
Polyolefins, a Success Story


Université de Lyon, Univ Lyon 1, CPE Lyon, CNRS, UMR 5265, C2P2 (Chemistry, Catalysis, Polymers & Processes), Bat. 308F, 43 Bd du 11 Novembre 1918, 69616 Villeurbanne, France
*
Author to whom correspondence should be addressed.
Polymers 2017, 9(6), 185; https://0-doi-org.brum.beds.ac.uk/10.3390/polym9060185
Submission received: 21 April 2017
/
Revised: 19 May 2017
/
Accepted: 21 May 2017
/
Published: 24 May 2017
(This article belongs to the Special Issue Tailored Polymer Synthesis by Advanced Polymerization Techniques)
Abstract
:This paper reports the principal discoveries which have played a major role in the polyolefin field and have positioned polyolefins as the most produced plastics. The early development of polyolefins covering the production of LDPE (Low density polyethylene) at ICI (Imperial Chemical Industries) and the discovery of Phillips or Ziegler-Natta catalysts are highlighted in the first section. In the second part, the impact of the implementation of molecular catalysts on the research in polyolefins is discussed together with the most recent advances leading to high-performance tailor-made resins.

{kind=link}
{kind=link}
1. Introduction
Polyolefins, in particular polyethylene (PE) and isotactic polypropylene (i-PP), are widely used in our everyday life for an extremely wide range of applications; indeed, they account for more than 50% in weight of the produced polymers. More than 300 grades of commercially available polyolefins provide a wide range of mechanical properties [1]. With more than 178 million tons having been produced in 2015 [2,3], polyolefins remain at the top of the global production of synthetic polymers [4]. From the point of view of cost, PE and i-PP rank as the least expensive polymers to produce, and stand out because they are far less toxic compared to many other commodity plastics [5]. As discussed by Mülhaupt et al. [6], polyolefins meet the requirements of sustainable development and green polymer chemistry [7,8]. The development of solvent-free catalytic processes, the design of lightweight engineering plastics, the recyclability and the low carbon footprint of polyolefins are the main arguments for this statement. Polyethylene is the simplest polyolefin, its general formula is (–CH2–CH2–)n, and it is generally composed of a mixture of interconnected crystalline and amorphous parts which can exhibit short and long chain branching. Even if polyethylene seems to be an unsophisticated material in a first glance, it turns to be possible to tailor the polymer to include elaborate structures based on the organization of the three basic groups, methyl, methylene and methine. This also includes the preparation of blends.
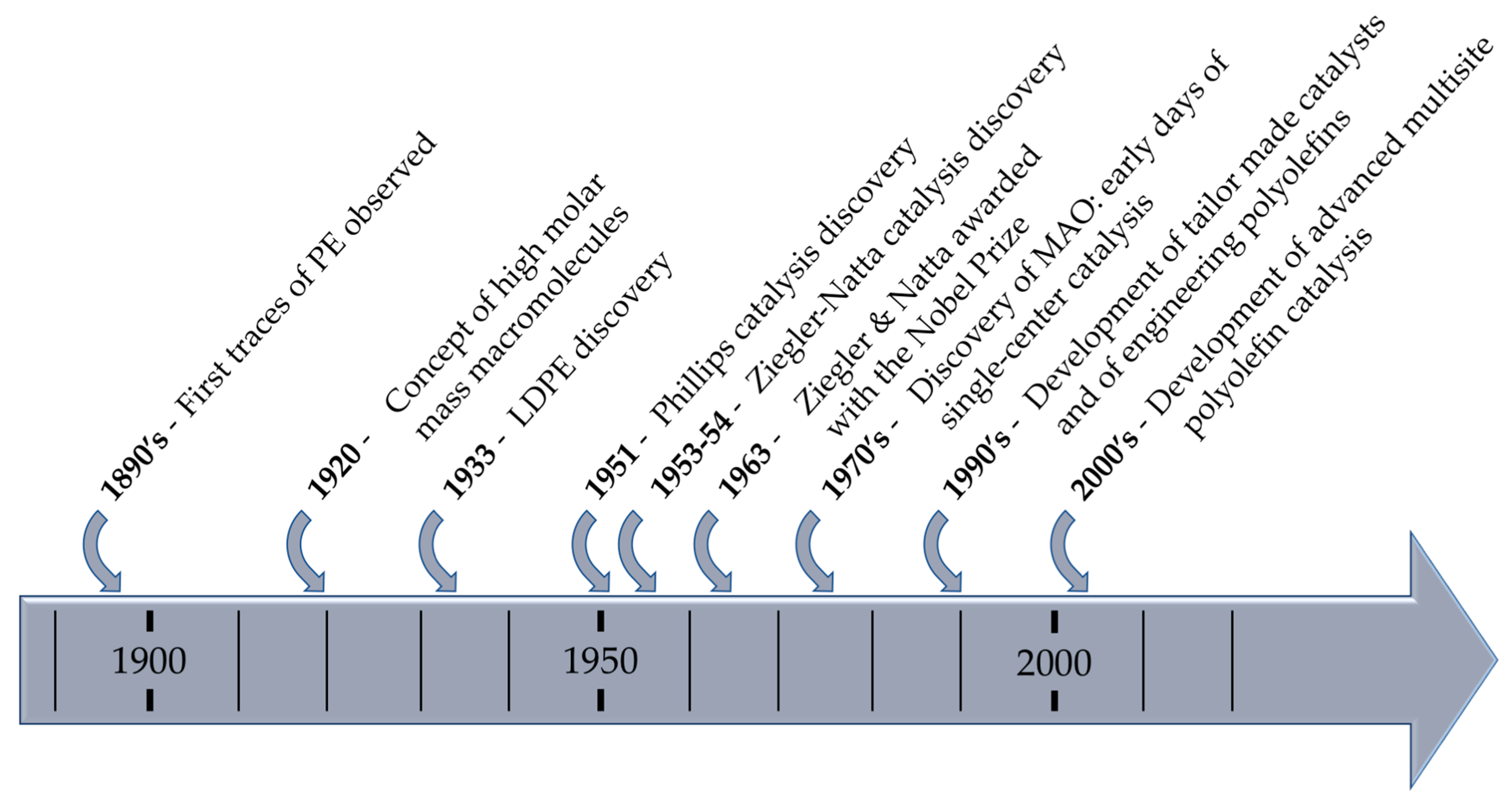
Polyolefins are either produced by a free radical process (Low density polyethylene: LDPE) or using coordination catalysis (Low linear density polyethylene: LLDPE, high density polyethylene: HDPE, and i-PP). The present paper highlights the chronology of the development of the polyolefin production and the recent advances leading to performance polyolefins available today (Scheme 1).
2. Polyolefins, the Beginning of the Story: How Does It All Start?
Although the first trace of polyethylene was reported at the end of the 19th century as a side product formed during the thermal decomposition of diazomethane [9], it was only identified later as a polymer.
In the 1920s Hermann Staudinger set the basis for a new field of chemistry by introducing the concept of high molar mass macromolecules, and defining the polymerization process as linking together individual small monomer molecules by the formation of covalent bonds [10,11,12]. It was only a few years later, in 1933 at the Imperial Chemical Industries (ICI) site in Winnington (Northwich, UK) that Fawcett and Gibson discovered the overnight formation of a small amount (about 1 g) of an unexpected white powder while running some condensation experiments with highly pressurized ethylene and benzaldehyde [13,14]. When Fawcett and Gibson tried to repeat this experiment without benzaldehyde, the reactor exploded. It took two additional years to finally repeat the production of polyethylene, and yet again, the success of the test was obtained by chance. Indeed, while repeating this experiment, one of their autoclaves had a leak which allowed some traces of oxygen to contaminate the ethylene batch. At high temperature, the molecular oxygen decomposed to provide free radicals thereby enabling the formation of polyethylene [15]. In this free radical process, intra- and intermolecular chain transfer led to the formation of LDPE containing short and long chain branching. A process employing high temperature (200 to 300 °C) and high pressure (1000 to 4000 bars) was patented by ICI in 1937 [16], and the first factory production plant started operating in 1939 with a capacity of 100 ton/year [14]. Nowadays, LDPE is still widely produced with conditions close to those elaborated by Fawcett, Gibson and Perrin, and largely used in the food industry because it combines excellent mechanical properties and food friendly advantages (non-toxic and metal free materials).
Technological advances continued during the 1940s; several groups developed some efforts in order to find a catalytic process for the production of polyethylene. Mayo at U.S. Rubber Company and Fischer at BASF AG produced HDPE. The U.S. Rubber company did not believe in HDPE’s commercial potential [17], while BASF AG showed interest in Fischer’s catalysis work involving titanium tetrafluoride and aluminum powder [13]. However, during this troubled period, Fischer was asked to refocus his research in order to participate in the war effort by developing new types of fuel for the army. This considerably slowed down his advances in polyolefin catalysis, but finally his catalytic system was patented in 1953, ten years after its discovery [18].
It was in the early 1950s that two major breakthroughs happened in the field of catalytic polymerization of olefins. These discoveries enabled to run polymerization under milder temperature and pressure conditions than those previously used for LDPE production. The first major progress concerned the work done by Hogan and Banks at Phillips Petroleum Company, in Bartlesville, Oklahoma. Hogan and Banks research focused on the conversion of gaseous olefin to liquid oligomers, to produce fuel. For this purpose researchers at Phillips used to work with nickel oxides supported on silica or alumina [19,20,21]. In June 1951 while investigating the effect of a mixture of nickel oxides and chromium oxide compounds supported on silica/alumina, they unexpectedly plugged their reactor with solid HDPE [22]. They finely tuned their catalyst to the point where only chromium oxides were supported on silica. They found that they were able to polymerize ethylene with this new catalyst, and in October 1951 they also managed to polymerize propylene. They filed a patent application which was approved in 1953 [23,24,25]. Subsequently, they developed a commercial process by licensing this invention no later than 4 years after the discovery. Nowadays Phillips catalysts still represent 40 to 50% of the global production of HDPE, and the Phillips process remains quite popular because of its faculty to offer small amounts of long chain branching (LCB) via macromer insertion [26,27,28,29]. The existence of LCB offers major advantages in terms of mechanical properties, as well as easily controlled molding parameters [30].
The second major breakthrough occurring in the 1950s was the work of Ziegler at the Mülheim Max Planck Institute in Germany. Ziegler had been developing new types of organometallic compounds for a carbon-carbon bond formation purpose since the 1920s. At this time Ziegler’s favorite monomers were olefins, styrene and dienes. For example in 1928 he performed the polymerization of butadiene using alkyllithium as initiator [31]. However, the production of polyethylene using this method was limited because of the decomposition of lithium alkyls into lithium hydride and olefins. Ziegler found a way to prepare LiAlH4 as well as a direct pathway to produce alkylaluminum compounds directly from aluminum, hydrogen and olefins. He used those organometallic reagents to develop the so called “Aufbaureaktion” which enabled the preparation of 1-olefins and aliphatic alcohol (alfol synthesis), as well as high purity alumina via the oxidation of the alkylaluminum compounds. Due to fortuitous circumstances Ziegler realized that the presence of nickel changed the path of his “Aufbaureaktion” by favoring the chain termination of the reaction, leading to the formation of 1-butene. This phenomenon was called “the nickel effect”. They explained the presence of nickel in their Inox autoclave from either a previous hydrogenation reaction, or an aggressive cleaning of the reaction vessel. However, the identification of this phenomenon stood out as the beginning of an important metal screening performed by Ziegler’s students, in order to identify the behavior of different metal complexes while associated with some alkylaluminum compounds [32]. From August to October 1953 the association of triethylaluminum with several transition metal complexes, such as chromium, vanadium, manganese and platinum was evaluated. At that time, the most significant result was obtained by combining zirconium acetylacetonate with triethylaluminum, leading to a conversion above 90% of the introduced ethylene. Based on these results, Ziegler filed a patent application just 3 days after repeating the experiment. This patent application claimed “a process for the production of a high molar mass polyethylene useful as a plastic” involving the association of triethylaluminum with several complexes involving metals from group 4 (Ti, Zr, Hf), 5 (V, Nb, Ta) and 6 (Cr, Mo) [33]. In November 1953 titanium tetrachloride mixed with triethylaluminum was discovered to triple the productivity of the polymerization, and they were able to run the polymerization of ethylene at lower temperatures and with pressure of ethylene below 55 bars. This outcome prompted Ziegler to file a second patent in December 1953, claiming more gentle conditions than in the previous patent [34]. In March 1954, Natta successfully performed the stereospecific polymerization of propylene using a Ziegler catalyst (titanium trichloride/triethylaluminum). The isolated isotactic polypropylene was characterized by X-ray crystallography [35]. Natta patented the process for production of isotactic polypropylene [36,37,38,39]. Ziegler also filed a patent application for the production of i-PP, but it was 10 days too late [15]. Even if Natta was not the first to produce polypropylene, he nonetheless introduced the concept of stereoregularity and participated actively in the development of the polyolefin field [40]. In 1957 the first plant producing isotactic polypropylene using Ziegler-Natta technology opened in Ferrara, Italy [41]. In the 1960s, Ziegler-Natta catalysts were extensively studied in order to maximize the activity and to improve the stereospecificity of propylene polymerization. In particular, the catalysts were supported on various surfaces, especially on MgCl2. The impact of the work in the field of olefin polymerization done by Ziegler and Natta was rewarded with the Noble Prize in Chemistry in 1963 [42]. Because of the importance of Ziegler’s invention, many companies tried to invalidate the patent filed by Ziegler in 1953, arguing that Fischer’s work at BASF described suitable conditions to form Ziegler’s catalysts. However a US court decision of 1967 proved them wrong and maintained Ziegler’s protection [43].
3. Single-Center Technology
In the mid. 1970s, another “incident” led to the discovery of a chemical species which changed the world of polymerization catalysis [44]. Indeed, at the Institute of Technical and Macromolecular Chemistry at the University of Hamburg, Sinn and Kaminsky were investigating side reactions occurring while using Ziegler’s catalysts [45,46]. They monitored their experiments by NMR, and in order to facilitate the interpretation of the NMR, the reaction between trimethylaluminum and bis(cyclopentadienyl)titanium dimethyl was chosen as model. In the presence of ethylene, and after five hours of reaction, moisture from the air passed thought the plastic lid causing the unexpected formation of PE. A following experiment at larger scale clearly confirmed the presence of polyethylene. Researchers suspected an unidentified species to be responsible of this phenomenon, so they tried to screen multiple possibilities such as a side reaction of AlMe3 with chlorides, oxygen or water. After eliminating the two first hypothesis, it turned out that water reacting with trimethylaluminum formed a new compound which enhanced catalytic activity when associated with a titanocene [47]. This new compound called methylaluminoxane, referred as MAO, is commonly represented as (–Al–O(–Me)–)n. The overall composition of MAO includes some free AlMe3 and it is generally admitted to say that MAO is a cluster of aluminum, oxygen and methyl groups with the general formulae [–(CH3)xAlO(3-x)/2−] [48]. The presence of free AlMe3 in commercial MAO plays a crucial role on the catalytic activity and chain transfer to aluminum via the formation of dormant heterobimetallic species [49].
Recently, computational exploration has allowed researchers to outline several energetically favored MAO structures, and deeper investigations enabled them to demonstration that MAO dissociation led to a [AlMe2]+ cation, which was suspected to play a major role in the process of metallocene activation [50,51]. The discovery of MAO was the starting point for the wider development of metallocene catalysts [52]. Jordan et al. identified the active site responsible for olefin polymerization while using an activated metallocene as a cationic species [53,54]. Beyond group 4 metallocenes, it was shown that MAO can be used to activate a wide range of molecular complexes of early and late transition metals, leading to a new generation of catalysts with tailor-made properties. These catalysts, based on well-defined precursors, are also named “single-center catalysts” [55]. As an example, in 1991, Exxon-Mobil launched its Exxpol™ Technology for production of polyolefins using a metallocene catalyst [56]. In addition to the gain of productivity enabled by MAO activation of the metallocene, these catalysts were also used to prepare stereoregular polyolefins [55,57,58] such as isotactic polypropylene (i-PP) [59,60], syndiotactic polypropylene (s-PP) [61], hemi-isotactic polypropylene (hi-PP) [62] and stereoblock polypropylene [63,64,65,66]. The relation between the symmetry of the metallocene center and the polypropylene microstructure can be established using Ewen’s stereocontrol rules [67]. These tailor-made catalysts for propylene polymerization constituted the second breakthrough generated by MAO discovery. Hemi-metallocenes were also developed in order to fit to specific need in terms of polyolefin properties [68]. In particular ansa-cyclopentadienyl-amido titanium complexes [69,70,71] were designed and implemented for ethylene polymerization, and were referred as constrained geometry catalysts (CGC) [72,73,74,75]. Dow’s INSITE™ CGC catalyst technology was launched in 1992 [56]. CGC catalysts enabled better insertion of co-monomer (especially α-olefins) than regular metallocenes, because of a less hindered coordination sphere, coupled to a Cp(centroid)-Ti-N with a smaller bite angle [76]. This class of complexes offered enhanced thermal stability, allowing them to be used at high temperature in the Dowlex™ solution process [77]. A vast range of non-metallocene molecular catalysts were developed during the last three decades leading to an outstanding variety of accessible polyolefin materials [68,78,79,80,81,82].
Subsequently, molecular activators able to activate metallocene (or post metallocene) complexes under a cationic form were discovered [83,84]. For example the tris(pentafluorophenyl)borane which was synthesized a decade before the discovery of MAO [85,86], was independently identified in the early 1990 by Marks [87,88] and Ewen [89,90] as an activator for group 4 metallocene complexes. The activation of metallocene complexes with tetraperfluoroarylborate salt ([Ph3C]+[B(C6F5)4]− [91,92] or [HNRR’2]+[B(C6F5)4]− [93,94,95]) led to the formation of dissociated ion pair species which featured increased activity in olefin polymerization. It is worth mentioning the role of dissociation of the catalyst ion pair (inner and outer ion pairs) in polymerization [96,97].
As in the case of classical Ziegler-Natta catalyst, the presence of main group organometallic species (AlR3, MgR2) or ZnEt2 in polymerization medium led to chain transfer to metal. Interestingly a fast and reversible chain transfer provided a unique method for controlled coordination polymerization of olefins [98,99,100,101].
4. Developments in Polyolefins Driven by Molecular Catalysts
For two decades since MAO metallocene activation, many improvements in single-site technology were realized as described above. Besides the activity gain and the access to unique stereoregular types of polypropylene (s-PP; hi-PP or stereoblock-PP), molecular catalyst evolution allowed the development of specialty polyolefins. For instance, ethylene/CO copolymers (polyketones—Carilon™) [102,103,104] and cycloolefin copolymers (COC) [105] were marketed by respectively Shell chemicals (discontinued in 2000) and Ticona. Dow launched new plastomers (AFFINITY™), polyolefin elastomers (ENGAGE™) and performance EPDM (NORDEL™) based on their INSITE™ technology [56]. The same company discovered non metallocene hafnium catalysts (pyridyl-amido hafnium catalyst) for the stereospecific polymerization of propylene at high temperature [81]. Propylene/ethylene copolymers were produced in a solution process and commercialized as plastomers and elastomers (VERSIFY™).
Bis(phenoxy-imine) group 4 catalysts (FI catalysts) are another major class of molecular catalysts introduced by Mitsui Chemicals. These catalysts showed high efficiency for olefin polymerization and were also very versatile [82,106,107]. Changing the substituent of the phenoxy-imine ligands, the metal (Ti and Zr) or the activator can lead to the production of a wide range of polyolefins, including ultrahigh molecular weight polyethylene, vinyl-terminated polyethylene that was turned to functional polyethylene, and isotactic or syndiotactic polypropylene. Note that Dow Chemicals used a FI catalyst in association with their pyridyl-amido hafnium catalyst and ZnEt2 to produce multiblock copolymers (see below). Intense research efforts at DSM (acquired by Lanxess in 2011, now Arlanxeo) led to the development of cyclopentadienyl-amidinato titanium catalysts for the production of high-performance EPDM (Keltan Ace™) [68]. The copolymerization of olefins with conjugated dienes was a challenge since these two classes of monomers polymerized via different mechanisms. Neodymium metallocene catalysts showed unprecedented copolymerization of ethylene with butadiene leading to a new class of elastomers [108,109,110]. Recently, Perrin et al. used computational investigation to demonstrate that a heterobimetallic and a monomeric intermediate were formed after insertion of ethylene and butadiene respectively [111]. More generally, progress in computational investigation using DFT as modeling method led to accurate prediction of the behavior of olefin polymerization catalysts [112].
For a long time, conventional multi-site catalysts (Ziegler-Natta) were opposed to molecular catalysts namely because of the lack of control in terms of chemical composition distribution leading to branching in the lower molar mass fraction of LLDPE. Importantly, researchers turned this paradigm around by switching from conventional multi-site to advanced multi-site catalysts via ingenious combinations of catalysts enabling the production of polyolefins with tailor-made properties. We can define two classes of multi-site catalysts (i) hybrid catalysts which combine individual effects of each component and (ii) concurrent tandem catalysts which lie on cooperative effect between each component. Hybrid catalysts were developed to produce high-performance bimodal resins where branching were preferentially located in the high molar mass fraction. These resins represent a new class of high-performance polyolefins. Single-center technology provides polyolefins with a low dispersity (Ð around 2), but in many instances it is essential to offer a resin which presents a broader molecular weight distribution in order to limit the shear forces involved during extrusion. In addition, mechanical properties can be largely improved by introducing branching in the high molar mass fraction. It is the concept of reverse comonomer distribution. This topic was comprehensively reviewed by Friederichs et al. [113] and Mülhaupt et al. [6]. It is worth pointing out that bimodal resins can not only be produced using combination of catalysts in a single reactor [114], but also by an engineering approach, in a cascade of (continuous) reactors [115,116,117], or multizone reactors [118,119]. The preparation of bimodal resins using hybrid catalysts has been evaluated since the early times of single site catalysts. Indeed Ewen and Welborn investigated the combination of different metallocenes in solution process [120,121]. In the case of particle-forming processes (slurry and gas phase), tremendous advances were achieved by combining supported catalysts (Ziegler-Natta catalysts, Phillips catalysts, supported molecular catalysts). The Prodigy BMC catalyst, commercialized by Univation Technologies, constitutes one of the better known classes of hybrid catalysts obtained by the combination of two metallocene complexes [122,123,124]. In 2016, Mülhaupt reported the synthesis of a tri-site catalyst supported on clay. This catalyst involved two different types of Cr based centers (CrBIP and CrQCp) associated to either a Zr based center (ZrCp) or to a Fe based center (FeBIP). The polymerization of ethylene with this tri-site catalyst afforded a tri-modal resin presenting extraordinary enhanced properties [125]. Independently of the techniques used to produce mutlimodal resins, this challenge led to a major competition between polyolefin producers and many advances were reported, in particular by Univation Technologies, Ineos, Chevron Phillips Chemical and LyondellBasell [6,113]. Multimodal resins produced in this manner offer significant gains in terms of processability, stiffness and crack resistance.
Tandem catalysts were developed for the preparation of branched polyolefins with ethylene using the combination of oligomerization and polymerization catalysts [126,127,128,129,130,131]. Interestingly, the combination of oligomerization catalysts leading to macromers with a CGC catalysts permitted the production of branch-block thermoplastic elastomers [132]. Recently, binuclear catalysts based on an olefin polymerization catalyst part (typically a Ti based CGC complex) linked to an ethylene trimerization catalyst part (typically a SNS-Cr based complex) were successfully synthesized by Marks et al. From ethylene feed and activation of this cooperative Ti-SNS-Cr catalysts by MAO, the resulting resin displayed only butyl branches (LLDPE) [133,134].
In a different manner, synergetic effects were also disclosed leading to unique performance polyolefins. Researchers at the Dow Chemical Company introduced the concept of chain shuttling combining two catalysts (a FI zirconium and a pyridyl-amido hafnium catalyst) with different comonomer responses and a chain shuttling agent (ZnEt2) transferring the growing chain from one metal to the other one. Olefin block copolymers (OBCs) bearing soft and hard polyolefin sequences were marketed (INFUSE™) and led to a new area of research [135,136,137,138].
Most of single-site catalyst’s development were first optimized for homogeneous catalysis, for example Dow’s INSITE™ technology which involved CGC catalysts was developed initially for solution process at high temperatures [56,139,140,141,142,143]. Today, solution processes, such as “Compact Process” from Borealis, “Dowlex” from Dow Chemicals, or “Sclairtech” from Nova Chemicals enable the production of a wide variety of commodity or specialty polyolefins [144]. Nevertheless, solution processes are energy consuming (high temperature) and require large amount of solvent. Most of the major industrial processes for olefin polymerization are heterogeneous (slurry and gas phase processes). In these processes, the polymer crystallizes during the polymerization and it is very important to control the polymer particle morphology. This requires the use of solid catalysts which can fragment and lead to spherical polyolefin particles. Thus, the use of molecular catalysts in particle forming processes requires the immobilization of the catalyst on a support such as silica or MgCl2. This step is far from being trivial and requires many efforts in order to combine a high efficiency, with the production of spherical particles and resins with expected properties, without a significant generation of heat, nor active species leaching. This major research area was remarkably reviewed in a comprehensive book edited by Severn and Chadwick [1] and in review articles [144,145].
5. Conclusions
To conclude, even though Polyolefins were discovered and identified many years ago, this research field is constantly evolving. Multiple side-events led to major breakthroughs, enabling us to finely adjust polyolefins’ properties. More than 60 years after the first catalytic processes, the fields of polyolefin catalysis remains a very active field of research, which has been greatly transformed over time, passing from intrinsically multisite catalysts, to new advanced multi-site catalysts, leading year after year to more advanced materials. In the future, polyolefins will keep its leading role in the plastic industry and will continue to offer a wide pallet of solutions worldwide. Finally, we could paraphrase Vincenzo Busico highlighting 10 years ago the emergence of the concept of degenerative chain transfer, and in particular chain shuttling polymerization for the production of olefin block copolymers, the “success” story continues [146].
Acknowledgments
The authors are grateful for the invitation to contribute the present review article in the Special issue entitled “Tailored Polymer Synthesis by Advanced Polymerization Techniques”.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Severn, J.R.; Chadwick, J.C. (Eds.) Tailor-Made Polymers; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008. [Google Scholar]
- AlMa’adeed, M.A.-A.; Krupa, I. Introduction. In Polyolefin Compounds and Materials; AlMa’adeed, M.A.-A., Krupa, I., Eds.; Springer Series on Polymer and Composite Materials; Springer International Publishing: Berlin/Heidelberg, Germany, 2016; pp. 1–11. [Google Scholar]
- Hutley, T.J.; Ouederni, M. Polyolefins—The History and Economic Impact. In Polyolefin Compounds and Materials; AlMa’adeed, M.A.-A., Krupa, I., Eds.; Springer Series on Polymer and Composite Materials; Springer International Publishing: Berlin/Heidelberg, Germany, 2016; pp. 13–50. [Google Scholar]
- Stalzer, M.M.; Delferro, M.; Marks, T.J. Supported Single-Site Organometallic Catalysts for the Synthesis of High-Performance Polyolefins. Catal. Lett. 2014, 145, 3–14. [Google Scholar] [CrossRef]
- Tabone, M.D.; Cregg, J.J.; Beckman, E.J.; Landis, A.E. Sustainability Metrics: Life Cycle Assessment and Green Design in Polymers. Environ. Sci. Technol. 2010, 44, 8264–8269. [Google Scholar] [CrossRef] [PubMed]
- Stürzel, M.; Mihan, S.; Mülhaupt, R. From Multisite Polymerization Catalysis to Sustainable Materials and All-Polyolefin Composites. Chem. Rev. 2016, 116, 1398–1433. [Google Scholar] [CrossRef] [PubMed]
- Anastas, P.T.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Mülhaupt, R. Green Polymer Chemistry and Bio-based Plastics: Dreams and Reality. Macromol. Chem. Phys. 2013, 214, 159–174. [Google Scholar] [CrossRef]
- Pechmann, H.V. Ueber Diazomethan und Nitrosoacylamine. Berichte der Deutschen Chemischen Gesellschaft 1898, 31, 2640–2646. [Google Scholar] [CrossRef]
- Staudinger, H. Über Polymerisation. Berichte der Deutschen Chemischen Gesellschaft B 1920, 53, 1073–1085. [Google Scholar] [CrossRef]
- Staudinger, H.; Fritschi, J. Über Isopren und Kautschuk. 5. Mitteilung. Über die Hydrierung des Kautschuks und über seine Konstitution. Helv. Chim. Acta 1922, 5, 785–806. [Google Scholar] [CrossRef]
- Staudinger, H. Über die Konstitution des Kautschuks (6. Mitteilung). Berichte der Deutschen Chemischen Gesellschaft B 1924, 57, 1203–1208. [Google Scholar] [CrossRef]
- Seymour, R.B.; Cheng, T. (Eds.) History of Polyolefins; Springer: Dordrecht, The Netherlands, 1985. [Google Scholar]
- Utracki, L.A. Polymer Blends; iSmithers Rapra Publishing: Shawbury, UK, 2000. [Google Scholar]
- Seymour, R.B.; Cheng, T. (Eds.) Advances in Polyolefins; Springer: Boston, MA, USA, 1987. [Google Scholar]
- Fawcett, E.W.; Gibson, R.O. Improvements in or Relating to the Polymerisation of Ethylene. Patent GB471590, 6 September 1937. [Google Scholar]
- McMillan, F.M. The Chain Straighteners: Fruitful Innovation; the Discovery of Linear and Stereoregular Synthetic Polymers; Macmillan: New York, NY, USA, 1979. [Google Scholar]
- Fischer, M.D. Verfahren zur Herstellung von festen Polymerisaten aus AEthylen oder aethylenreichenGasen. Patent DE874215 C, 20 April 1953. [Google Scholar]
- Bailey, G.C.; Reid, J.A. Catalytic Polymerization of Olefins. U.S. Patent 2381198, 7 August 1945. [Google Scholar]
- Bailey, G.C.; Reid, J.A. SiO2-Al2O3-NiO Catalyst and Its Preparation. U.S. Patent 2581228, 1 January 1952. [Google Scholar]
- Bailey, G.C.; Reid, J.A. Catalytic Polymerization of Olefins. U.S. Patent 2606940, 12 August 1952. [Google Scholar]
- Sailors, H.R.; Hogan, J.P. History of Polyolefins. J. Macromol. Sci. Part. Chem. 1981, 15, 1377–1402. [Google Scholar] [CrossRef]
- Hogan, J.P.; Banks, R.L. Polymers and Production Thereof. U.S. Patent 2825721, 4 March 1958. [Google Scholar]
- Hogan, J.P.; Banks, R.L. Improvements in or Relating to Polymerization of Olefins. Patent GB790195, 10 October 1957. [Google Scholar]
- Hogan, J.P.; Banks, R.L. Perfectionnements aux Polymères et à Leur Production par Action Catalytique. Patent FR1144587, 15 October 1957. [Google Scholar]
- Hogan, J.P. Ethylene polymerization catalysis over chromium oxide. J. Polym. Sci. 1970, 8, 2637–2652. [Google Scholar] [CrossRef]
- Hogan, J.P. Ch.6—Catalysis of the Phillips Petroleum Company Polyethylene Process. In Applied Industrial Catalysis; Academic Press: New York, NY, USA, 1983; pp. 149–176. [Google Scholar]
- McDaniel, M.P. Supported Chromium Catalysts for Ethylene Polymerization. In Advances in Catalysis; Academic Press: New York, NY, USA, 1985; Volume 33, pp. 47–98. [Google Scholar]
- McDaniel, M.P.; Rohlfing, D.C.; Benham, E.A. Long Chain Branching in Polyethylene from the Phillips Chromium Catalyst. Polym. React. Eng. 2003, 11, 101–132. [Google Scholar] [CrossRef]
- Janzen, J.; Colby, R.H. Diagnosing long-chain branching in polyethylenes. J. Mol. Struct. 1999, 485–486, 569–584. [Google Scholar] [CrossRef]
- Ziegler, K.; Bähr, K. Über den vermutlichen Mechanismus der Polymerisationen durch Alkalimetalle (Vorläufige Mitteilung). Berichte der Deutschen Chemischen Gesellschaft B 1928, 61, 253–263. [Google Scholar] [CrossRef]
- Ziegler, K. A Forty Years’ Stroll through the Realms of Organometallic Chemistry. In Advances in Organometallic Chemistry; Academic Press: New York, NY, USA, 1968; Volume 6, pp. 1–17. [Google Scholar]
- Ziegler, K.; Heinz, B.; Erhard, H.; Heinz, M. High Molecular Polyethylenes. Patent DE973626, 14 April 1960. [Google Scholar]
- Ziegler, K.; Breil, H.; Martin, H. High molecular polyethylenes. Patent GB799392, 21 March 1957. [Google Scholar]
- Natta, G.; Pino, P.; Corradini, P.; Danusso, F.; Mantica, E.; Mazzanti, G.; Moraglio, G. Crystalline high polymers of α-olefins. J. Am. Chem. Soc. 1955, 77, 1708–1710. [Google Scholar] [CrossRef]
- Natta, G.; Pino, P.; Mazzanti, G. Prevailingly to Substantially Atactic Crude Polymers and Methods for Producing the Same. U.S. Patent 3261820, 19 July 1966. [Google Scholar]
- Natta, G.; Pasquon, I. Process for Polymerizing Unsaturated Hydrocarbons to Crystalline Polymers of Regulated Molecular Weight. U.S. Patent 3245973, 12 April 1966. [Google Scholar]
- Natta, G.; Crespi, G. Block Polymers of Alpha-Olefines, Processes for Producing the Same, and Mixtures Thereof with Isotactic Polyolefines. U.S. Patent 3175999, 30 March 1965. [Google Scholar]
- Natta, G.; Pino, P.; Mazzanti, G. Isotactic Polypropylene. U.S. Patent 3112300, 26 November 1963. [Google Scholar]
- Corradini, P. The impact of the discovery of stereoregular polymers in macromolecular science. Macromol. Symp. 1995, 89, 1–11. [Google Scholar] [CrossRef]
- Galli, P. Forty years of industrial developments in the field of isotactic polyolefins. Macromol. Symp. 1995, 89, 13–26. [Google Scholar] [CrossRef]
- The Nobel Prize in Chemistry 1963. Available online: http://www.nobelprize.org/nobel_prizes/chemistry/laureates/1963/ (accessed on 15 September 2015).
- Martin, H. Polymers, Patents, Profits: A Classic Case Study for Patent Infighting; John Wiley & Sons: New York, NY, USA, 2007. [Google Scholar]
- Kaminsky, W. Discovery of Methylaluminoxane as Cocatalyst for Olefin Polymerization. Macromolecules 2012, 45, 3289–3297. [Google Scholar] [CrossRef]
- Sinn, H.; Hinck, H.; Bandermann, F.; Grützmacher, H.F. Bildung von Poly(methyl-methylen-aluminium). Angew. Chem. 1968, 80, 190. [Google Scholar] [CrossRef]
- Kaminsky, W.; Vollmer, H.-J.; Heins, E.; Sinn, H. Die Bildung von Dimetalloalkylenen, eine unvermeidliche Nebenreaktion homogener ZIiegler-Katalysatoren. Makromol. Chem. 1974, 175, 443–456. [Google Scholar] [CrossRef]
- Andresen, A.; Cordes, H.-G.; Herwig, J.; Kaminsky, W.; Merck, A.; Mottweiler, R.; Pein, J.; Sinn, H.; Vollmer, H.-J. Halogen-Free Soluble Ziegler Catalysts for the Polymerization of Ethylene. Control of Molecular Weight by Choice of Temperature. Angew. Chem. Int. Ed. Engl. 1976, 15, 630–632. [Google Scholar] [CrossRef]
- Imhoff, D.W.; Simeral, L.S.; Sangokoya, S.A.; Peel, J.H. Characterization of Methylaluminoxanes and Determination of Trimethylaluminum Using Proton NMR. Organometallics 1998, 17, 1941–1945. [Google Scholar] [CrossRef]
- Ehm, C.; Cipullo, R.; Budzelaar, P.H.M.; Busico, V. Role(s) of TMA in polymerization. Dalton Trans. 2016, 45, 6847–6855. [Google Scholar] [CrossRef] [PubMed]
- Ghiotto, F.; Pateraki, C.; Tanskanen, J.; Severn, J.R.; Luehmann, N.; Kusmin, A.; Stellbrink, J.; Linnolahti, M.; Bochmann, M. Probing the Structure of Methylalumoxane (MAO) by a Combined Chemical, Spectroscopic, Neutron Scattering, and Computational Approach. Organometallics 2013, 32, 3354–3362. [Google Scholar] [CrossRef]
- Hirvi, J.T.; Bochmann, M.; Severn, J.R.; Linnolahti, M. Formation of Octameric Methylaluminoxanes by Hydrolysis of Trimethylaluminum and the Mechanisms of Catalyst Activation in Single-Site α-Olefin Polymerization Catalysis. ChemPhysChem 2014, 15, 2732–2742. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, W. The discovery of metallocene catalysts and their present state of the art. J. Polym. Sci. Part. A Polym. Chem. 2004, 42, 3911–3921. [Google Scholar] [CrossRef]
- Jordan, R.F.; Dasher, W.E.; Echols, S.F. Reactive cationic dicyclopentadienyl zirconium(IV) complexes. J. Am. Chem. Soc. 1986, 108, 1718–1719. [Google Scholar] [CrossRef]
- Jordan, R.F.; Bajgur, C.S.; Willett, R.; Scott, B. Ethylene polymerization by a cationic dicyclopentadienyl zirconium(IV) alkyl complex. J. Am. Chem. Soc. 1986, 108, 7410–7411. [Google Scholar] [CrossRef]
- Resconi, L.; Cavallo, L.; Fait, A.; Piemontesi, F. Selectivity in Propene Polymerization with Metallocene Catalysts. Chem. Rev. 2000, 100, 1253–1346. [Google Scholar] [CrossRef] [PubMed]
- Chum, P.S.; Swogger, K.W. Olefin polymer technologies—History and recent progress at The Dow Chemical Company. Prog. Polym. Sci. 2008, 33, 797–819. [Google Scholar] [CrossRef]
- Corradini, P.; Guerra, G.; Cavallo, L. Do New Century Catalysts Unravel the Mechanism of Stereocontrol of Old Ziegler—Natta Catalysts? Acc. Chem. Res. 2004, 37, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Busico, V.; Cipullo, R. Microstructure of polypropylene. Prog. Polym. Sci. 2001, 26, 443–533. [Google Scholar] [CrossRef]
- Ewen, J.A. Mechanisms of stereochemical control in propylene polymerizations with soluble Group 4B metallocene/methylalumoxane catalysts. J. Am. Chem. Soc. 1984, 106, 6355–6364. [Google Scholar] [CrossRef]
- Kaminsky, W.; Külper, K.; Brintzinger, H.H.; Wild, F.R.W.P. Polymerisation von Propen und Buten mit einem chiralen Zirconocen und Methylaluminoxan als Cokatalysator. Angew. Chem. 1985, 97, 507–508. [Google Scholar] [CrossRef]
- Ewen, J.A.; Jones, R.L.; Razavi, A.; Ferrara, J.D. Syndiospecific propylene polymerizations with Group IVB metallocenes. J. Am. Chem. Soc. 1988, 110, 6255–6256. [Google Scholar] [CrossRef] [PubMed]
- Ewen, J.A.; Elder, M.J.; Jones, R.L.; Haspeslagh, L.; Atwood, J.L.; Bott, S.G.; Robinson, K. Metallocene/polypropylene structural relationships: Implications on polymerization and stereochemical control mechanisms. Makromol. Chem. Macromol. Symp. 1991, 48–49, 253–295. [Google Scholar] [CrossRef]
- Coates, G.W.; Waymouth, R.M. Oscillating Stereocontrol: A Strategy for the Synthesis of Thermoplastic Elastomeric Polypropylene. Science 1995, 267, 217–219. [Google Scholar] [CrossRef] [PubMed]
- Wild, F.R.W.P.; Zsolnai, L.; Huttner, G.; Brintzinger, H.H. Ansa-Metallocene Derivatives: IV. Synthesis and molecular structures of chiral ansa-titanocene derivatives with bridged tetrahydroindenyl ligands. J. Organomet. Chem. 1982, 232, 233–247. [Google Scholar] [CrossRef]
- Schnutenhaus, H.; Brintzinger, H.H. 1,1’-Trimethylenebis(η5−3-tert-butylcyclopentadienyl)-titanium(IV)Dichloride, a Chiral ansa-Titanocene Derivative. Angew. Chem. Int. Ed. Engl. 1979, 18, 777–778. [Google Scholar] [CrossRef]
- Brintzinger, H.H.; Fischer, D.; Mülhaupt, R.; Rieger, B.; Waymouth, R.M. Stereospecific Olefin Polymerization with Chiral Metallocene Catalysts. Angew. Chem. Int. Ed. Engl. 1995, 34, 1143–1170. [Google Scholar] [CrossRef]
- Ewen, J.A. Symmetry rules and reaction mechanisms of Ziegler–Natta catalysts1. J. Mol. Catal. Chem. 1998, 128, 103–109. [Google Scholar] [CrossRef]
- Baier, M.C.; Zuideveld, M.A.; Mecking, S. Post-Metallocenes in the Industrial Production of Polyolefins. Angew. Chem. Int. Ed. 2014, 53, 9722–9744. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, P.J.; Bunel, E.; Schaefer, W.P.; Bercaw, J.E. Scandium complex [{(η5-C5Me4)Me2Si(η1-NCMe3)}(PMe3)ScH]2: A unique example of a single-component α-olefin polymerization catalyst. Organometallics 1990, 9, 867–869. [Google Scholar] [CrossRef]
- Gielens, E.E.; Tiesnitsch, J.Y.; Hessen, B.; Teuben, J.H. Titanium Hydrocarbyl Complexes with a Linked Cyclopentadienyl—Alkoxide Ancillary Ligand; Participation of the Ligand in an Unusual Activation of a (Trimethylsilyl)methyl Group. Organometallics 1998, 17, 1652–1654. [Google Scholar] [CrossRef]
- Piers, W.E.; Shapiro, P.J.; Bunel, E.E.; Bercaw, J.E. Coping with Extreme Lewis Acidity: Strategies for the Synthesis of Stable, Mononuclear Organometallic Derivatives of Scandium. Synlett 1990, 1990, 74–84. [Google Scholar] [CrossRef]
- Chen, Y.-X.; Fu, P.-F.; Stern, C.L.; Marks, T.J. A Novel Phenolate “Constrained Geometry” Catalyst System. Efficient Synthesis, Structural Characterization, and α-Olefin Polymerization Catalysis. Organometallics 1997, 16, 5958–5963. [Google Scholar] [CrossRef]
- Cano, J.; Kunz, K. How to synthesize a constrained geometry catalyst (CGC)—A survey. J. Organomet. Chem. 2007, 692, 4411–4423. [Google Scholar] [CrossRef]
- McKnight, A.L.; Waymouth, R.M. Group 4 ansa-Cyclopentadienyl-Amido Catalysts for Olefin Polymerization. Chem. Rev. 1998, 98, 2587–2598. [Google Scholar] [CrossRef] [PubMed]
- Babinec Susan, S.; Blanchard, M.; Guest, M.J.; Walther, B.; Chaudhary, B.I.; Barry, R.P. Compositions of Interpolymers of Alpha-Olefin Monomers with One or More Vinyl or Vinylidene Aromatic Monomers. European Patent WO9920685, 2 August 2000. [Google Scholar]
- Braunschweig, H.; Breitling, F.M. Constrained geometry complexes—Synthesis and applications. Coord. Chem. Rev. 2006, 250, 2691–2720. [Google Scholar] [CrossRef]
- Soares, J.B.P.; McKenna, T.F.L. Polyolefin Reaction Engineering; John Wiley & Sons: New York, NY, USA, 2013. [Google Scholar]
- Gibson, V.C.; Spitzmesser, S.K. Advances in Non-Metallocene Olefin Polymerization Catalysis. Chem. Rev. 2003, 103, 283–316. [Google Scholar] [CrossRef] [PubMed]
- Zuccaccia, C.; Macchioni, A.; Busico, V.; Cipullo, R.; Talarico, G.; Alfano, F.; Boone, H.W.; Frazier, K.A.; Hustad, P.D.; Stevens, J.C.; et al. Intra- and Intermolecular NMR Studies on the Activation of Arylcyclometallated Hafnium Pyridyl-Amido Olefin Polymerization Precatalysts. J. Am. Chem. Soc. 2008, 130, 10354–10368. [Google Scholar] [CrossRef] [PubMed]
- Heurtefeu, B.; Vaultier, F.; Leino, R.; Boisson, C.; Cramail, H. Single-Site Catalysts. In Encyclopedia of Polymer Science and Technology; John Wiley & Sons, Inc.: New York, NY, USA, 2014; Volume 12, pp. 551–602. [Google Scholar]
- Boussie, T.R.; Diamond, G.M.; Goh, C.; Hall, K.A.; LaPointe, A.M.; Leclerc, M.K.; Murphy, V.; Shoemaker, J.A.W.; Turner, H.; Rosen, R.K.; et al. Nonconventional Catalysts for Isotactic Propene Polymerization in Solution Developed by Using High-Throughput-Screening Technologies. Angew. Chem. Int. Ed. 2006, 45, 3278–3283. [Google Scholar] [CrossRef] [PubMed]
- Makio, H.; Kashiwa, N.; Fujita, T. FI Catalysts: A New Family of High Performance Catalysts for Olefin Polymerization. Adv. Synth. Catal. 2002, 344, 477–493. [Google Scholar] [CrossRef]
- Chen, E.Y.-X.; Marks, T.J. Cocatalysts for Metal-Catalyzed Olefin Polymerization: Activators, Activation Processes, and Structure-Activity Relationships. Chem. Rev. 2000, 100, 1391–1434. [Google Scholar] [CrossRef] [PubMed]
- Bochmann, M. The Chemistry of Catalyst Activation: The Case of Group 4 Polymerization Catalysts. Organometallics 2010, 29, 4711–4740. [Google Scholar] [CrossRef]
- Massey, A.G.; Park, A.J. Perfluorophenyl derivatives of the elements: I. Tris(pentafluorophenyl)boron. J. Organomet. Chem. 1964, 2, 245–250. [Google Scholar] [CrossRef]
- Massey, A.G.; Park, A.J. Perfluorophenyl derivatives of the elements: VII. further studies on tris(pentafluorophenyl)boron. J. Organomet. Chem. 1966, 5, 218–225. [Google Scholar] [CrossRef]
- Yang, X.; Stern, C.L.; Marks, T.J. Cation-like homogeneous olefin polymerization catalysts based upon zirconocene alkyls and tris(pentafluorophenyl)borane. J. Am. Chem. Soc. 1991, 113, 3623–3625. [Google Scholar] [CrossRef]
- Yang, X.; Stern, C.L.; Marks, T.J. Cationic Zirconocene Olefin Polymerization Catalysts Based on the Organo-Lewis Acid Tris(pentafluorophenyl)borane. A Synthetic, Structural, Solution Dynamic, and Polymerization Catalytic Study. J. Am. Chem. Soc. 1994, 116, 10015–10031. [Google Scholar] [CrossRef]
- Ewen, J.A.; Elder, M.J. Metallocene Catalysts with Lewis Acids and Aluminum Alkyls. Patent EP0427697, 8 May 1996. [Google Scholar]
- Ewen, J.A.; Elder, M.J. Metallocene Catalysts with Lewis Acids and Aluminum Alkyls. U.S. Patent 5561092, 1 October 1996. [Google Scholar]
- Chien, J.C.W.; Tsai, W.M.; Rausch, M.D. Isospecific polymerization of propylene catalyzed by rac-ethylenebis(indenyl)methylzirconium cation. J. Am. Chem. Soc. 1991, 113, 8570–8571. [Google Scholar] [CrossRef]
- Ewen, J.A.; Elder, M.J. Preparation of metallocene catalysts for polymerization of olefins. Patent EP0426637, 5 April 1995. [Google Scholar]
- Turner, H.W. Catalysts, method of preparing these catalysts and method of using said catalysts. Patent EP0277004, 3 August 1988. [Google Scholar]
- Hlatky, G.; Upton, D.; Turner, H. Supported Ionic Metallocene Catalysts for Olefin Polymerization. Patent WO9109882, 11 July 1991. [Google Scholar]
- Yang, X.; Stern, C.; Marks, T.J. Models for organometallic molecule-support complexes. Very large counterion modulation of cationic actinide alkyl reactivity. Organometallics 1991, 10, 840–842. [Google Scholar] [CrossRef]
- Macchioni, A. Ion Pairing in Transition-Metal Organometallic Chemistry. Chem. Rev. 2005, 105, 2039–2074. [Google Scholar] [CrossRef] [PubMed]
- Ciancaleoni, G.; Fraldi, N.; Budzelaar, P.H.M.; Busico, V.; Cipullo, R.; Macchioni, A. Structure—Activity Relationship in Olefin Polymerization Catalysis: Is Entropy the Key? J. Am. Chem. Soc. 2010, 132, 13651–13653. [Google Scholar] [CrossRef] [PubMed]
- Valente, A.; Mortreux, A.; Visseaux, M.; Zinck, P. Coordinative Chain Transfer Polymerization. Chem. Rev. 2013, 113, 3836–3857. [Google Scholar] [CrossRef] [PubMed]
- Sita, L.R. Ex Uno Plures (“Out of One, Many”): New Paradigms for Expanding the Range of Polyolefins through Reversible Group Transfers. Angew. Chem. Int. Ed. 2009, 48, 2464–2472. [Google Scholar] [CrossRef] [PubMed]
- Kempe, R. How to Polymerize Ethylene in a Highly Controlled Fashion? Chem. Eur. J. 2007, 13, 2764–2773. [Google Scholar] [CrossRef] [PubMed]
- D’Agosto, F.; Boisson, C. A RAFT Analogue Olefin Polymerization Technique Using Coordination Chemistry. Aust. J. Chem. 2010, 63, 1155–1158. [Google Scholar] [CrossRef]
- Drent, E. Process for the Preparation of Polyketones. Patent EP0121965, 17 October 1984. [Google Scholar]
- Drent, E.; Van Broekhoven, J.A.M.; Doyle, M.J. Efficient palladium catalysts for the copolymerization of carbon monoxide with olefins to produce perfectly alternating polyketones. J. Organomet. Chem. 1991, 417, 235–251. [Google Scholar] [CrossRef]
- Drent, E.; Mul, W.P.; Smaardijk, A.A. Polyketones. In Encyclopedia of Polymer Science and Technology; John Wiley & Sons, Inc.: New York, NY, USA, 2002. [Google Scholar]
- Goodall, B.L. Cycloaliphatic Polymers. In Encyclopedia of Materials: Science and Technology, 2nd ed.; Buschow, K.H.J., Cahn, R.W., Flemings, M.C., Ilschner, B., Kramer, E.J., Mahajan, S., Veyssière, P., Eds.; Elsevier: Oxford, UK, 2001; pp. 1959–1962. [Google Scholar]
- Makio, H.; Fujita, T. Development and Application of FI Catalysts for Olefin Polymerization: Unique Catalysis and Distinctive Polymer Formation. Acc. Chem. Res. 2009, 42, 1532–1544. [Google Scholar] [CrossRef] [PubMed]
- Mitani, M.; Saito, J.; Ishii, S.; Nakayama, Y.; Makio, H.; Matsukawa, N.; Matsui, S.; Mohri, J.; Furuyama, R.; Terao, H. FI Catalysts: New olefin polymerization catalysts for the creation of value-added polymers. Chem. Rec. 2004, 4, 137–158. [Google Scholar] [CrossRef] [PubMed]
- Thuilliez, J.; Ricard, L.; Nief, F.; Boisson, F.; Boisson, C. Ansa-Bis(fluorenyl)neodymium Catalysts for Cyclocopolymerization of Ethylene with Butadiene. Macromolecules 2009, 42, 3774–3779. [Google Scholar] [CrossRef]
- Llauro, M.F.; Monnet, C.; Barbotin, F.; Monteil, V.; Spitz, R.; Boisson, C. Investigation of Ethylene/Butadiene Copolymers Microstructure by 1H and 13C NMR. Macromolecules 2001, 34, 6304–6311. [Google Scholar] [CrossRef]
- Belaid, I.; Monteil, V.; Boisson, C. Handbook of Transition Metal Polymerization Catalysts; Hoff, R., Tthers, R., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 2017; Chapter 20. [Google Scholar]
- Ribeiro, R.; Ruivo, R.; Nsiri, H.; Norsic, S.; D’Agosto, F.; Perrin, L.; Boisson, C. Deciphering the Mechanism of Coordinative Chain Transfer Polymerization of Ethylene Using Neodymocene Catalysts and Dialkylmagnesium. ACS Catal. 2016, 6, 851–860. [Google Scholar] [CrossRef]
- Zaccaria, F.; Ehm, C.; Budzelaar, P.H.M.; Busico, V. Accurate Prediction of Copolymerization Statistics in Molecular Olefin Polymerization Catalysis: The Role of Entropic, Electronic, and Steric Effects in Catalyst Comonomer Affinity. ACS Catal. 2017, 7, 1512–1519. [Google Scholar] [CrossRef]
- Friederichs, N.; Ghalit, N.; Xu, W. Chptr8—Supported Multicomponent Single-Site α-Olefin Polymerization Catalysts. In Tailor-Made Polymers; Severn, J.R., Chadwick, J.C., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2008; pp. 211–237. [Google Scholar]
- Liu, H.-T.; Davey, C.R.; Shirodkar, P.P. Bimodal polyethylene products from UNIPOL™ single gas phase reactor using engineered catalysts. Macromol. Symp. 2003, 195, 309–316. [Google Scholar] [CrossRef]
- Ruff, M.; Paulik, C. Controlling Polyolefin Properties by In-Reactor Blending, 1–Polymerization Process, Precise Kinetics, and Molecular Properties of UHMW-PE Polymers. Macromol. React. Eng. 2012, 6, 302–317. [Google Scholar] [CrossRef]
- Ruff, M.; Paulik, C. Controlling Polyolefin Properties by In-Reactor Blending: 2. Particle Design. Macromol. React. Eng. 2013, 7, 71–83. [Google Scholar] [CrossRef]
- Ruff, M.; Lang, R.W.; Paulik, C. Controlling Polyolefin Properties by In-Reactor Blending: 3. Mechanical Properties. Macromol. React. Eng. 2013, 7, 328–343. [Google Scholar] [CrossRef]
- Mei, G.; Herben, P.; Cagnani, C.; Mazzucco, A. The Spherizone Process: A New PP Manufacturing Platform. Macromol. Symp. 2006, 245–246, 677–680. [Google Scholar] [CrossRef]
- Dorini, M.; Mei, G. Basell Spherizone Technology. In Sustainable Industrial Chemistry; Cavani, F., Centi, G., Perathoner, S., Trifiró, F., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 563–578. [Google Scholar]
- Ewen, J.A.; Welborn, J. Process and Catalyst for Producing Polyethylene Having a Broad Molecular Weight Distribution. U.S. Patent 4530914, 23 July 1985. [Google Scholar]
- Ewen, J.A.; Welborn, J. Process and Catalyst for Producing Reactor Blend Polyolefins. U.S. Patent 4937299, 26 June 1990. [Google Scholar]
- Lue, C.-T.; Crowther, D.J. Mixed Catalysts for Use in a Polymerization Process. U.S. Patent 6492472, 10 December 2002. [Google Scholar]
- Vaughan, G.A.; Szul, J.F.; Mckee, M.G.; Farley, J.M.; Lue, C.-T.; Kao, S.-C. Mixed Metallocene Catalyst Systems Containing a Poor Comonomer Incorporator and a Good Comonomer Incorporator. U.S. Patent 7141632, 28 November 2006. [Google Scholar]
- Liu, H.-T.; Mure, C.R. Polyethylene Compositions. U.S. Patent 8378029, 19 February 2013. [Google Scholar]
- Stürzel, M.; Hees, T.; Enders, M.; Thomann, Y.; Blattmann, H.; Mülhaupt, R. Nanostructured Polyethylene Reactor Blends with Tailored Trimodal Molar Mass Distributions as Melt-Processable All-Polymer Composites. Macromolecules 2016, 49, 8048–8060. [Google Scholar] [CrossRef]
- Denger, C.; Haase, U.; Fink, G. Simultaneous oligomerization and polymerization of ethylene. Makromol. Chem. Rapid Commun. 1991, 12, 697–701. [Google Scholar] [CrossRef]
- Ye, Z.; AlObaidi, F.; Zhu, S. A Tandem Catalytic System for the Synthesis of Ethylene–Hex-1-ene Copolymers from Ethylene Stock. Macromol. Rapid Commun. 2004, 25, 647–652. [Google Scholar] [CrossRef]
- Bianchini, C.; Frediani, M.; Giambastiani, G.; Kaminsky, W.; Meli, A.; Passaglia, E. Amorphous Polyethylene by Tandem Action of Cobalt and Titanium Single-Site Catalysts. Macromol. Rapid Commun. 2005, 26, 1218–1223. [Google Scholar] [CrossRef]
- Komon, Z.J.A.; Bu, X.; Bazan, G.C. Synthesis of Butene−Ethylene and Hexene−Butene−Ethylene Copolymers from Ethylene via Tandem Action of Well-Defined Homogeneous Catalysts. J. Am. Chem. Soc. 2000, 122, 1830–1831. [Google Scholar] [CrossRef]
- Komon, Z.J.A.; Diamond, G.M.; Leclerc, M.K.; Murphy, V.; Okazaki, M.; Bazan, G.C. Triple Tandem Catalyst Mixtures for the Synthesis of Polyethylenes with Varying Structures. J. Am. Chem. Soc. 2002, 124, 15280–15285. [Google Scholar] [CrossRef] [PubMed]
- Karbach, F.F.; Macko, T.; Duchateau, R. Preparation of Ethylene/1-Hexene Copolymers from Ethylene Using a Fully Silica-Supported Tandem Catalyst System. Macromolecules 2016, 49, 1229–1241. [Google Scholar] [CrossRef]
- Markel, E.J.; Weng, W.; Peacock, A.J.; Dekmezian, A.H. Metallocene-Based Branch−Block Thermoplastic Elastomers. Macromolecules 2000, 33, 8541–8548. [Google Scholar] [CrossRef]
- Liu, S.; Motta, A.; Delferro, M.; Marks, T.J. Synthesis, Characterization, and Heterobimetallic Cooperation in a Titanium–Chromium Catalyst for Highly Branched Polyethylenes. J. Am. Chem. Soc. 2013, 135, 8830–8833. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Motta, A.; Mouat, A.R.; Delferro, M.; Marks, T.J. Very Large Cooperative Effects in Heterobimetallic Titanium-Chromium Catalysts for Ethylene Polymerization/Copolymerization. J. Am. Chem. Soc. 2014, 136, 10460–10469. [Google Scholar] [CrossRef] [PubMed]
- Arriola, D.J.; Carnahan, E.M.; Hustad, P.D.; Kuhlman, R.L.; Wenzel, T.T. Catalytic Production of Olefin Block Copolymers via Chain Shuttling Polymerization. Science 2006, 312, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Hustad, P.D.; Kuhlman, R.L.; Arriola, D.J.; Carnahan, E.M.; Wenzel, T.T. Continuous Production of Ethylene-Based Diblock Copolymers Using Coordinative Chain Transfer Polymerization. Macromolecules 2007, 40, 7061–7064. [Google Scholar] [CrossRef]
- Hustad, P.D. Frontiers in Olefin Polymerization: Reinventing the World’s Most Common Synthetic Polymers. Science 2009, 325, 704–707. [Google Scholar] [CrossRef] [PubMed]
- Hustad, P.D.; Marchand, G.R.; Garcia-Meitin, E.I.; Roberts, P.L.; Weinhold, J.D. Photonic Polyethylene from Self-Assembled Mesophases of Polydisperse Olefin Block Copolymers. Macromolecules 2009, 42, 3788–3794. [Google Scholar] [CrossRef]
- Arora, A.; Fosfuri, A.; Gambardella, A. Markets for Technology: The Economics of Innovation and Corporate Strategy; MIT Press: Cambridge, MA, USA, 2004. [Google Scholar]
- Chaudhary, B.I.; Barry, R.P. Extruded Non-Crosslinked Foams Made from Ethylene-Styrene Interpolymers and Blends with Polyethylene. J. Cell. Plast. 1999, 35, 531–549. [Google Scholar]
- Timmers, F.J. Pseudo-Random Copolymers Formed by Use of Constrained Geometry Addition Polymerization Catalysts. U.S. Patent 5703187, 30 December 1997. [Google Scholar]
- Chum, P.S.; Kruper, W.J.; Guest, M.J. Materials Properties Derived from INSITE Metallocene Catalysts. Adv. Mater. 2000, 12, 1759–1767. [Google Scholar] [CrossRef]
- Thayer, A.M. Metallocene Catalysts Initiate New Era in Polymer Synthesis. Chem. Eng. News Arch. 1995, 73, 15–20. [Google Scholar] [CrossRef]
- Severn, J.R.; Chadwick, J.C.; Duchateau, R.; Friederichs, N. “Bound but Not Gagged”Immobilizing Single-Site α-Olefin Polymerization Catalysts. Chem. Rev. 2005, 105, 4073–4147. [Google Scholar] [CrossRef] [PubMed]
- Severn, J.R.; Chadwick, J.C. Immobilisation of homogeneous olefin polymerisation catalysts. Factors influencing activity and stability. Dalton Trans. 2013, 42, 8979–8987. [Google Scholar] [CrossRef] [PubMed]
- Busico, V. Catalytic Olefin Polymerization is a Mature Field. Isn’t it? Macromol. Chem. Phys. 2007, 208, 26–29. [Google Scholar] [CrossRef]
Scheme 1.
Major advances in polyolefins development.

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sauter, D.W.; Taoufik, M.; Boisson, C. Polyolefins, a Success Story. Polymers 2017, 9, 185. https://0-doi-org.brum.beds.ac.uk/10.3390/polym9060185
AMA Style
Sauter DW, Taoufik M, Boisson C. Polyolefins, a Success Story. Polymers. 2017; 9(6):185. https://0-doi-org.brum.beds.ac.uk/10.3390/polym9060185
Chicago/Turabian StyleSauter, Dominique W., Mostafa Taoufik, and Christophe Boisson. 2017. "Polyolefins, a Success Story" Polymers 9, no. 6: 185. https://0-doi-org.brum.beds.ac.uk/10.3390/polym9060185
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.