; Rincόn, A.C.; Alonso, M.; Reboto, V.; Molina, M.I.T.; Rodríguez-cabello, J.C. Self-assembled particles of an elastin-like polymer as vehicles for controlled drug release. J. Control. Release 2005, 102, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Driessen, W.; Jiang, X.Q. Oligo(ethylene glycol)-based thermosensitive dendrimers and their tumor accumulation and penetration. J. Am. Chem. Soc. 2014, 136, 3145–3155. [Google Scholar] [CrossRef] [PubMed]
- Koga, T.; Nakamoto, K.; Odawara, K.; Matsuoka, T.; Higashi, N. Fabrication of thermo-responsive molecular layers from self-assembling llastin-like oligopeptides containing cell-binding domain for tissue engineering. Polymers 2015, 7, 134–146. [Google Scholar] [CrossRef]
- Wang, L.; Liu, G.H.; Wang, X.R.; Hu, J.M.; Zhang, G.Y.; Liu, S.Y. Acid-disintegratable polymersomes of pH-responsive amphiphilic diblock copolymers for intracellular drug delivery. Macromolecules 2015, 48, 7262–7272. [Google Scholar] [CrossRef]
- Hu, J.M.; Liu, G.H.; Wang, C.; Liu, T.; Zhang, G.Y.; Liu, S.Y. Spatiotemporal monitoring endocytic and cytosolic pH gradients with endosomal escaping pH-responsive micellar nanocarriers. Biomacromolecules 2014, 15, 4293–4301. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.; Shin, H.J.; Na, K.; Bae, Y.H. Poly(l-histidine)-PEG block copolymer micelles and pH-induced destabilization. J. Control. Release 2003, 90, 363–374. [Google Scholar] [CrossRef]
- Johnson, R.P.; Jeong, Y.I.; Choi, E.; Chung, C.W.; Kang, D.H.; Oh, S.O.; Suh, H.; Kim, I. Biocompatible poly(2-hydroxyethyl methacrylate)-b-poly(l-histidine) hybrid materials for pH-sensitive intracellular anticancer drug delivery. Adv. Funct. Mater. 2012, 22, 1058–1068. [Google Scholar] [CrossRef]
- Bellomo, E.G.; Wyrsta, M.D.; Pakstis, L.; Pochan, D.J.; Deming, T.J. Stimuli-responsive polypeptide vesicles by conformation-specific assembly. Nat. Mater. 2004, 3, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Holowka, E.P.; Sun, V.Z.; Kamei, D.T.; Deming, T.J. Polyarginine segments in block copolypeptides drive both vesicular assembly and intracellular delivery. Nat. Mater. 2007, 6, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.C.; Ryser, H.J.P. Cis-aconityl spacer between daunomycin and macromolecular carriers: A model of pH-sensitive linkage releasing drug from a lysosomotropic conjugate. BioChem. Biophys. Res. Commun. 1981, 102, 1048–1054. [Google Scholar] [CrossRef]
- Bae, Y.; Fukushima, S.; Harada, A.; Kataoka, K. Design of environment-sensitive supramolecular assemblies for intracellular drug delivery: Polymeric micelles that are responsive to intracellular pH change. Angew. Chem. 2003, 42, 4640–4643. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.Q.; Grailer, J.J.; Rowland, I.J.; Javadi, A.; Hurley, S.A.; Matson, V.Z.; Steeber, D.A.; Gong, S.Q. Multifunctional stable and pH responsive polymer vesicles formed by heterofunctional triblock copolymer for targeted anticancer drug delivery and ultrasensitive MR imaging. ACS Nano 2010, 5, 6805–6817. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Du, W.J.; Sun, G.R.; Wooley, K.L. pH-responsive shell cross-linked nanoparticles with hydrolytically labile cross-links. Macromolecules 2008, 41, 6605–6607. [Google Scholar] [CrossRef]
- Shim, M.S.; Kwon, Y.J. Acid-transforming polypeptide micelles for targeted nonviral gene delivery. Biomaterials 2010, 31, 3404–3413. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.X.; Gu, J.X.; Qu, X.Z.; Yang, Z.Z. Preparation of multifunctional drug carrier for tumor-specific uptake and enhanced intracellular delivery through the conjugation of weak acid labile linker. BioConjugate Chem. 2009, 20, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.W.; Flores, J.D.; McCormick, C.L. Reversible imine shell cross-linked micelles from aqueous RAFT synthesized thermoresponsive triblock copolymers as potential nanocarriers for “pH-triggered” drug release. Macromolecules 2011, 44, 1327–1334. [Google Scholar] [CrossRef]
- Gillies, E.R.; Jonsson, T.B.; Fréchet, J.M.J. Stimuli-responsive supramolecular assemblies of linear-dendritic copolymers. J. Am. Chem. Soc. 2004, 126, 11936–11943. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.Y.; Liu, Y.; Du, J.Z.; Cao, Z.T.; Xu, C.F.; Wang, J. Facile generation of tumor-pH-labile linkage-bridged block copolymers for chemotherapeutic delivery. Angew. Chem. 2016, 128, 1022–1026. [Google Scholar] [CrossRef]
- Xu, C.F.; Zhang, H.B.; Sun, C.Y.; Liu, Y.; Shen, S.; Yang, X.Z.; Zhu, Y.H.; Wang, J. Tumor acidity-sensitive linkage-bridged block copolymer for therapeutic siRNA delivery. Biomaterials 2016, 88, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.M.; Liu, T.; Zhang, G.Y.; Jin, F.; Liu, S.Y. Synergistically enhance magnetic resonance/fluorescence imaging performance of responsive polymeric nanoparticles under mildly acidic biological milieu. Macromol. Rapid Commun. 2013, 34, 749–758. [Google Scholar] [CrossRef] [PubMed]
- Itaka, K.J.; Ohba, S.; Miyata, K.; Kawaguchi, H.; Nakamura, K.; Takato, T.; Chung, U.; Kataoka, K. Bone regeneration by regulated in vivo gene transfer using biocompatible polyplex nanomicelles. Mol. Ther. 2007, 15, 1655–1662. [Google Scholar] [CrossRef] [PubMed]
- Akagi, D.; Oba, M.; Koyama, H.; Nishiyama, N.; Fukushima, S.; Miyata, T.; Nagawa, H.; Kataoka, K. Biocompatible micellar nanovectors achieve efficient gene transfer to vascular lesions without cytotoxicity and thrombus formation. Gene Ther. 2007, 14, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, S.G.; Miyata, K.J.; Nishiyama, N.B.; Kanayama, N.K.; Yamasaki, Y.C.; Kataoka, K.Z.N. PEGylated polyplex micelles from triblock catiomers with spatially ordered layering of condensed pDNA and buffering units for enhanced intracellular gene delivery. J. Am. Chem. Soc. 2005, 127, 2810–2811. [Google Scholar] [CrossRef] [PubMed]
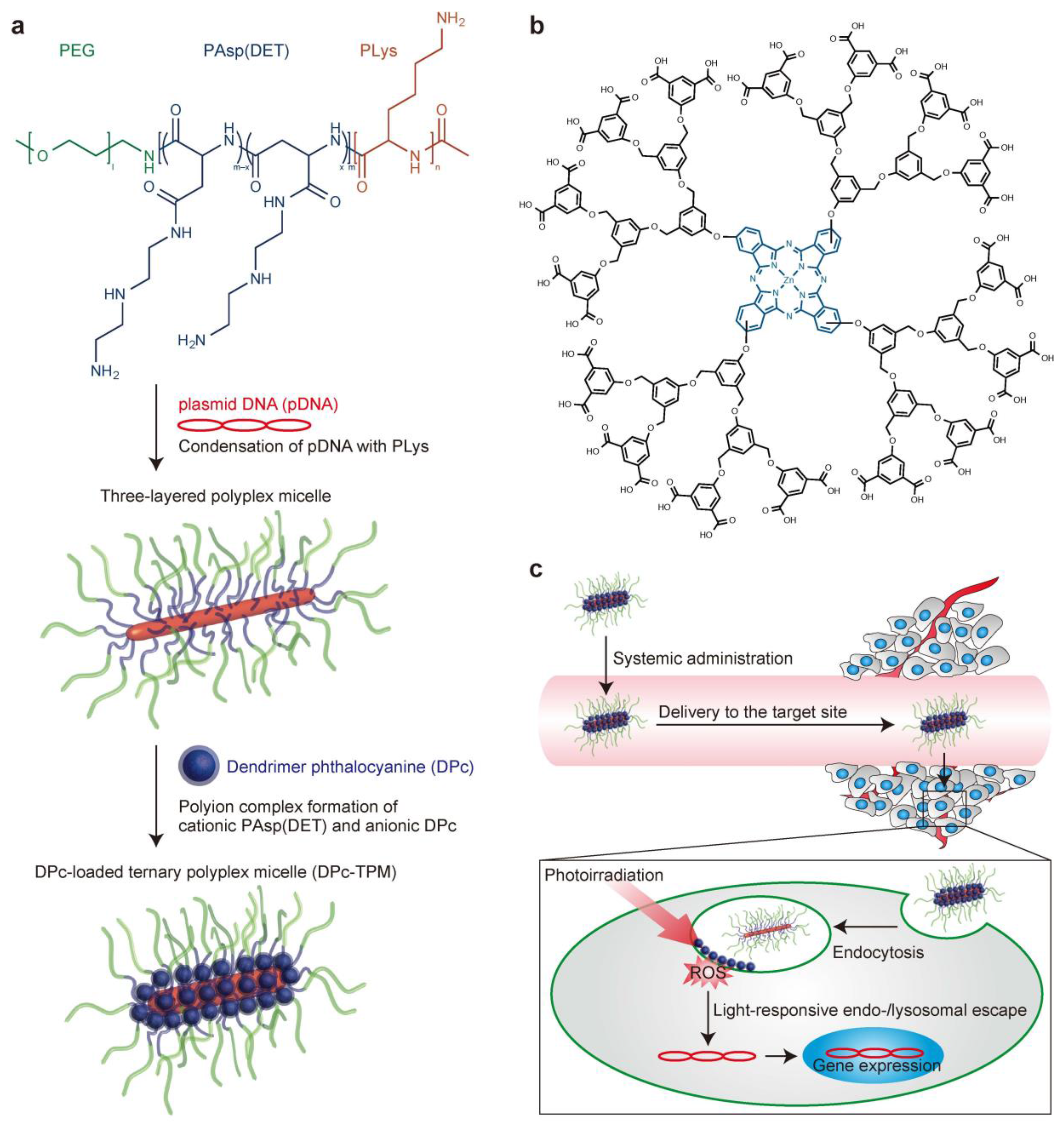
- Nomoto, T.; Fukushima, S.; Kumagai, M.; Machitani, K.; Arnida; Matsumoto, Y.; Oba, M.K.; Miyata, K.J.; Osada, K.S.; Nishiyama, N.B.; et al. Three-layered polyplex micelle as a multifunctional nanocarrier platform for light-induced systemic gene transfer. Nat. Commun. 2014, 3545–3554. [Google Scholar] [CrossRef] [PubMed]
- Lynn, D.M.; Langer, R. Degradable poly(β-amino esters): Synthesis, characterization, and self-assembly with plasmid DNA. J. Am. Chem. Soc. 2000, 122, 10761–10768. [Google Scholar] [CrossRef]
- Kim, M.S.; Lee, D.S.; Choi, E.K.; Park, H.J.; Kim, J.S. Modulation of poly(β-amino ester) pH-sensitive polymers by molecular weight control. Macromol. Res. 2005, 13, 147–151. [Google Scholar] [CrossRef]
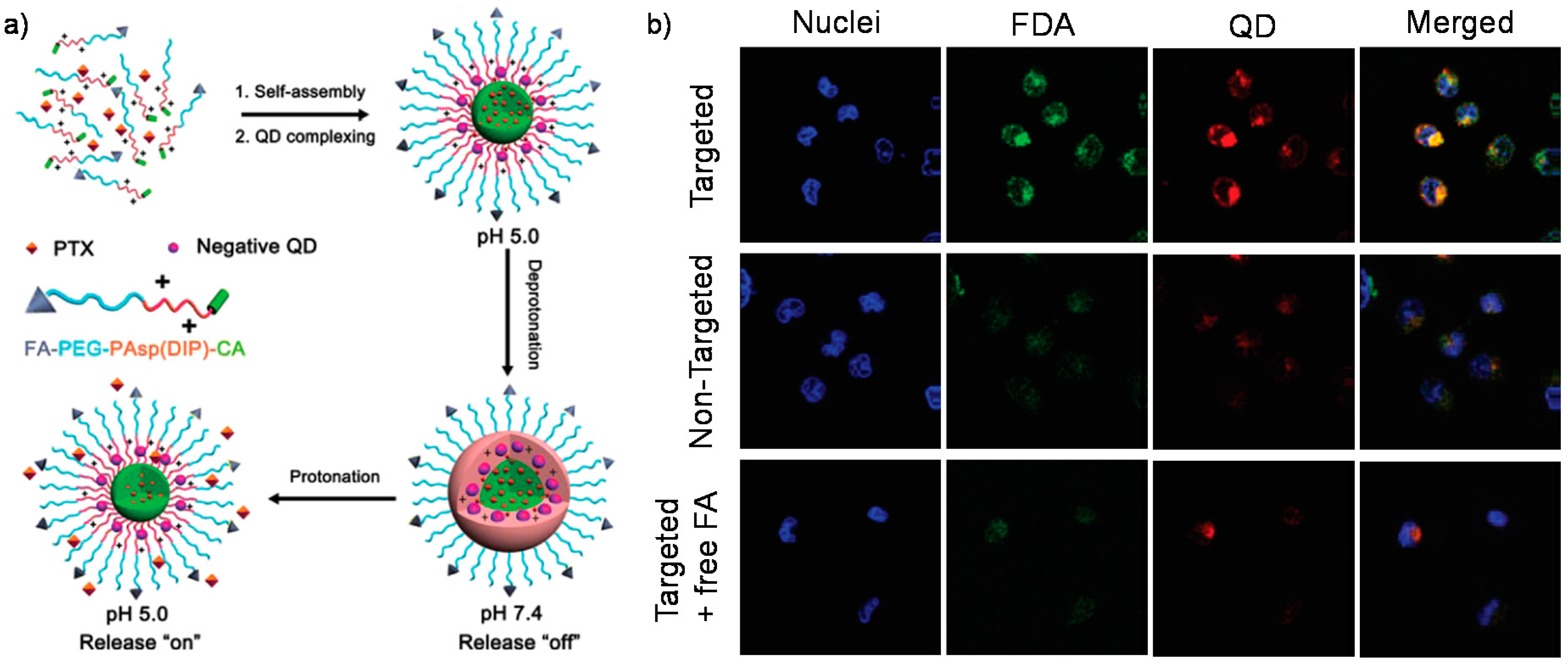
- Cheng, T.J.; Ma, R.J.; Zhang, Y.M.; Ding, Y.X.; Liu, J.J.; Ou, H.L.; An, Y.L.; Liu, J.F.; Shi, L.Q. A surface-adaptive nanocarrier to prolong circulation time and enhance cellular uptake. Chem. Commun. 2015, 51, 14985–14988. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.J.; Cheng, T.J.; Liu, J.F.; Liu, J.J.; Yang, C.H.; Chu, L.P.; Zhang, Y.M.; Ma, R.J.; Shi, L.Q. Self-regulated multifunctional collaboration of targeted nanocarriers for enhanced tumor therapy. Biomacromolecules 2014, 15, 3634–3642. [Google Scholar] [CrossRef] [PubMed]
- Turkbey, B.; Choyke, P.L. Multiparametric MRI and prostate cancer diagnosis and risk stratification. Curr. Opin. Urol. 2012, 22, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.W.; Cheng, D.; Gong, F.M.; Miao, X.M.; Shuai, X.T. Design of multifunctional micelle for tumor-targeted intracellular drug release and fluorescent imaging. Adv. Mater. 2012, 24, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Piedrafita, G.; Keller, M.A.; Ralser, M. The impact of non-enzymatic reactions and enzyme promiscuity on cellular metabolism during (oxidative) stress conditions. Biomolecules 2015, 5, 2101–2122. [Google Scholar] [CrossRef] [PubMed]
- Ghadiali, J.E.; Stevens, M.M. Enzyme-responsive nanoparticle systems. Adv. Mater. 2008, 20, 4359–4363. [Google Scholar] [CrossRef]
- Hahn, M.E.; Gianneschi, N.C. Enzyme-directed assembly and manipulation of organic nanomaterials. Chem. Commun. 2011, 47, 11814–11821. [Google Scholar] [CrossRef] [PubMed]
- Ulijn, R.V. Enzyme-responsive materials: A new class of smart biomaterials. J. Mater. Chem. 2006, 16, 2217. [Google Scholar] [CrossRef]
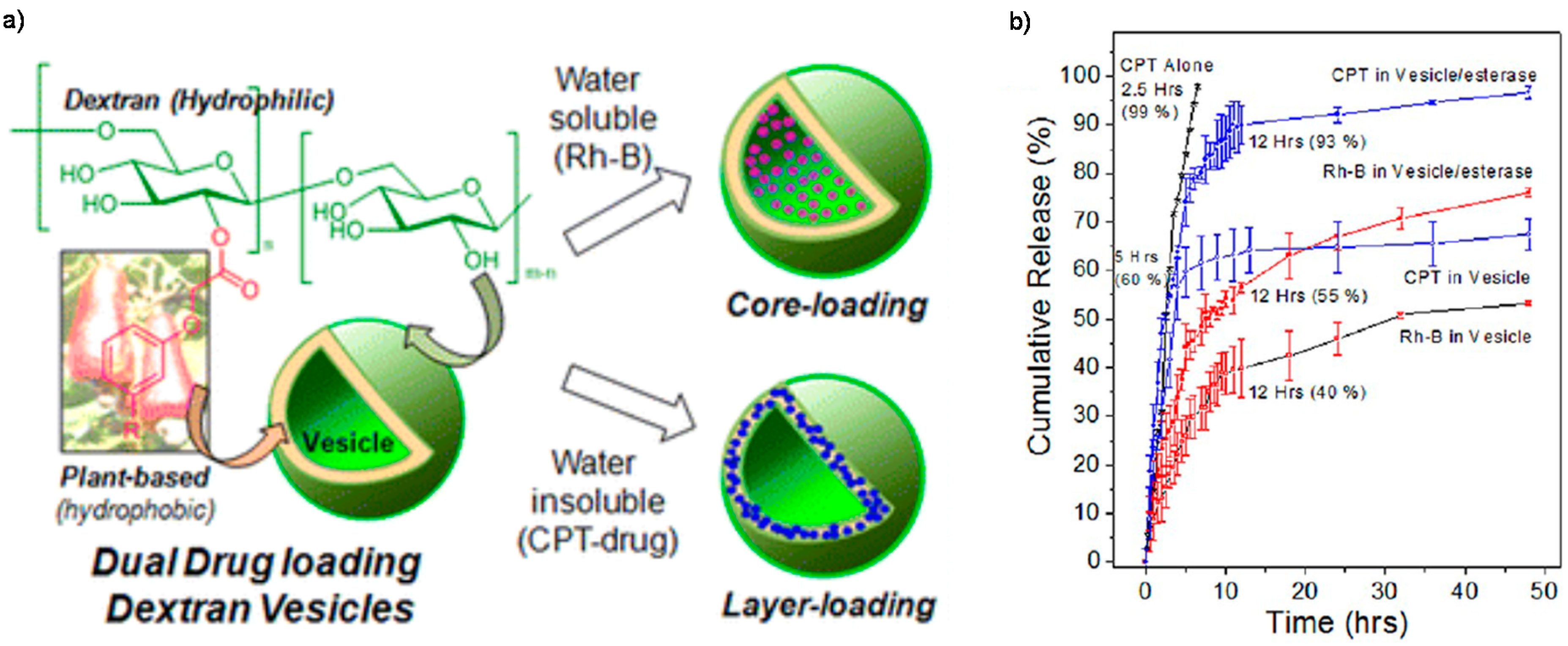
- Pramod, P.S.; Takamura, K.; Chaphekar, S.; Balasubramanian, N.; Jayakannan, M. Dextran vesicular carriers for dual encapsulation of hydrophilic and hydrophobic molecules and delivery into cells. Biomacromolecules 2012, 13, 3627–3640. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Liotta, L.A.; Kohn, E.C. The microenvironment of the tumor-host interface. Nature 2001, 411, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Vicent, M.J.; Greco, F.; Nicholson, R.I.; Paul, A.; Griffiths, P.C.; Duncan, R. Polymer therapeutics designed for a combination therapy of hormone-dependent cancer. Angew. Chem. 2005, 44, 4061–4066. [Google Scholar] [CrossRef] [PubMed]
- Chau, Y.; Frederick, E.T.; Robert, L. Synthesis and characterization of dextran-peptide-methotrexate conjugates for tumor targeting via mediation by matrix metalloproteinase II and matrix metalloproteinase IX. BioConjugate Chem. 2004, 15, 931–941. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Srivastava, A.; Galaev, I.Y.; Mattiasson, B. Smart polymers: Physical forms and bioengineering applications. Prog. Polym. Sci. 2007, 32, 1205–1237. [Google Scholar] [CrossRef]
- Shi, J.J.; Xiao, Z.Y.; Kamaly, N.; Farokhzad, O.C. Self-assembled targeted nanoparticles: Evolution of technologies and bench to bedside translation. Acc. Chem. Res. 2011, 44, 1123–1134. [Google Scholar] [CrossRef] [PubMed]
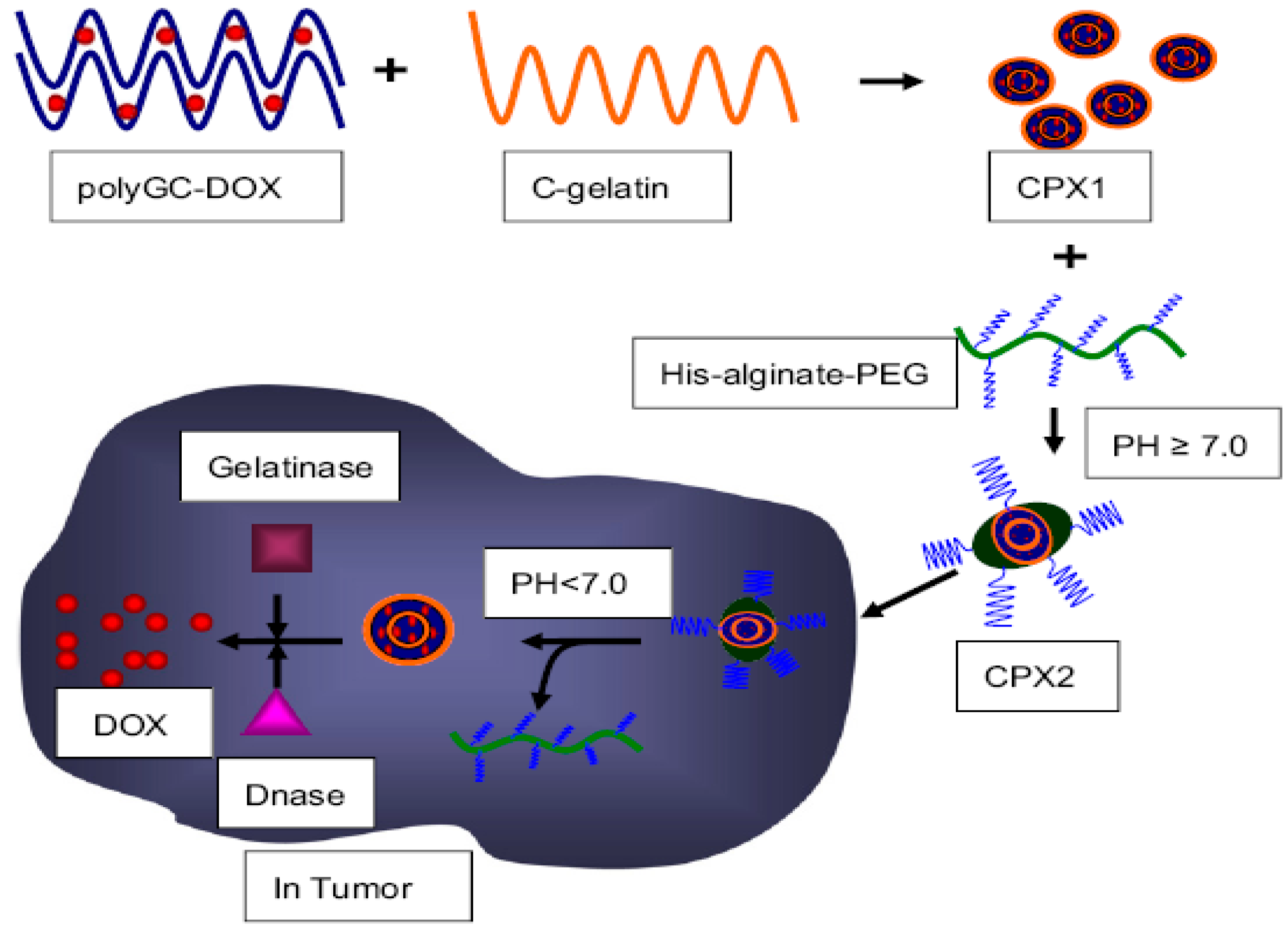
- Dong, L.; Xia, S.H.; Wu, K.; Huang, Z.; Chen, H.; Chen, J.N.; Zhang, J.F. A pH/Enzyme-responsive tumor-specific delivery system for doxorubicin. Biomaterials 2010, 31, 6309–6316. [Google Scholar] [CrossRef] [PubMed]
- Messersmith, T.J.P.B.; Messersmith, A.E. In situ crosslinking of a biomimetic peptide-PEG hydrogel via thermally triggered activation of factor XIII. Biomaterials 2002, 23, 2703–2710. [Google Scholar]
- Ehrbar, M.; Rizzi, S.C.; Schoenmakers, R.G.; Miguel, B.S.; Hubbell, J.A.; Weber, F.E.; Lutolf, M.P. Biomolecular hydrogels formed and degraded via site-specific enzymatic reactions. Biomacromolecules 2007, 8, 3000–3007. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.H.; Messersmith, P.B. Rational design of transglutaminase substrate peptides for rapid enzymatic formation of hydrogels. J. Am. Chem. Soc. 2003, 125, 14298–14299. [Google Scholar] [CrossRef] [PubMed]
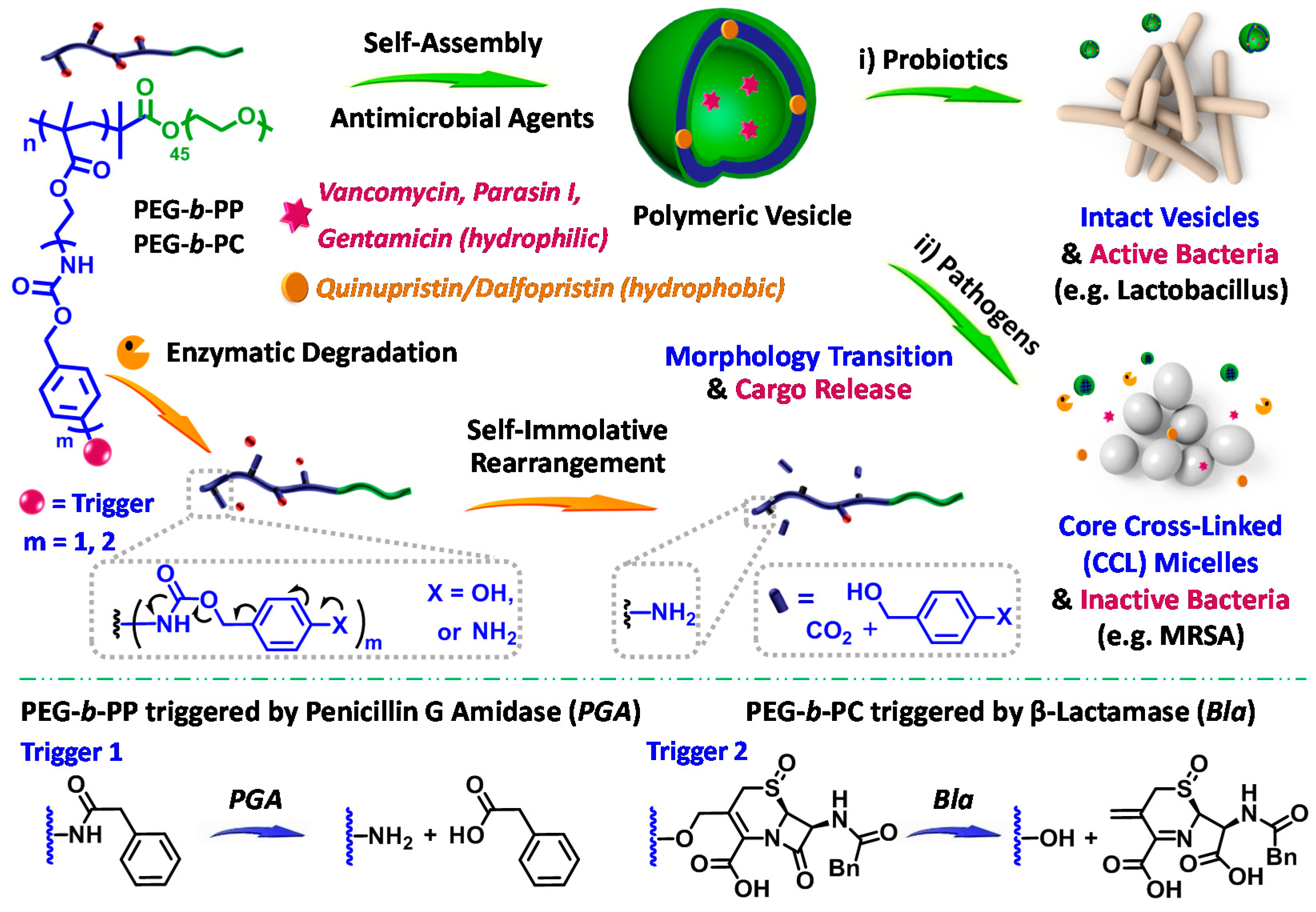
- Li, Y.M.; Liu, G.H.; Wang, X.R.; Hu, J.M.; Liu, S.Y. Enzyme-responsive polymeric vesicles for bacterial-strain-selective delivery of antimicrobial agents. Angew. Chem. 2016, 55, 1760–1764. [Google Scholar] [CrossRef] [PubMed]
- Simona, C.; Diana, V.; Jeffrey, A.H. PEG-SS-PPS: Reduction-sensitive disulfide block copolymer vesicles for intracellular drug delivery. Biomacromolecules 2007, 8, 1966–1972. [Google Scholar]
- Li, C.H.; Wu, T.; Hong, C.Y.; Zhang, G.Q.; Liu, S.Y. A general strategy to construct fluorogenic probes from charge-generation polymers (CGPs) and AIE-active fluorogens through triggered complexation. Angew. Chem. 2012, 51, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Baek, C.; Ha, T.L.; Kim, E.; Jeong, S.W.; Lee, S.G.; Lee, S.J.; Kim, H.C. Bioreducible micelles self-assembled from poly(ethylene glycol)-cholesteryl conjugate as a drug delivery platform. Polymers 2015, 7, 2245–2258. [Google Scholar] [CrossRef]
- Jiang, Y.Y.; Liu, G.H.; Wang, X.R.; Hu, J.M.; Zhang, G.Y.; Liu, S.Y. Cytosol-specific fluorogenic reactions for visualizing intracellular disintegration of responsive polymeric nanocarriers and triggered drug release. Macromolecules 2015, 48, 764–774. [Google Scholar] [CrossRef]
- Hu, J.M.; Wang, X.R.; Qian, Y.F.; Yu, Y.Q.; Jiang, Y.Y.; Zhang, G.Y.; Liu, S.Y. Cytoplasmic reactive cationic amphiphiles for efficient intracellular delivery and self-reporting smart release. Macromolecules 2015, 48, 5959–5968. [Google Scholar] [CrossRef]
- Li, Y.; Liu, T.; Zhang, G.Y.; Ge, Z.S.; Liu, S.Y. Tumor-targeted redox-responsive nonviral gene delivery nanocarriers based on neutral-cationic brush block copolymers. Macromol. Rapid Commun. 2014, 35, 466–473. [Google Scholar] [CrossRef] [PubMed]
- Duong, H.T.T.; Marquis, C.P.; Michael, W.; Davis, T.P.; Boyer, C. Acid degradable and biocompatible polymeric nanoparticles for the potential codelivery of therapeutic agents. Macromolecules 2011, 44, 8008–8019. [Google Scholar] [CrossRef]
- Cai, K.M.; He, X.; Song, Z.Y.; Yin, Q.; Zhang, Y.F.; Uckun, F.M.; Jiang, C.; Cheng, J.J. Dimeric drug polymeric nanoparticles with exceptionally high drug loading and quantitative loading efficiency. J. Am. Chem. Soc. 2015, 137, 3458–3461. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.L.; Hu, J.M.; Tian, J.; Ge, Z.S.; Zhang, G.Y.; Luo, K.F.; Liu, S.Y. Polyprodrug amphiphiles: Hierarchical assemblies for shape-regulated cellular internalization, trafficking, and drug delivery. J. Am. Chem. Soc. 2013, 135, 17617–17629. [Google Scholar] [CrossRef] [PubMed]
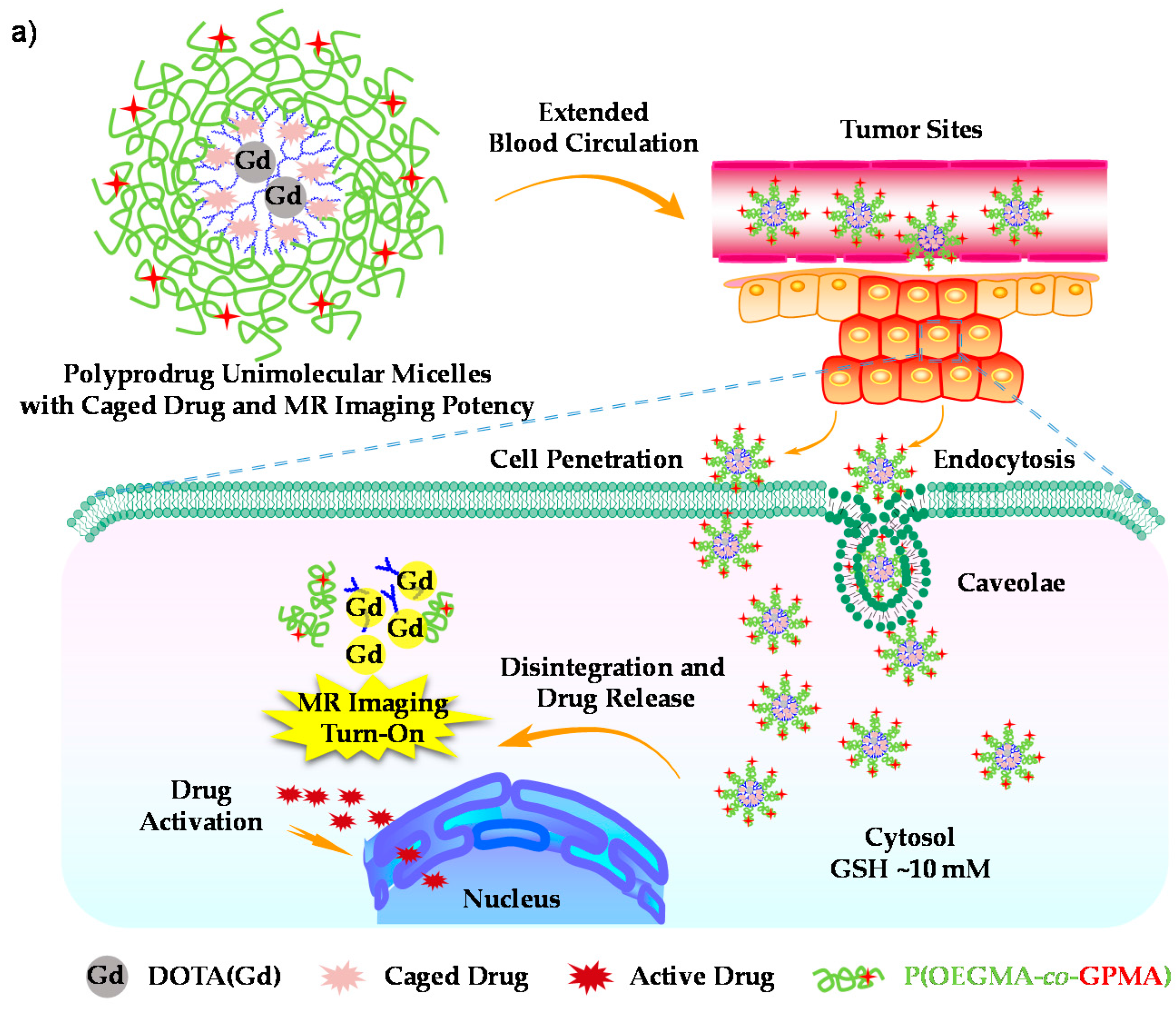
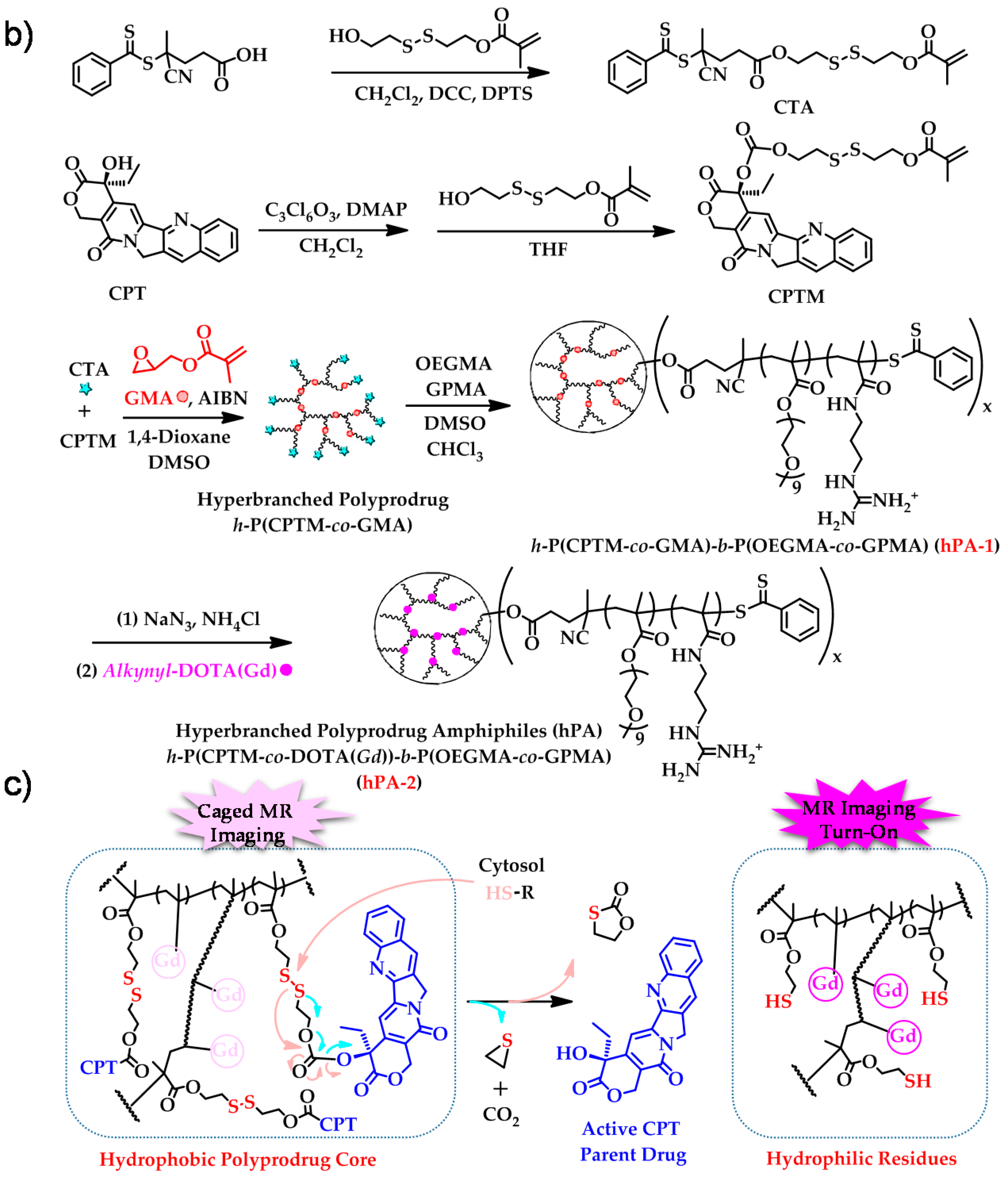
- Hu, X.L.; Liu, G.H.; Li, Y.; Wang, X.R.; Liu, S.Y. Cell-penetrating hyperbranched polyprodrug amphiphiles for synergistic reductive milieu-triggered drug release and enhanced magnetic resonance signals. J. Am. Chem. Soc. 2015, 137, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Fruehauf, J.P.; Meyskens, F.L. Reactive oxygen species: A breath of life or death. Clin. Cancer Res. 2007, 13, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Song, C.C.; Ji, R.; Du, F.S.; Liang, D.H.; Li, Z.C. Oxidation-accelerated hydrolysis of the ortho ester-containing acid-labile polymers. ACS Macro Lett. 2013, 2, 273–277. [Google Scholar] [CrossRef]
- Li, C.H.; Hu, J.M.; Liu, T.; Liu, S.Y. Stimuli-triggered off/on switchable complexation between a novel type of charge-generation polymer (CGP) and gold nanoparticles for the sensitive colorimetric detection of hydrogen peroxide and glucose. Macromolecules 2011, 44, 429–431. [Google Scholar] [CrossRef]
- Xu, H.P.; Cao, W.; Zhang, X. Selenium-containing polymers: Promising biomaterials for controlled release and enzyme mimics. Acc. Chem. Res 2013, 46, 1647–1658. [Google Scholar] [CrossRef] [PubMed]
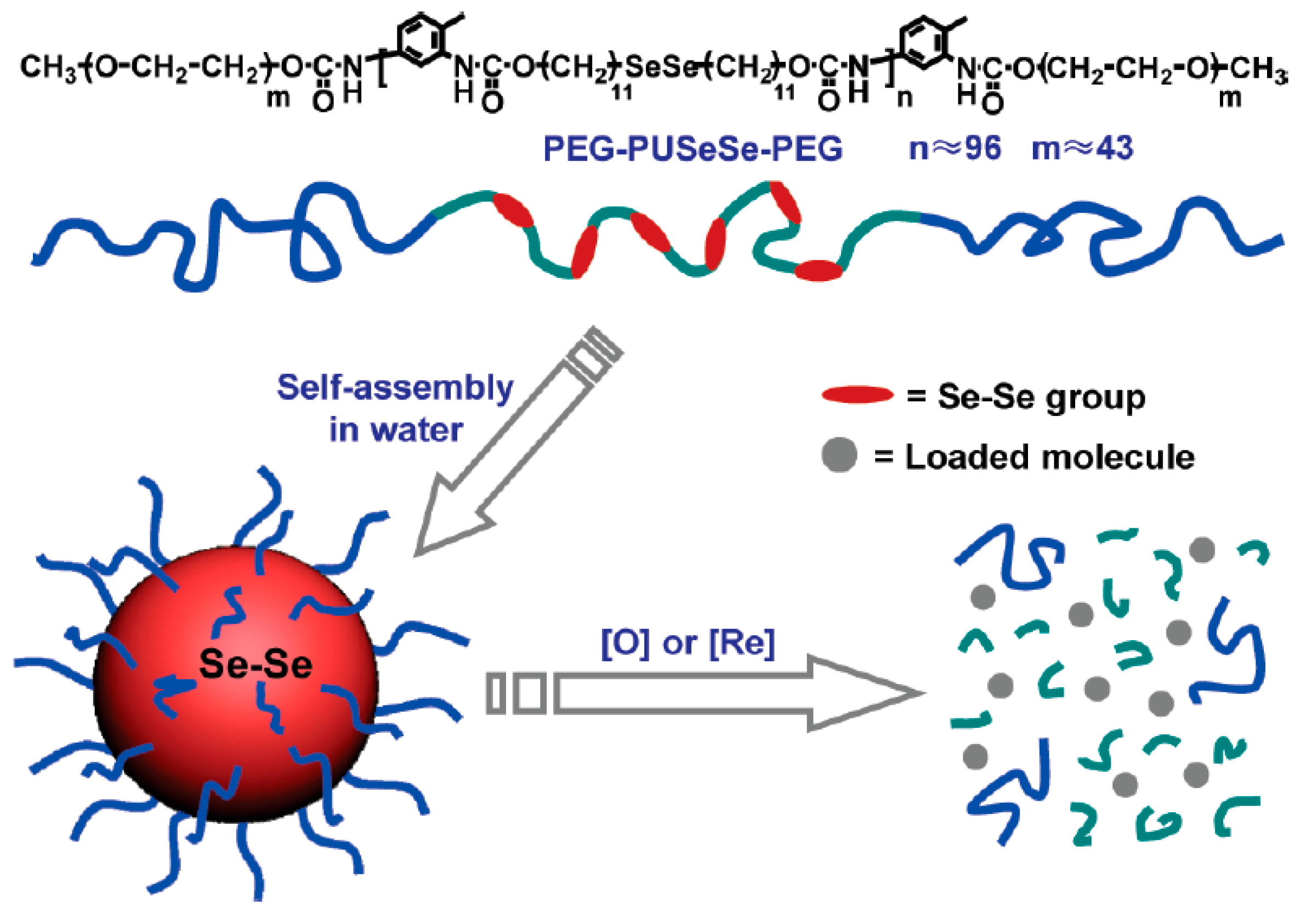
- Ma, N.; Li, Y.; Xu, H.P.; Wang, Z.Q.; Zhang, X. Dual redox responsive assemblies formed from diselenide block copolymers. J. Am. Chem. Soc. 2010, 132, 442–443. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.Y.; Yu, G.C.; Li, J.Y.; Huang, F.H. Photo-responsive self-assembly based on a water-soluble pillar[6]arene and an azobenzene-containing amphiphile in water. Chem. Commun. 2014, 50, 3606–3608. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.Y.; Wei, P.F.; Shi, B.B.; Huang, F.H. A pillar[6]arene-based [2]pseudorotaxane in solution and in the solid state and its photo-responsive self-assembly behavior in solution. Chem. Commun. 2016, 52, 513–516. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y. Light-responsive block copolymer micelles. Macromolecules 2012, 45, 3647–3657. [Google Scholar] [CrossRef]
- Li, Y.M.; Qian, Y.F.; Liu, T.; Zhang, G.Y.; Liu, S.Y. Light-triggered concomitant enhancement of magnetic resonance imaging contrast performance and drug release rate of functionalized amphiphilic diblock copolymer micelles. Biomacromolecules 2012, 13, 3877–3886. [Google Scholar] [CrossRef] [PubMed]
- Blasco, E.; Bernhard, V.K.J.S.; Kowollik, C.B.; Oriol, L. A novel photoresponsive azobenzene-containing miktoarm star polymer: Self-assembly and photoresponse properties. Macromolecules 2014, 47, 3693–3700. [Google Scholar] [CrossRef]
- Fomina, N.; Sankaranarayanan, J.; Almutairi, A. Photochemical mechanisms of light-triggered release from nanocarriers. Adv. Drug Delivery Rev. 2012, 64, 1005–1020. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.I.; Wu, W.; Oh, J.K.; Mueller, L.; Sherwood, G.; Peteanu, L.; Tomasz, K.L.W.; Ewski, K.S.M. Light-induced reversible formation of polymeric micelles. Angew. Chem. 2007, 46, 2453–2457. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zou, G.; Hu, J.M.; Liu, S.Y. Fabrication of photoswitchable and thermotunable multicolor fluorescent hybrid silica nanoparticles coated with dye-labeled poly(N-isopropylacrylamide) brushes. Chem. Mater. 2009, 21, 3788–3798. [Google Scholar] [CrossRef]
- Yin, J.; Hu, H.B.; Wu, Y.H.; Liu, S.Y. Thermo- and light-regulated fluorescence resonance energy transfer processes within dually responsive microgels. Polym. Chem. 2011, 2, 363–371. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, J.; Liu, F.; Kittredge, K.; Whitesell, J.K.; Fox, M.A. Competitive photochemical reactivity in a self-assembled monolayer on a colloidal gold cluster. J. Am. Chem. Soc. 2001, 123, 1464–1470. [Google Scholar] [CrossRef]
- Il’ichev, Y.V.; Schworer, M.A.; Wirz, J. Photochemical reaction mechanisms of 2-nitrobenzyl compounds: methyl ethers and caged ATP. J. Am. Chem. Soc. 2004, 126, 4581–4595. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Tong, X.; Morris, D.; Zhao, Y. Toward photocontrolled release using light-dissociable block copolymer micelles. Macromolecules 2006, 39, 4633–4640. [Google Scholar] [CrossRef]
- Zhao, H.; Sterner, E.S.; Coughlin, E.B.; Theato, P. o-Nitrobenzyl alcohol derivatives: Opportunities in polymer and materials science. Macromolecules 2012, 45, 1723–1736. [Google Scholar] [CrossRef]
- Zhang, Y.F.; Yin, Q.; Yin, L.C.; Ma, L.; Tang, L.; Cheng, J.J. Chain-shattering polymeric therapeutics with on-demand drug-release capability. Angew. Chem. 2013, 52, 6435–6439. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.R.; Liu, G.H.; Hu, J.M.; Zhang, G.Y.; Liu, S.Y. Concurrent block copolymer polymersome stabilization and bilayer permeabilization by stimuli-regulated “traceless” crosslinking. Angew. Chem. 2014, 53, 3138–3142. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.R.; Hu, J.M.; Liu, G.H.; Tian, J.; Wang, H.J.; Gong, M.; Liu, S.Y. Reversibly switching bilayer permeability and release modules of photochromic polymersomes stabilized by cooperative noncovalent interactions. J. Am. Chem. Soc. 2015, 137, 15262–15275. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Hu, J.M.; Zhang, G.Y.; Liu, S.Y. Rationally engineering phototherapy modules of eosin-conjugated responsive polymeric nanocarriers via intracellular endocytic pH gradients. BioConjugate Chem. 2015, 26, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Wang, X.R.; Hu, J.M.; Zhang, G.Y.; Liu, S.Y. Self-immolative polymersomes for high-efficiency triggered release and programmed enzymatic reactions. J. Am. Chem. Soc. 2014, 136, 7492–7497. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.H.; Zhang, G.F.; Hu, J.M.; Wang, X.R.; Zhu, M.Q.; Liu, S.Y. Hyperbranched self-immolative polymers (hSIPs) for programmed payload delivery and ultrasensitive detection. J. Am. Chem. Soc. 2015, 137, 11645–11655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Y.; Guo, R.; Yang, M.; Jiang, X.Q.; Liu, B.R. Thermo and pH dual-responsive nanoparticles for anti-cancer drug delivery. Adv. Mater. 2007, 19, 2988–2992. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C.W. Analysis of nanoparticle delivery to tumours. Nat. Rev. Mater. 2016, 10, 16014. [Google Scholar] [CrossRef]
- Truong, N.P.; Quinn, J.F.; Whittaker, M.R.; Davis, T.P. Polymeric filomicelles and nanoworms: Two decades of synthesis and application. Polym. Chem. 2016, 7, 4295–4312. [Google Scholar] [CrossRef]
- Morris, M.C.; Depollier, J.; Mery, J.; Heitz, F.; Divita, G.N. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Biotechnol. 2001, 19, 1173–1176. [Google Scholar] [CrossRef] [PubMed]
- Daniels, D.S.; Schepartz, A. Intrinsically cell-permeable miniature proteins based on a minimal cationic PPII motif. J. Am. Chem. Soc. 2007, 129, 14578–14579. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.A.; Daniels, D.S.; Coplin, A.E.; Jordan, G.E.; McGregor, L.M.; Schepartz, A. Minimally cationic cell-permeable miniature proteins via α-helical arginine display. J. Am. Chem. Soc. 2008, 130, 2948–2949. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.Y.; Yin, L.C.; Kim, K.H.; Cheng, J.J. Helical poly(arginine) mimics with superior cell-penetrating and molecular transporting properties. Chem. Sci. 2013, 4, 3839–3844. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.Q.; Nagai, K.; Banno, M.; Okoshi, K.; Onitsuka, K.; Yashima, E. Enantiomer-selective and helix-sense-selective living block copolymerization of isocyanide enantiomers initiated by single-handed helical poly(phenyl isocyanide)s. J. Am. Chem. Soc. 2009, 131, 6708–6718. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.Y.; He, Y.G.; Chen, W.W.; Liu, N.; Zhu, Y.Y.; Ding, Y.S.; Yin, J.; Wu, Z.Q. Enantiomer-selective polypeptide-b-poly(phenyl isocyanide) hybrid rod-rod copolymers: One-pot synthesis, self-assembly, and cell imaging. Macromol. Rapid Commun. 2015, 36, 1511–1520. [Google Scholar] [CrossRef] [PubMed]
- He, Y.G.; Shi, S.Y.; Liu, N.; Zhu, Y.Y.; Ding, Y.S.; Yin, J.; Wu, Z.Q. Fabrication of SERS-active conjugated copolymers/gold nanoparticles composite films by interface-directed assembly. RSC Adv. 2015, 5, 39697–39704. [Google Scholar] [CrossRef]
- Li, W.; He, Y.G.; Shi, S.Y.; Liu, N.; Zhu, Y.Y.; Ding, Y.S.; Yin, J.; Wu, Z.Q. Fabrication of multi-charges generable poly(phenyl isocyanide)-block-poly(3-hexylthiophene) rod-rod conjugated copolymer. Polym. Chem. 2015, 6, 2348–2355. [Google Scholar] [CrossRef]
- He, Y.G.; Shi, S.Y.; Liu, N.; Zhu, Y.Y.; Ding, Y.S.; Yin, J.; Wu, Z.Q. Tetraphenylethene-functionalized conjugated helical poly(phenyl isocyanide) with tunable light emission, assembly morphology, and specific applications. Macromolecules 2016, 49, 48–58. [Google Scholar] [CrossRef]















© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, J.; Chen, Y.; Zhang, Z.-H.; Han, X. Stimuli-Responsive Block Copolymer-Based Assemblies for Cargo Delivery and Theranostic Applications. Polymers 2016, 8, 268. https://0-doi-org.brum.beds.ac.uk/10.3390/polym8070268
Yin J, Chen Y, Zhang Z-H, Han X. Stimuli-Responsive Block Copolymer-Based Assemblies for Cargo Delivery and Theranostic Applications. Polymers. 2016; 8(7):268. https://0-doi-org.brum.beds.ac.uk/10.3390/polym8070268
Chicago/Turabian StyleYin, Jun, Yu Chen, Zhi-Huang Zhang, and Xin Han. 2016. "Stimuli-Responsive Block Copolymer-Based Assemblies for Cargo Delivery and Theranostic Applications" Polymers 8, no. 7: 268. https://0-doi-org.brum.beds.ac.uk/10.3390/polym8070268