
Daisychain: Search and Interactive Visualisation of Homologs in Genome Assemblies

 , , , , and
, , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
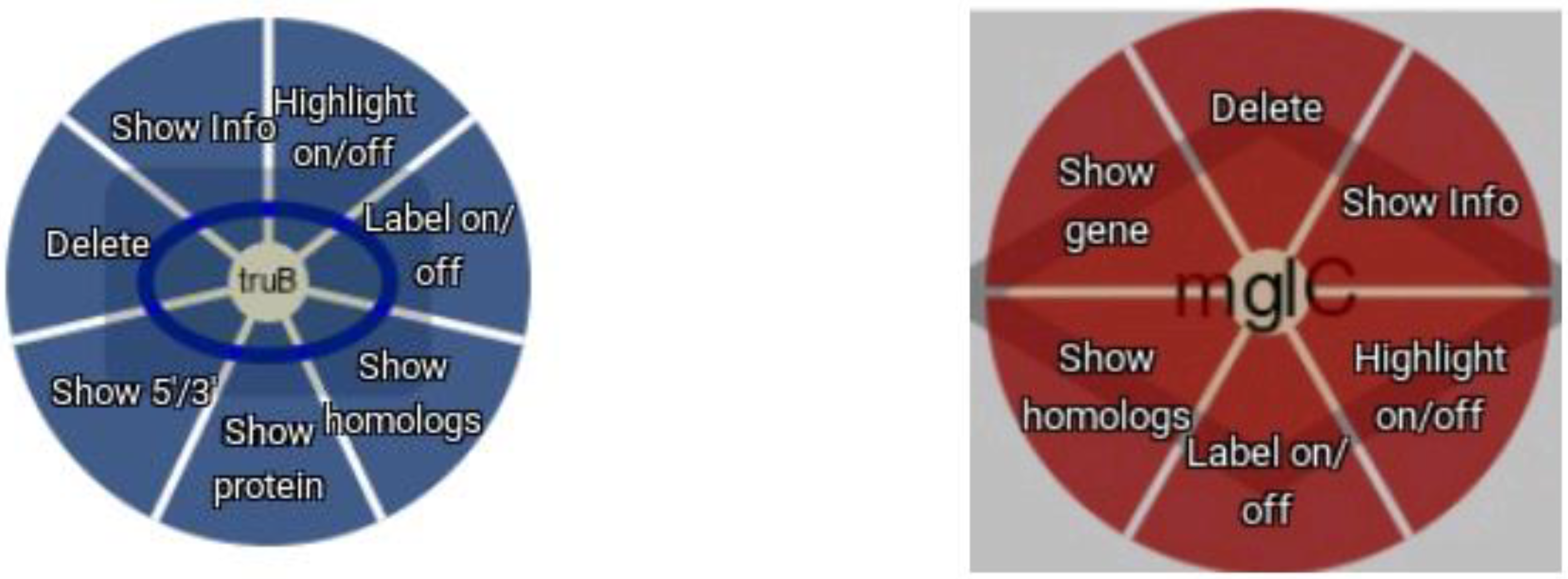{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- NCBI, R.C. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2016, 44, D7–D19. [Google Scholar] [CrossRef] [Green Version]
- Kriventseva, E.V.; Tegenfeldt, F.; Petty, T.J.; Waterhouse, R.M.; Simão, F.A.; Pozdnyakov, I.A.; Ioannidis, P.; Zdobnov, E.M. OrthoDB v8: Update of the hierarchical catalog of orthologs and the underlying free software. Nucleic Acids Res. 2015, 43, D250–D256. [Google Scholar] [CrossRef] [PubMed]
- Sonnhammer, E.L.L.; Östlund, G. InParanoid 8: Orthology analysis between 273 proteomes, mostly eukaryotic. Nucleic Acids Res. 2015, 43, D234–D239. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. SHOOT: Phylogenetic gene search and ortholog inference. bioRxiv 2021. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [Green Version]
- Fischer, S.; Brunk, B.P.; Chen, F.; Gao, X.; Harb, O.S.; Iodice, J.B.; Shanmugam, D.; Roos, D.S.; Stoeckert, C.J. Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr. Protoc. Bioinform. 2011, 35, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a Versatile Software Package for Scalable and Robust Microbial Pangenome Analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Moreira, B.; Cantalapiedra, C.P.; Garcia-Pereira, M.J.; Gordon, S.P.; Vogel, J.P.; Igartua, E.; Casas, A.M.; Vinuesa, P. Analysis of Plant Pan-Genomes and Transcriptomes with GET_HOMOLOGUES-EST, a Clustering Solution for Sequences of the Same Species. Front. Plant Sci. 2017, 8, 184. [Google Scholar] [CrossRef] [Green Version]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalhoub, B.; Denoeud, F.; Liu, S.; Parkin, I.A.; Tang, H.; Wang, X.; Chiquet, J.; Belcram, H.; Tong, C.; Samans, B.; et al. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 2014, 345, 950–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, F.; Fan, G.; Hu, Q.; Zhou, Y.; Guan, M.; Tong, C.; Li, J.; Du, D.; Qi, C.; Jiang, L.; et al. The high-quality genome of Brassica napus cultivar ‘ZS11’ reveals the introgression history in semi-winter morphotype. Plant J. 2017, 92, 452–468. [Google Scholar] [CrossRef] [Green Version]
- Bayer, P.E.; Hurgobin, B.; Golicz, A.A.; Chan, C.K.; Yuan, Y.; Lee, H.; Renton, M.; Meng, J.; Li, R.; Long, Y.; et al. Assembly and comparison of two closely related Brassica napus genomes. Plant Biotechnol. J. 2017, 15, 1602–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.-M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Chen, X.; Tong, C.; Zhang, X.; Song, A.; Hu, M.; Dong, W.; Chen, F.; Wang, Y.; Tu, J.; Liu, S.; et al. A high-quality Brassica napus genome reveals expansion of transposable elements, subgenome evolution and disease resistance. Plant Biotechnol. J. 2021, 19, 615–630. [Google Scholar] [CrossRef]
- Bayer, P.E.; Scheben, A.; Golicz, A.A.; Yuan, Y.; Faure, S.; Lee, H.; Chawla, H.S.; Anderson, R.; Bancroft, I.; Raman, H.; et al. Modelling of gene loss propensity in the pangenomes of three Brassica species suggests different mechanisms between polyploids and diploids. Plant Biotechnol. J. 2021, 19, 2488–2500. [Google Scholar] [CrossRef] [PubMed]
- Horesh, G.; Blackwell, G.A.; Tonkin-Hill, G.; Corander, J.; Heinz, E.; Thomson, N.R. A comprehensive and high-quality collection of Escherichia coli genomes and their genes. Microb. Genom. 2021, 7, 000499. [Google Scholar] [CrossRef]
- Golicz, A.A.; Bayer, P.E.; Bhalla, P.L.; Batley, J.; Edwards, D. Pangenomics comes of age: From bacteria to plant and animal applications. Trends Genet. 2020, 36, 132–145. [Google Scholar] [CrossRef]
- Bayer, P.E.; Golicz, A.A.; Scheben, A.; Batley, J.; Edwards, D. Plant pan-genomes are the new reference. Nat. Plants 2020, 6, 914–920. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: Implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golicz, A.A.; Bayer, P.E.; Barker, G.C.; Edger, P.P.; Kim, H.; Martinez, P.A.; Chan, C.K.K.; Severn-Ellis, A.; McCombie, W.R.; Parkin, I.A.P.; et al. The pangenome of an agronomically important crop plant Brassica oleracea. Nat. Commun. 2016, 7, 13390. [Google Scholar] [CrossRef] [PubMed]
- Hurgobin, B.; Golicz, A.A.; Bayer, P.E.; Chan, C.K.K.; Tirnaz, S.; Dolatabadian, A.; Schiessl, S.V.; Samans, B.; Montenegro, J.D.; Parkin, I.A. Homoeologous exchange is a major cause of gene presence/absence variation in the amphidiploid Brassica napus. Plant Biotechnol. J. 2018, 16, 1265–1274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Du, H.; Li, P.; Shen, Y.; Peng, H.; Liu, S.; Zhou, G.A.; Zhang, H.; Liu, Z.; Shi, M.; et al. Pan-Genome of Wild and Cultivated Soybeans. Cell 2020, 182, 162–176.e13. [Google Scholar] [CrossRef] [PubMed]
- Bayer, P.E.; Valliyodan, B.; Hu, H.; Marsh, J.I.; Yuan, Y.; Vuong, T.D.; Patil, G.; Song, Q.; Batley, J.; Varshney, R.K. Sequencing the USDA core soybean collection reveals gene loss during domestication and breeding. Plant Genome 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bayer, P.; Ruperao, P.; Saxena, R.; Khan, A.; Golicz, A.; Nguyen, H.; Batley, J.; Edwards, D.; Varshney, R. Trait associations in the pangenome of pigeon pea (Cajanus cajan). Plant Biotechnol. J. 2020, 18, 1946–1954. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.; Golicz, A.A.; Lu, K.; Dossa, K.; Zhang, Y.; Chen, J.; Wang, L.; You, J.; Fan, D.; Edwards, D. Insight into the evolution and functional characteristics of the pan-genome assembly from sesame landraces and modern cultivars. Plant Biotechnol. J. 2019, 17, 881–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Q.; Feng, Q.; Lu, H.; Li, Y.; Wang, A.; Tian, Q.; Zhan, Q.; Lu, Y.; Zhang, L.; Huang, T.; et al. Pan-genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 2018, 50, 278–284. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Chebotarov, D.; Kudrna, D.; Llaca, V.; Lee, S.; Rajasekar, S.; Mohammed, N.; Al-Bader, N.; Sobel-Sorenson, C.; Parakkal, P. A platinum standard pan-genome resource that represents the population structure of Asian rice. Sci. Data 2020, 7, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Rijzaani, H.; Bayer, P.E.; Rouard, M.; Doležel, J.; Batley, J.; Edwards, D. The pangenome of banana highlights differences between genera and genomes. Plant Genome 2021, e20100. [Google Scholar] [CrossRef]
- Montenegro, J.D.; Golicz, A.A.; Bayer, P.E.; Hurgobin, B.; Lee, H.; Chan, C.K.K.; Visendi, P.; Lai, K.; Doležel, J.; Batley, J. The pangenome of hexaploid bread wheat. Plant J. 2017, 90, 1007–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Rawlings, C.; Hassani-Pak, K. KnetMaps: A BioJS component to visualize biological knowledge networks [version 1; peer review: 3 approved, 1 approved with reservations]. F1000Research 2018, 7, 1651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franz, M.; Lopes, C.T.; Huck, G.; Dong, Y.; Sumer, O.; Bader, G.D. Cytoscape.js: A graph theory library for visualisation and analysis. Bioinformatics 2016, 32, 309–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Dongen, S.M. Graph Clustering by Flow Simulation. Ph.D. Thesis, Universiteit Utrecht, Utrecht, The Netherlands, 2000. [Google Scholar]
- Cantila, A.Y.; Saad, N.S.M.; Amas, J.C.; Edwards, D.; Batley, J. Recent Findings Unravel Genes and Genetic Factors Underlying Leptosphaeria maculans Resistance in Brassica napus and Its Relatives. Int. J. Mol. Sci. 2020, 22, 313. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schliebs, O.; Chan, C.-K.K.; Bayer, P.E.; Petereit, J.; Singh, A.; Hassani-Pak, K.; Batley, J.; Edwards, D. Daisychain: Search and Interactive Visualisation of Homologs in Genome Assemblies. Agronomy 2021, 11, 2587. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy11122587
Schliebs O, Chan C-KK, Bayer PE, Petereit J, Singh A, Hassani-Pak K, Batley J, Edwards D. Daisychain: Search and Interactive Visualisation of Homologs in Genome Assemblies. Agronomy. 2021; 11(12):2587. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy11122587
Chicago/Turabian StyleSchliebs, Oliver, Chon-Kit Kenneth Chan, Philipp E. Bayer, Jakob Petereit, Ajit Singh, Keywan Hassani-Pak, Jacqueline Batley, and David Edwards. 2021. "Daisychain: Search and Interactive Visualisation of Homologs in Genome Assemblies" Agronomy 11, no. 12: 2587. https://0-doi-org.brum.beds.ac.uk/10.3390/agronomy11122587