MMP-9 Signaling Pathways That Engage Rho GTPases in Brain Plasticity

 , ,
, ,  ,
, 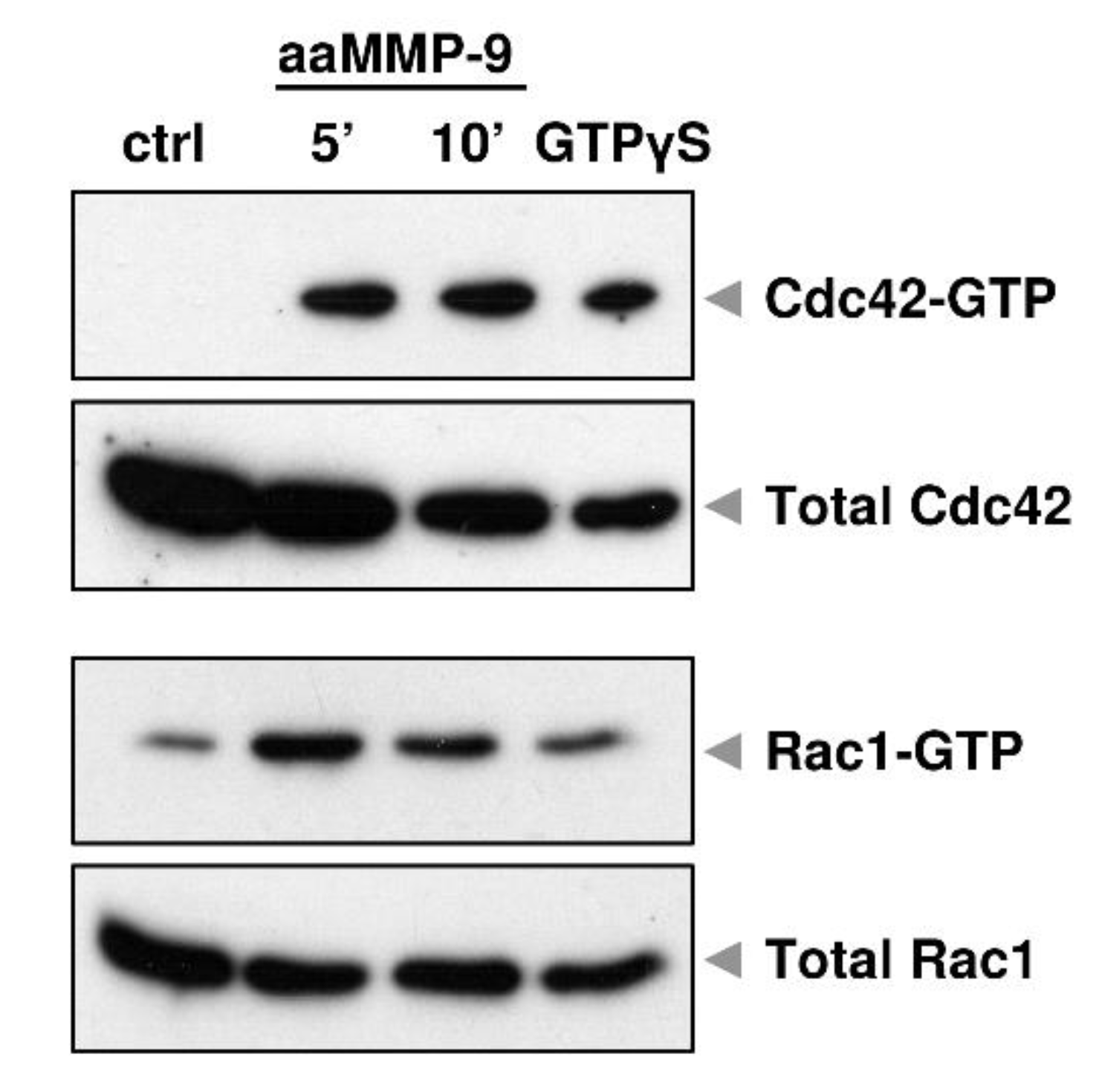{kind=link}
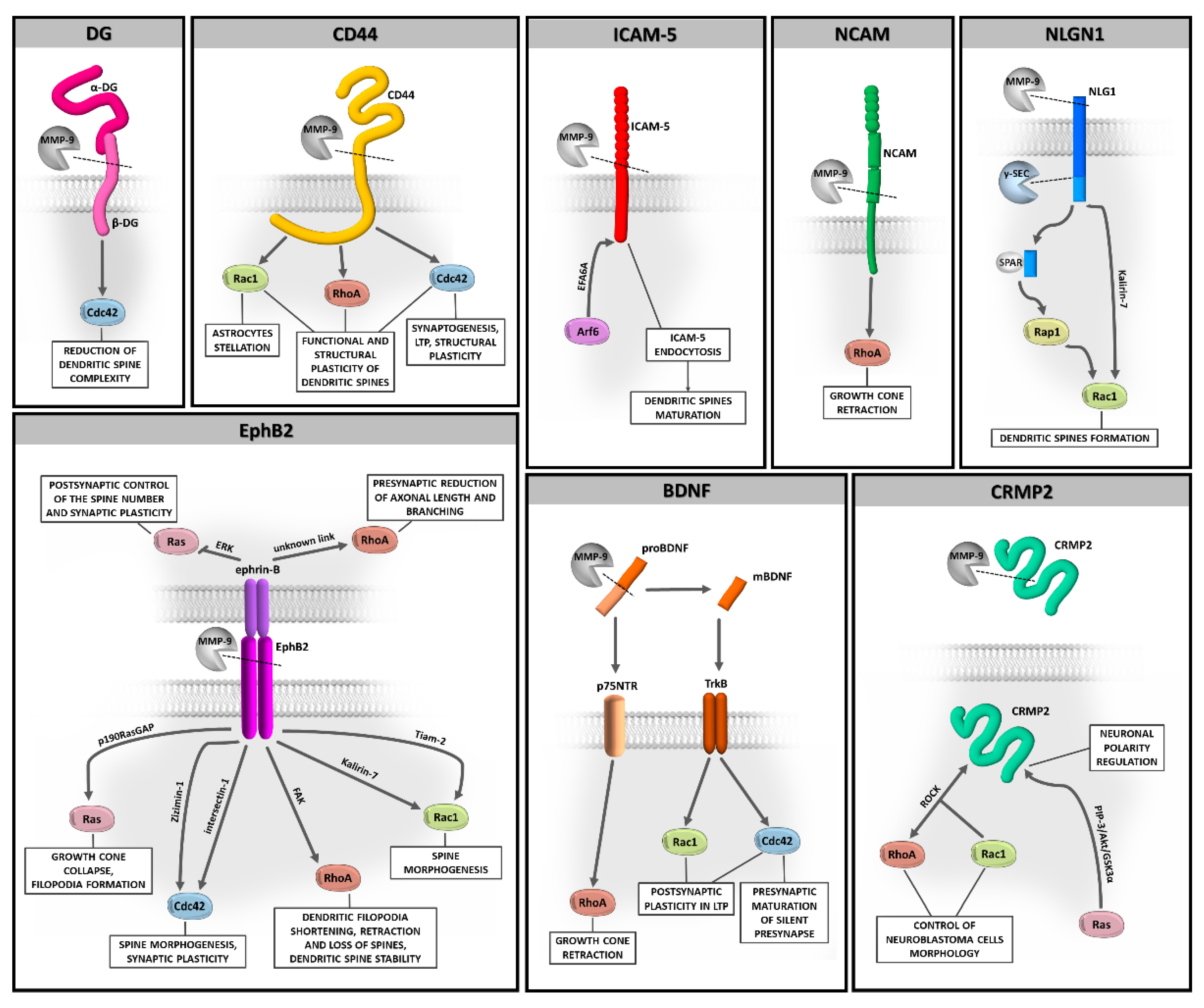{kind=link}
Abstract
:1. Introduction
2. Regulation of MMP-9 Substrates by Small Rho GTPases
2.1. DG
2.2. CD44
2.3. ICAM-5
2.4. NLGN1
2.5. BDNF
2.6. NCAM
2.7. EphB2
2.8. CRMP2
3. Post-Translational Modifications of Rho GTPases Involved in Neuronal Plasticity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPA | α-amino-3-hydroxy-5-methylisoxazole-propionic acid |
| BDNF | brain-derived neurotrophic factor |
| cLTP | chemically induced long-term potentiation |
| CNS | central nervous system |
| CRMP2 | collapsin response mediator protein 2 |
| DG | dystroglycan |
| ECM | extracellular matrix |
| EphB2 | ephrin type-B receptor 2 |
| ER | endoplasmic reticulum |
| ERK | extracellular-signal-regulated kinase |
| ERM | ezrin, radixin, moesin |
| FAK | focal adhesion kinase |
| GABAA | γ-aminobutyric acid type A |
| GAP | GTPase activating protein |
| GDI | guanine nucleotide dissociation inhibitor |
| GEF | guanine nucleotide exchange factor |
| ICAM-5 | intracellular adhesion molecule-5 |
| L-LTP | late-phase long-term potentiation |
| LTP | long-term potentiation |
| MAPK | mitogen-activated protein kinase |
| MMP | matrix metalloproteinase |
| NCAM | neural cell adhesion molecule |
| NLGN1 | neuroligin 1 |
| NMDA | N-methyl-D-aspartic acid |
| PAK | p21-activated kinase |
| PKA | protein kinase A |
| PM | plasma membrane |
| PTM | post-translational modifications |
| PTZ | pentylenetetrazole |
| ROCK | Rho-associated protein kinase |
| sLTP | structural long-term potentiation |
| TrkB | tyrosine kinase B |
References
- Huntley, G.W. Synaptic circuit remodelling by matrix metalloproteinases in health and disease. Nat. Rev. Neurosci. 2012, 13, 743–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsien, R.Y. Very long-term memories may be stored in the pattern of holes in the perineuronal net. Proc. Natl. Acad. Sci. USA 2013, 110, 12456–12461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dityatev, A. Remodeling of extracellular matrix and epileptogenesis. Epilepsia 2010, 51, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Ethell, I.M.; Ethell, D.W. Matrix metalloproteinases in brain development and remodeling: Synaptic functions and targets. J. Neurosci. Res. 2007, 85, 2813–2823. [Google Scholar] [CrossRef] [PubMed]
- Michaluk, P.; Kolodziej, L.R.; Mioduszewska, B.; Wilczynski, G.M.; Dzwonek, J.; Jaworski, J.; Gorecki, D.C.; Ottersen, O.P.; Kaczmarek, L. β-Dystroglycan as a Target for MMP-9, in Response to Enhanced Neuronal Activity. J. Biol. Chem. 2007, 282, 16036–16041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bajor, M.; Michaluk, P.; Gulyássy, P.; Kékesi, K.A.; Juhász, G.; Kaczmarek, L. Synaptic cell adhesion molecule-2 and collapsin response mediator protein-2 are novel members of the matrix metalloproteinase-9 degradome. J. Neurochem. 2012, 122, 775–788. [Google Scholar] [CrossRef] [Green Version]
- Meighan, S.E.; Meighan, P.C.; Choudhury, P.; Davis, C.J.; Olson, M.L.; Zornes, P.A.; Wright, J.W.; Harding, J.W. Effects of extracellular matrix-degrading proteases matrix metalloproteinases 3 and 9 on spatial learning and synaptic plasticity. J. Neurochem. 2006, 96, 1227–1241. [Google Scholar] [CrossRef] [PubMed]
- Nagy, V.; Bozdagi, O.; Matynia, A.; Balcerzyk, M.; Okulski, P.; Dzwonek, J.; Costa, R.M.; Silva, A.J.; Kaczmarek, L.; Huntley, G.W. Matrix Metalloproteinase-9 Is Required for Hippocampal Late-Phase Long-Term Potentiation and Memory. J. Neurosci. 2006, 26, 1923–1934. [Google Scholar] [CrossRef]
- Okulski, P.; Jay, T.M.; Jaworski, J.; Duniec, K.; Dzwonek, J.; Konopacki, F.A.; Wilczynski, G.M.; Sánchez-Capelo, A.; Mallet, J.; Kaczmarek, L. TIMP-1 Abolishes MMP-9-Dependent Long-lasting Long-term Potentiation in the Prefrontal Cortex. Biol. Psychiatry 2007, 62, 359–362. [Google Scholar] [CrossRef]
- Peixoto, R.T.; Kunz, P.A.; Kwon, H.; Mabb, A.M.; Sabatini, B.L.; Philpot, B.D.; Ehlers, M.D. Transsynaptic Signaling by Activity-Dependent Cleavage of Neuroligin-1. Neuron 2012, 76, 396–409. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.B.; Bozdagi, O.; Nikitczuk, J.S.; Zhai, Z.W.; Zhou, Q.; Huntley, G.W. Extracellular Proteolysis by Matrix Metallopro-teinase-9 Drives Dendritic Spine Enlargement and Long-Term Potentiation Coordinately. Proc. Natl. Acad. Sci. USA 2008, 105, 19520–19525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaluk, P.; Wawrzyniak, M.; Alot, P.; Szczot, M.; Wyrembek, P.; Mercik, K.; Medvedev, N.; Wilczek, E.; De Roo, M.; Zuschratter, W.; et al. Influence of matrix metalloproteinase MMP-9 on dendritic spine morphology. J. Cell Sci. 2011, 124, 3369–3380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szepesi, Z.; Bijata, M.; Ruszczycki, B.; Kaczmarek, L.; Włodarczyk, J. Matrix Metalloproteinases Regulate the Formation of Dendritic Spine Head Protrusions during Chemically Induced Long-Term Potentiation. PLoS ONE 2013, 8, e63314. [Google Scholar] [CrossRef] [PubMed]
- Szepesi, Z.; Hosy, E.; Ruszczycki, B.; Bijata, M.; Pyskaty, M.; Bikbaev, A.; Heine, M.; Choquet, D.; Kaczmarek, L.; Włodarczyk, J. Synaptically Released Matrix Metalloproteinase Activity in Control of Structural Plasticity and the Cell Surface Distribution of GluA1-AMPA Receptors. PLoS ONE 2014, 9, e98274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stawarski, M.; Stefaniuk, M.; Wlodarczyk, J. Matrix Metalloproteinase-9 Involvement in the Structural Plasticity of Dendritic Spines. Front. Neuroanat. 2014, 8, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and Their Regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Kjøller, L.; Hall, A. Signaling to Rho GTPases. Exp. Cell Res. 1999, 253, 166–179. [Google Scholar] [CrossRef]
- Heasman, S.J.; Ridley, A.J. Mammalian Rho GTPases: New insights into their functions from in vivo studies. Nat. Rev. Mol. Cell Biol. 2008, 9, 690–701. [Google Scholar] [CrossRef]
- Niftullayev, S.; Lamarche-Vane, N. Regulators of Rho GTPases in the Nervous System: Molecular Implication in Axon Guid-ance and Neurological Disorders. Int. J. Mol. Sci. 2019, 20, 1497. [Google Scholar] [CrossRef] [Green Version]
- Martino, A.; Ettorre, M.; Musilli, M.; Lorenzetto, E.; Buffelli, M.; Diana, G. Rho GTPase-dependent plasticity of dendritic spines in the adult brain. Front. Cell. Neurosci. 2013, 7, 62. [Google Scholar] [CrossRef] [Green Version]
- Shinde, S.R.; Maddika, S. Post Translational Modifications of Rab GTPases. Small GTPases 2018, 9, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bijata, M.; Włodarczyk, J.; Figiel, I. Dystroglycan controls dendritic morphogenesis of hippocampal neurons in vitro. Front. Cell. Neurosci. 2015, 9, 199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ervasti, J.M.; Campbell, K.P. Membrane organization of the dystrophin-glycoprotein complex. Cell 1991, 66, 1121–1131. [Google Scholar] [CrossRef]
- Ibraghimov-Beskrovnaya, O.; Ervasti, J.M.; Leveille, C.J.; Slaughter, C.A.; Sernett, S.W.; Campbell, K.P. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nat. Cell Biol. 1992, 355, 696–702. [Google Scholar] [CrossRef] [PubMed]
- Gee, S.H.; Montanaro, F.; Lindenbaum, M.H.; Carbonetto, S. Dystroglycan-α, a dystrophin-associated glycoprotein, is a functional agrin receptor. Cell 1994, 77, 675–686. [Google Scholar] [CrossRef]
- Talts, J.F.; Andac, Z.; Gohring, W.; Brancaccio, A.; Timpl, R. Binding of the G Domains of Laminin Alpha1 and Alpha2 Chains and Perlecan to Heparin, Sulfatides, Alpha-Dystroglycan and Several Extracellular Matrix Proteins. EMBO J. 1999, 18, 863–870. [Google Scholar] [CrossRef]
- Sugita, S.; Saito, F.; Tang, J.; Satz, J.; Campbell, K.; Südhof, T.C. A stoichiometric complex of neurexins and dystroglycan in brain. J. Cell Biol. 2001, 154, 435–446. [Google Scholar] [CrossRef] [Green Version]
- Barresi, R.; Campbell, K.P. Dystroglycan: From Biosynthesis to Pathogenesis of Human Disease. J. Cell Sci. 2006, 119, 199–207. [Google Scholar] [CrossRef] [Green Version]
- Williamson, R.A.; Henry, M.D.; Daniels, K.J.; Hrstka, R.F.; Lee, J.C.; Sunada, Y.; Ibraghimov-Beskrovnaya, O.; Campbell, K.P. Dystroglycan Is Essential for Early Embryonic Development: Disruption of Reichert’s Membrane in Dag1-Null Mice. Hum. Mol. Genet. 1997, 6, 831–841. [Google Scholar] [CrossRef] [Green Version]
- Satz, J.S.; Ostendorf, A.P.; Hou, S.; Turner, A.; Kusano, H.; Lee, J.C.; Turk, R.; Nguyen, H.; Ross-Barta, S.E.; Westra, S.; et al. Distinct Functions of Glial and Neuronal Dystroglycan in the Developing and Adult Mouse Brain. J. Neurosci. 2010, 30, 14560–14572. [Google Scholar] [CrossRef] [Green Version]
- Russo, K.; Di Stasio, E.; Macchia, G.; Rosa, G.; Brancaccio, A.; Petrucci, T.C. Characterization of the Beta-Dystroglycan-Growth Factor Receptor 2 (Grb2) Interaction. Biochem. Biophys. Res. Commun. 2000, 274, 93–98. [Google Scholar] [CrossRef]
- Spence, H.J.; Dhillon, A.S.; James, M.; Winder, S.J. Dystroglycan, a scaffold for the ERK–MAP kinase cascade. EMBO Rep. 2004, 5, 484–489. [Google Scholar] [CrossRef] [Green Version]
- Zaccaria, M.; Di Tommaso, F.; Brancaccio, A.; Paggi, P.; Petrucci, T. Dystroglycan distribution in adult mouse brain: A light and electron microscopy study. Neuroscience 2001, 104, 311–324. [Google Scholar] [CrossRef]
- Levi, S.; Grady, R.M.; Henry, M.D.; Campbell, K.P.; Sanes, J.R.; Craig, A.M. Dystroglycan Is Selectively Associated with In-hibitory GABAergic Synapses but Is Dispensable for Their Differentiation. J. Neurosci. 2002, 22, 4274–4285. [Google Scholar] [CrossRef] [Green Version]
- Briatore, F.; Patrizi, A.; Viltono, L.; Sassoè-Pognetto, M.; Wulff, P. Quantitative Organization of GABAergic Synapses in the Molecular Layer of the Mouse Cerebellar Cortex. PLoS ONE 2010, 5, e12119. [Google Scholar] [CrossRef] [Green Version]
- Pribiag, H.; Peng, H.; Shah, W.A.; Stellwagen, D.; Carbonetto, S. Dystroglycan mediates homeostatic synaptic plasticity at GABAergic synapses. Proc. Natl. Acad. Sci. USA 2014, 111, 6810–6815. [Google Scholar] [CrossRef] [Green Version]
- Ganguly, K.; Rejmak, E.; Mikosz, M.; Nikolaev, E.; Knapska, E.; Kaczmarek, L. Matrix Metalloproteinase (MMP) 9 Transcription in Mouse Brain Induced by Fear Learning. J. Biol. Chem. 2013, 288, 20978–20991. [Google Scholar] [CrossRef] [Green Version]
- Yeghiazaryan, M.; Rutkowska-Wlodarczyk, I.; Konopka, A.; Wilczyński, G.M.; Melikyan, A.; Korkotian, E.; Kaczmarek, L.; Figiel, I. DP-b99 Modulates Matrix Metalloproteinase Activity and Neuronal Plasticity. PLoS ONE 2014, 9, e99789. [Google Scholar] [CrossRef]
- Batchelor, C.L.; Higginson, J.R.; Chen, Y.-J.; Vanni, C.; Eva, A.; Winder, S.J. Recruitment of Dbl by Ezrin and Dystroglycan Drives Membrane Proximal Cdc42 Activation and Filopodia Formation. Cell Cycle 2007, 6, 353–363. [Google Scholar] [CrossRef] [Green Version]
- Kochlamazashvili, G.; Henneberger, C.; Bukalo, O.; Dvoretskova, E.; Senkov, O.; Lievens, P.M.-J.; Westenbroek, R.; Engel, A.K.; Catterall, W.A.; Rusakov, D.A.; et al. The Extracellular Matrix Molecule Hyaluronic Acid Regulates Hippocampal Synaptic Plasticity by Modulating Postsynaptic L-Type Ca2+ Channels. Neuron 2010, 67, 116–128. [Google Scholar] [CrossRef] [Green Version]
- Wlodarczyk, J.; Mukhina, I.; Kaczmarek, L.; Dityatev, A. Extracellular Matrix Molecules, Their Receptors, and Secreted Pro-teases in Synaptic Plasticity. Dev. Neurobiol. 2011, 71, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Bourguignon, L.Y.W.; Gilad, E.; Rothman, K.; Peyrollier, K. Hyaluronan-CD44 Interaction with IQGAP1 Promotes Cdc42 and ERK Signaling, Leading to Actin Binding, Elk-1/Estrogen Receptor Transcriptional Activation, and Ovarian Cancer Progression. J. Biol. Chem. 2005, 280, 11961–11972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dzwonek, J.; Wilczyński, G.M. CD44: Molecular interactions, signaling and functions in the nervous system. Front. Cell. Neurosci. 2015, 9, 175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chetty, C.; Vanamala, S.K.; Gondi, C.S.; Dinh, D.H.; Gujrati, M.; Rao, J.S. MMP-9 induces CD44 cleavage and CD44 mediated cell migration in glioblastoma xenograft cells. Cell. Signal. 2012, 24, 549–559. [Google Scholar] [CrossRef] [Green Version]
- Bijata, M.; Labus, J.; Guseva, D.; Stawarski, M.; Butzlaff, M.; Dzwonek, J.; Schneeberg, J.; Bohm, K.; Michaluk, P.; Rusakov, D.A.; et al. Synaptic Remodeling Depends on Signaling between Ser-otonin Receptors and the Extracellular Matrix. Cell Rep. 2017, 19, 1767–1782. [Google Scholar] [CrossRef] [Green Version]
- Roszkowska, M.; Skupien, A.; Wójtowicz, T.; Konopka, A.; Gorlewicz, A.; Kisiel, M.; Bekisz, M.; Ruszczycki, B.; Dolezyczek, H.; Rejmak, E.; et al. CD44: A novel synaptic cell adhesion molecule regulating structural and functional plasticity of dendritic spines. Mol. Biol. Cell 2016, 27, 4055–4066. [Google Scholar] [CrossRef]
- Haber, M.; Zhou, L.; Murai, K.K. Cooperative Astrocyte and Dendritic Spine Dynamics at Hippocampal Excitatory Synapses. J. Neurosci. 2006, 26, 8881–8891. [Google Scholar] [CrossRef]
- Theodosis, D.T.; Poulain, D.A.; Oliet, S.H. Activity-Dependent Structural and Functional Plasticity of Astrocyte-Neuron In-teractions. Physiol. Rev. 2008, 88, 983–1008. [Google Scholar] [CrossRef] [Green Version]
- Volterra, A.; Meldolesi, J. Astrocytes, from brain glue to communication elements: The revolution continues. Nat. Rev. Neurosci. 2005, 6, 626–640. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Halassa, M.M.; Haydon, P.G. Integrated Brain Circuits: Astrocytic Networks Modulate Neuronal Activity and Behavior. Annu. Rev. Physiol. 2010, 72, 335–355. [Google Scholar] [CrossRef] [Green Version]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Araque, A.; Carmignoto, G.; Haydon, P.G.; Oliet, S.H.R.; Robitaille, R.; Volterra, A. Gliotransmitters Travel in Time and Space. Neuron 2014, 81, 728–739. [Google Scholar] [CrossRef] [Green Version]
- Ullian, E.M.; Sapperstein, S.K.; Christopherson, K.S.; Barres, B.A. Control of Synapse Number by Glia. Science 2001, 291, 657–661. [Google Scholar] [CrossRef]
- Bernardinelli, Y.; Randall, J.; Janett, E.; Nikonenko, I.; Konig, S.; Jones, E.V.; Flores, C.E.; Murai, K.K.; Bochet, C.G.; Holtmaat, A.; et al. Activity-Dependent Structural Plasticity of Perisynaptic Astrocytic Domains Promotes Excitatory Synapse Stability. Curr. Biol. 2014, 24, 1679–1688. [Google Scholar] [CrossRef] [Green Version]
- Konopka, A.; Zeug, A.; Skupien, A.; Kaza, B.; Mueller, F.; Chwedorowicz, A.; Ponimaskin, E.; Wilczynski, G.M.; Dzwonek, J. Cleavage of Hyaluronan and CD44 Adhesion Molecule Regulate Astrocyte Morphology via Rac1 Signalling. PLoS ONE 2016, 11, e0155053. [Google Scholar] [CrossRef] [Green Version]
- Gahmberg, C.G.; Ning, L.; Paetau, S. ICAM-5: A neuronal dendritic adhesion molecule involved in immune and neuronal functions. Adv. Neurobiol. 2014, 8, 117–132. [Google Scholar] [CrossRef]
- Yoshihara, Y.; Oka, S.; Nemoto, Y.; Watanabe, Y.; Nagata, S.; Kagamiyama, H.; Mori, K. An ICAM-Related Neuronal Glycoprotein, Telencephalin, with Brain Segment-Specific Expression. Neuron 1994, 12, 541–553. [Google Scholar] [CrossRef]
- Matsuno, H.; Okabe, S.; Mishina, M.; Yanagida, T.; Mori, K.; Yoshihara, Y. Telencephalin Slows Spine Maturation. J. Neurosci. 2006, 26, 1776–1786. [Google Scholar] [CrossRef] [Green Version]
- Furutani, Y.; Matsuno, H.; Kawasaki, M.; Sasaki, T.; Mori, K.; Yoshihara, Y. Interaction between Telencephalin and ERM Family Proteins Mediates Dendritic Filopodia Formation. J. Neurosci. 2007, 27, 8866–8876. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Stefanidakis, M.; Ning, L.; Van Lint, P.; Nyman-Huttunen, H.; Libert, C.; Itohara, S.; Mishina, M.; Rauvala, H.; Gahmberg, C.G. Activation of NMDA receptors promotes dendritic spine development through MMP-mediated ICAM-5 cleavage. J. Cell Biol. 2007, 178, 687–700. [Google Scholar] [CrossRef] [Green Version]
- Conant, K.; Wang, Y.; Szklarczyk, A.; Dudak, A.; Mattson, M.P.; Lim, S.T. Matrix metalloproteinase-dependent shedding of intercellular adhesion molecule-5 occurs with long-term potentiation. Neuroscience 2010, 166, 508–521. [Google Scholar] [CrossRef] [Green Version]
- Kelly, E.A.; Russo, A.S.; Jackson, C.D.; Lamantia, C.E.; Majewska, A. Proteolytic regulation of synaptic plasticity in the mouse primary visual cortex: Analysis of matrix metalloproteinase 9 deficient mice. Front. Cell. Neurosci. 2015, 9, 368. [Google Scholar] [CrossRef] [Green Version]
- Reinhard, S.M.; Razak, K.; Ethell, I.M. A delicate balance: Role of MMP-9 in brain development and pathophysiology of neurodevelopmental disorders. Front. Cell. Neurosci. 2015, 9, 280. [Google Scholar] [CrossRef] [Green Version]
- Murase, S.; Lantz, C.L.; Quinlan, E.M. Light Reintroduction after Dark Exposure Reactivates Plasticity in Adults via Peri-synaptic Activation of MMP-9. eLife 2017, 6, e27345. [Google Scholar] [CrossRef]
- Murase, S.; Winkowski, D.; Liu, J.; Kanold, P.O.; Quinlan, E.M. Homeostatic Regulation of Perisynaptic Matrix Metallopro-teinase 9 (MMP9) Activity in the Amblyopic Visual Cortex. eLife 2019, 8, e52503. [Google Scholar] [CrossRef]
- Kelly, E.A.; Tremblay, M.-È.; Gahmberg, C.G.; Tian, L.; Majewska, A. Subcellular localization of intercellular adhesion molecule-5 (telencephalin) in the visual cortex is not developmentally regulated in the absence of matrix metalloproteinase-9. J. Comp. Neurol. 2013, 522, 676–688. [Google Scholar] [CrossRef] [Green Version]
- Kelly, E.A.; Tremblay, M.-È.; Gahmberg, C.G.; Tian, L.; Majewska, A. Interactions between intercellular adhesion molecule-5 positive elements and their surroundings in the rodent visual cortex. Commun. Integr. Biol. 2013, 6, e27315. [Google Scholar] [CrossRef]
- Raemaekers, T.; Peric, A.; Baatsen, P.; Sannerud, R.; Declerck, I.; Baert, V.; Michiels, C.; Annaert, W. ARF6-Mediated Endo-somal Transport of Telencephalin Affects Dendritic Filopodia-to-Spine Maturation. EMBO J. 2012, 31, 3252–3269. [Google Scholar] [CrossRef] [Green Version]
- Choi, S.; Ko, J.; Lee, J.-R.; Lee, H.W.; Kim, K.; Chung, H.S.; Kim, H.; Kim, E. ARF6 and EFA6A Regulate the Development and Maintenance of Dendritic Spines. J. Neurosci. 2006, 26, 4811–4819. [Google Scholar] [CrossRef] [Green Version]
- Dean, C.; Dresbach, T. Neuroligins and neurexins: Linking cell adhesion, synapse formation and cognitive function. Trends Neurosci. 2006, 29, 21–29. [Google Scholar] [CrossRef]
- Südhof, T.C. Neuroligins and neurexins link synaptic function to cognitive disease. Nat. Cell Biol. 2008, 455, 903–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.-Y.; Ichtchenko, K.; Südhof, T.C.; Brose, N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc. Natl. Acad. Sci. USA 1999, 96, 1100–1105. [Google Scholar] [CrossRef] [Green Version]
- Chubykin, A.A.; Atasoy, D.; Etherton, M.R.; Brose, N.; Kavalali, E.T.; Gibson, J.R.; Sudhof, T.C. Activity-Dependent Validation of Excitatory versus Inhibitory Synapses by Neuroligin-1 versus Neuroligin-2. Neuron 2007, 54, 919–931. [Google Scholar] [CrossRef] [Green Version]
- Ichtchenko, K.; Hata, Y.; Nguyen, T.; Ullrich, B.; Missler, M.; Moomaw, C.; Sudhof, T.C. Neuroligin 1: A Splice Site-Specific Ligand for Beta-Neurexins. Cell 1995, 81, 435–443. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Sudhof, T.C. Binding Properties of Neuroligin 1 and Neurexin 1beta Reveal Function as Heterophilic Cell Adhesion Molecules. J. Biol. Chem. 1997, 272, 26032–26039. [Google Scholar] [CrossRef] [Green Version]
- Irie, M.; Hata, Y.; Takeuchi, M.; Ichtchenko, K.; Toyoda, A.; Hirao, K.; Takai, Y.; Rosahl, T.W.; Sudhof, T.C. Binding of Neu-roligins to PSD-95. Science 1997, 277, 1511–1515. [Google Scholar] [CrossRef]
- Jeong, J.; Pandey, S.; Li, Y.; Badger, J.D.; Lu, W.; Roche, K.W. PSD-95 Binding Dynamically Regulates NLGN1 Trafficking and Function. Proc. Natl. Acad. Sci. USA 2019, 116, 12035–12044. [Google Scholar] [CrossRef] [Green Version]
- Varoqueaux, F.; Aramuni, G.; Rawson, R.L.; Mohrmann, R.; Missler, M.; Gottmann, K.; Zhang, W.; Südhof, T.C.; Brose, N. Neuroligins Determine Synapse Maturation and Function. Neuron 2006, 51, 741–754. [Google Scholar] [CrossRef] [Green Version]
- Krueger-Burg, D.; Tuffy, L.P.; Papadopoulos, T.; Brose, N. The role of neurexins and neuroligins in the formation, maturation, and function of vertebrate synapses. Curr. Opin. Neurobiol. 2012, 22, 412–422. [Google Scholar] [CrossRef]
- Jedlicka, P.; Muellerleile, J.; Schwarzacher, S.W. Synaptic Plasticity and Excitation-Inhibition Balance in the Dentate Gyrus: Insights fromIn VivoRecordings in Neuroligin-1, Neuroligin-2, and Collybistin Knockouts. Neural Plast. 2018, 2018, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeidan, A.; Ziv, N.E. Neuroligin-1 Loss Is Associated with Reduced Tenacity of Excitatory Synapses. PLoS ONE 2012, 7, e42314. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jung, S.-Y.; Lee, Y.K.; Park, S.; Choi, J.-S.; Lee, C.J.; Kim, H.-S.; Choi, Y.-B.; Scheiffele, P.; Bailey, C.H.; et al. Neuroligin-1 is required for normal expression of LTP and associative fear memory in the amygdala of adult animals. Proc. Natl. Acad. Sci. USA 2008, 105, 9087–9092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jedlicka, P.; Vnencak, M.; Krueger, D.D.; Jungenitz, T.; Brose, N.; Schwarzacher, S.W. Neuroligin-1 regulates excitatory synaptic transmission, LTP and EPSP-spike coupling in the dentate gyrus in vivo. Brain Struct. Funct. 2015, 220, 47–58. [Google Scholar] [CrossRef]
- Fang, M.; Wei, J.-L.; Tang, B.; Liu, J.; Chen, L.; Tang, Z.-H.; Luo, J.; Chen, G.-J.; Wang, X.-F. Neuroligin-1 Knockdown Suppresses Seizure Activity by Regulating Neuronal Hyperexcitability. Mol. Neurobiol. 2016, 53, 270–284. [Google Scholar] [CrossRef]
- Millson, A.; Lagrave, D.; Willis, M.J.; Rowe, L.R.; Lyon, E.; South, S.T. Chromosomal loss of 3q26.3-3q26.32, involving a partial neuroligin 1 deletion, identified by genomic microarray in a child with microcephaly, seizure disorder, and severe intellectual disability. Am. J. Med. Genet. 2011, 158, 159–165. [Google Scholar] [CrossRef]
- Nakanishi, M.; Nomura, J.; Ji, X.; Tamada, K.; Arai, T.; Takahashi, E.; Bucan, M.; Takumi, T. Functional Significance of Rare Neuroligin 1 Variants Found in Autism. PLoS Genet. 2017, 13, e1006940. [Google Scholar] [CrossRef] [Green Version]
- Trobiani, L.; Meringolo, M.; Diamanti, T.; Bourne, Y.; Marchot, P.; Martella, G.; Dini, L.; Pisani, A.; De Jaco, A.; Bonsi, P. The neuroligins and the synaptic pathway in Autism Spectrum Disorder. Neurosci. Biobehav. Rev. 2020, 119, 37–51. [Google Scholar] [CrossRef]
- Gjorlund, M.D.; Carlsen, E.M.M.; Konig, A.B.; Dmytrieva, O.; Petersen, A.V.; Jacobsen, J.; Berezin, V.; Perrier, J.F.; Owczarek, S. Soluble Ectodomain of Neuroligin 1 Decreases Synaptic Activity by Activating Metabotropic Glutamate Receptor 2. Front. Mol. Neurosci. 2017, 10, 116. [Google Scholar] [CrossRef]
- Liu, A.; Zhou, Z.; Dang, R.; Zhu, Y.; Qi, J.; He, G.; Leung, C.; Pak, D.; Jia, Z.; Xie, W. Neuroligin 1 regulates spines and synaptic plasticity via LIMK1/cofilin-mediated actin reorganization. J. Cell Biol. 2016, 212, 449–463. [Google Scholar] [CrossRef]
- Tu, Y.K.; Duman, J.G.; Tolias, K.F. The Adhesion-GPCR BAI1 Promotes Excitatory Synaptogenesis by Coordinating Bidirec-tional Trans-Synaptic Signaling. J. Neurosci. 2018, 38, 8388–8406. [Google Scholar] [CrossRef] [PubMed]
- Paskus, J.D.; Tian, C.; Fingleton, E.; Shen, C.; Chen, X.; Li, Y.; Myers, S.A.; Badger, J.D.; Bemben, M.A.; Herring, B.E.; et al. Synaptic Kalirin-7 and Trio Interactomes Reveal a GEF Protein-Dependent Neuroligin-1 Mechanism of Action. Cell Rep. 2019, 29, 2944–2952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabelli, R.J.; Hohn, A.; Shatz, C.J. Inhibition of Ocular Dominance Column Formation by Infusion of NT-4/5 or BDNF. Science 1995, 267, 1662–1666. [Google Scholar] [CrossRef] [PubMed]
- Lu, B. BDNF and Activity-Dependent Synaptic Modulation. Learn. Mem. 2003, 10, 86–98. [Google Scholar] [CrossRef] [Green Version]
- Poo, M.-M. Neurotrophins as synaptic modulators. Nat. Rev. Neurosci. 2001, 2, 24–32. [Google Scholar] [CrossRef]
- Kang, H.; Welcher, A.A.; Shelton, D.; Schuman, E.M. Neurotrophins and Time: Different Roles for TrkB Signaling in Hippo-campal Long-Term Potentiation. Neuron 1997, 19, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Lohof, A.M.; Ip, N.Y.; Poo, M.-M. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nat. Cell Biol. 1993, 363, 350–353. [Google Scholar] [CrossRef]
- Fang, H.; Chartier, J.; Sodja, C.; Desbois, A.; Ribecco-Lutkiewicz, M.; Walker, P.R.; Sikorska, M. Transcriptional Activation of the Human Brain-derived Neurotrophic Factor Gene Promoter III by Dopamine Signaling in NT2/N Neurons. J. Biol. Chem. 2003, 278, 26401–26409. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.-R.; Walther, D.; Drgon, T.; Polesskaya, O.; Lesnick, T.G.; Strain, K.J.; De Andrade, M.; Bower, J.H.; Maraganore, D.M.; Uhl, G.R. Human brain derived neurotrophic factor (BDNF) genes, splicing patterns, and assessments of associations with substance abuse and Parkinson’s Disease. Am. J. Med. Genet. Neuropsychiatr. Genet. 2005, 134 B, 93–103. [Google Scholar] [CrossRef]
- Pruunsild, P.; Kazantseva, A.; Aid, T.; Palm, K.; Timmusk, T. Dissecting the human BDNF locus: Bidirectional transcription, complex splicing, and multiple promoters. Genomics 2007, 90, 397–406. [Google Scholar] [CrossRef] [Green Version]
- Aid, T.; Kazantseva, A.; Piirsoo, M.; Palm, K.; Timmusk, T. Mouse and ratBDNF gene structure and expression revisited. J. Neurosci. Res. 2007, 85, 525–535. [Google Scholar] [CrossRef]
- Timmusk, T.; Palm, K.; Metsis, M.; Reintam, T.; Paalme, V.; Saarma, M.; Persson, H. Multiple promoters direct tissue-specific expression of the rat BDNF gene. Neuron 1993, 10, 475–489. [Google Scholar] [CrossRef]
- Yang, J.; Siao, C.-J.; Nagappan, G.; Marinic, T.; Jing, D.; McGrath, K.; Chen, Z.-Y.; Mark, W.; Tessarollo, L.; Lee, F.S.; et al. Neuronal release of proBDNF. Nat. Neurosci. 2009, 12, 113–115. [Google Scholar] [CrossRef]
- Ernfors, P.; Ibanez, C.F.; Ebendal, T.; Olson, L.; Persson, H. Molecular cloning and neurotrophic activities of a protein with structural similarities to nerve growth factor: Developmental and topographical expression in the brain. Proc. Natl. Acad. Sci. USA 1990, 87, 5454–5458. [Google Scholar] [CrossRef] [Green Version]
- Katoh-Semba, R.; Takeuchi, I.K.; Semba, R.; Kato, K. Distribution of Brain-Derived Neurotrophic Factor in Rats and Its Changes with Development in the Brain. J. Neurochem. 2002, 69, 34–42. [Google Scholar] [CrossRef]
- Edelmann, E.; Lessmann, V.; Brigadski, T. Pre- and postsynaptic twists in BDNF secretion and action in synaptic plasticity. Neuropharmacology 2014, 76, 610–627. [Google Scholar] [CrossRef]
- Andreska, T.; Aufmkolk, S.; Sauer, M.; Blum, R. High abundance of BDNF within glutamatergic presynapses of cultured hippocampal neurons. Front. Cell. Neurosci. 2014, 8, 107. [Google Scholar] [CrossRef] [Green Version]
- Dieni, S.; Matsumoto, T.; Dekkers, M.; Rauskolb, S.; Ionescu, M.S.; Deogracias, R.; Gundelfinger, E.D.; Kojima, M.; Nestel, S.; Frotscher, M.; et al. BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. J. Cell Biol. 2012, 196, 775–788. [Google Scholar] [CrossRef] [Green Version]
- Brigadski, T.; Hartmann, M.; Lessmann, V. Differential Vesicular Targeting and Time Course of Synaptic Secretion of the Mammalian Neurotrophins. J. Neurosci. 2005, 25, 7601–7614. [Google Scholar] [CrossRef] [Green Version]
- Harward, S.C.; Hedrick, N.G.; Hall, C.E.; Parra-Bueno, P.; Milner, T.A.; Pan, E.; Laviv, T.; Hempstead, B.L.; Yasuda, R.; McNamara, J.O. Autocrine BDNF-TrkB Signalling within a Single Dendritic Spine. Nature 2016, 538, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Coull, J.A.M.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; De Koninck, Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nat. Cell Biol. 2005, 438, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Su, C.; Cunningham, R.L.; Rybalchenko, N.; Singh, M. Progesterone Increases the Release of Brain-Derived Neurotrophic Factor from Glia via Progesterone Receptor Membrane Component 1 (Pgrmc1)-Dependent ERK5 Signaling. Endocrinology 2012, 153, 4389–4400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amooeian, V.G.; Rashidi, E. Dysfunction in Brain-Derived Neurotrophic Factor Signaling Pathway and Susceptibility to Schizophrenia, Parkinson’s and Alzheimer’s Diseases. Curr. Gene Ther. 2018, 18, 45–63. [Google Scholar] [CrossRef]
- Castrén, E.; Hen, R. Neuronal plasticity and antidepressant actions. Trends Neurosci. 2013, 36, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Qin, X.Y.; Feng, J.C.; Cao, C.; Wu, H.T.; Loh, Y.P.; Cheng, Y. Association of Peripheral Blood Levels of Brain-Derived Neu-rotrophic Factor with Autism Spectrum Disorder in Children: A Systematic Review and Meta-Analysis. JAMA Pediatr. 2016, 170, 1079–1086. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhang, L.; Zhu, T.; Huang, J.; Qu, Y.; Mu, D. Peripheral brain-derived neurotrophic factor in autism spectrum disorder: A systematic review and meta-analysis. Sci. Rep. 2016, 6, 31241. [Google Scholar] [CrossRef] [Green Version]
- Mowla, S.J.; Farhadi, H.F.; Pareek, S.; Atwal, J.K.; Morris, S.J.; Seidah, N.G.; Murphy, R.A. Biosynthesis and Post-translational Processing of the Precursor to Brain-derived Neurotrophic Factor. J. Biol. Chem. 2001, 276, 12660–12666. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-Y.; Liu, F.; Chen, Y.; Guo, W.-C.; Zhang, Z.-H. Proprotein Convertase 1/3-Mediated down-Regulation of Brain-Derived Neurotrophic Factor in Cortical Neurons Induced by Oxygen-Glucose Deprivation. Neural Regen. Res. 2020, 15, 1066. [Google Scholar]
- Wetsel, W.C.; Rodriguiz, R.M.; Guillemot, J.; Rousselet, E.; Essalmani, R.; Kim, I.H.; Bryant, J.C.; Marcinkiewicz, J.; Desjardins, R.; Day, R.; et al. Disruption of the expression of the proprotein convertase PC7 reduces BDNF production and affects learning and memory in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 17362–17367. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.; Kermani, P.; Teng, K.K.; Hempstead, B.L. Regulation of Cell Survival by Secreted Proneurotrophins. Science 2001, 294, 1945–1948. [Google Scholar] [CrossRef] [Green Version]
- Pang, P.T.; Teng, H.K.; Zaitsev, E.; Woo, N.T.; Sakata, K.; Zhen, S.; Teng, K.K.; Yung, W.H.; Hempstead, B.L.; Lu, B. Cleavage of ProBDNF by TPA/Plasmin Is Essential for Long-Term Hippocampal Plasticity. Science 2004, 306, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Nagappan, G.; Zaitsev, E.; Senatorov, V.V.; Yang, J.; Hempstead, B.L.; Lu, B. Control of extracellular cleavage of ProBDNF by high frequency neuronal activity. Proc. Natl. Acad. Sci. USA 2009, 106, 1267–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Harte-Hargrove, L.C.; Siao, C.-J.; Marinic, T.; Clarke, R.; Ma, Q.; Jing, D.; Lafrancois, J.J.; Bath, K.G.; Mark, W.; et al. proBDNF Negatively Regulates Neuronal Remodeling, Synaptic Transmission, and Synaptic Plasticity in Hippocampus. Cell Rep. 2014, 7, 796–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleitas, C.; Piñol-Ripoll, G.; Marfull, P.; Rocandio, D.; Ferrer, I.; Rampon, C.; Egea, J.; Espinet, C. ProBDNF Is Modified by Advanced Glycation End Products in Alzheimer’s Disease and Causes Neuronal Apoptosis by Inducing P75 Neurotrophin Receptor Processing 11 Medical and Health Sciences 1109 Neurosciences. Mol. Brain 2018, 11, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koshimizu, H.; Kiyosue, K.; Hara, T.; Hazama, S.; Suzuki, S.; Uegaki, K.; Nagappan, G.; Zaitsev, E.; Hirokawa, T.; Tatsu, Y.; et al. Multiple functions of precursor BDNF to CNS neurons: Negative regulation of neurite growth, spine formation and cell survival. Mol. Brain 2009, 2, 27. [Google Scholar] [CrossRef] [Green Version]
- Rösch, H.; Schweigreiter, R.; Bonhoeffer, T.; Barde, Y.-A.; Korte, M. The neurotrophin receptor p75NTR modulates long-term depression and regulates the expression of AMPA receptor subunits in the hippocampus. Proc. Natl. Acad. Sci. USA 2005, 102, 7362–7367. [Google Scholar] [CrossRef] [Green Version]
- Woo, N.H.; Teng, H.K.; Siao, C.-J.; Chiaruttini, C.; Pang, P.T.; Milner, T.A.; Hempstead, B.L.; Lu, B. Activation of p75NTR by proBDNF facilitates hippocampal long-term depression. Nat. Neurosci. 2005, 8, 1069–1077. [Google Scholar] [CrossRef]
- Korte, M.; Carroll, P.; Wolf, E.; Brem, G.; Thoenen, H.; Bonhoeffer, T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc. Natl. Acad. Sci. USA 1995, 92, 8856–8860. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, H.; Nakade, J.; Tachibana, M.; Ibi, D.; Someya, E.; Koike, H.; Kamei, H.; Nabeshima, T.; Itohara, S.; Takuma, K.; et al. Matrix Metalloproteinase-9 Contributes to Kindled Seizure Development in Pentylene-tetrazole-Treated Mice by Converting pro-BDNF to Mature BDNF in the Hippocampus. J. Neurosci. 2011, 31, 12963–12971. [Google Scholar] [CrossRef]
- Shen, W.; Wu, B.; Zhang, Z.; Dou, Y.; Rao, Z.-R.; Chen, Y.-R.; Duan, S. Activity-Induced Rapid Synaptic Maturation Mediated by Presynaptic Cdc42 Signaling. Neuron 2006, 50, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Park, K.J.; Grosso, C.A.; Aubert, I.; Kaplan, D.R.; Miller, F.D. p75NTR-dependent, myelin-mediated axonal degeneration regulates neural connectivity in the adult brain. Nat. Neurosci. 2010, 13, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Colicos, M.A.; Barker, P.A.; Kennedy, T.E. p75 Neurotrophin Receptor Expression Is Induced in Apoptotic Neurons After Seizure. J. Neurosci. 1999, 19, 6887–6896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokaia, Z.; Andsberg, G.; Martinez-Serrano, A.; Lindvall, O. Focal cerebral ischemia in rats induces expression of p75 neurotrophin receptor in resistant striatal cholinergic neurons. Neuroscience 1998, 84, 1113–1125. [Google Scholar] [CrossRef]
- Murakoshi, H.; Wang, H.; Yasuda, R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nat. Cell Biol. 2011, 472, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, N.G.; Harward, S.C.; Hall, C.E.; Murakoshi, H.; McNamara, J.O.; Yasuda, R. Rho GTPase Complementation Underlies BDNF-Dependent Homo- and Heterosynaptic Plasticity. Nature 2016, 538, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Finne, J.; Finne, U.; Deagostini-Bazin, H.; Goridis, C. Occurrence of α2–8 linked polysialosyl units in a neural cell adhesion molecule. Biochem. Biophys. Res. Commun. 1983, 112, 482–487. [Google Scholar] [CrossRef]
- Panicker, A.K.; Buhusi, M.; Thelen, K.; Maness, P.F. Cellular Signalling Mechanisms of Neural Cell Adhesion Molecules. Front. Biosci. 2003, 8, d900–d911. [Google Scholar]
- Becker, C.G.; Artola, A.; Gerardy-Schahn, R.; Becker, T.; Welzl, H.; Schachner, M. The Polysialic Acid Modification of the Neural Cell Adhesion Molecule Is Involved in Spatial Learning and Hippocampal Long-Term Potentiation. J. Neurosci. Res. 1996, 45, 143–152. [Google Scholar] [CrossRef]
- Senkov, O.; Sun, M.; Weinhold, B.; Gerardy-Schahn, R.; Schachner, M.; Dityatev, A. Polysialylated Neural Cell Adhesion Molecule Is Involved in Induction of Long-Term Potentiation and Memory Acquisition and Consolidation in a Fear-Conditioning Paradigm. J. Neurosci. 2006, 26, 10888–109898. [Google Scholar] [CrossRef]
- Stoenica, L.; Senkov, O.; Gerardy-Schahn, R.; Weinhold, B.; Schachner, M.; Dityatev, A. In Vivo Synaptic Plasticity in the Dentate Gyrus of Mice Deficient in the Neural Cell Adhesion Molecule NCAM or Its Polysialic Acid. Eur. J. Neurosci. 2006, 23, 2255–2264. [Google Scholar] [CrossRef]
- Suzuki, M.; Angata, K.; Nakayama, J.; Fukuda, M. Polysialic Acid and Mucin TypeO-Glycans on the Neural Cell Adhesion Molecule Differentially Regulate Myoblast Fusion. J. Biol. Chem. 2003, 278, 49459–49468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stork, O.; Welzl, H.; Wolfer, D.; Schuster, T.; Mantei, N.; Stork, S.; Hoyer, D.; Lipp, H.-P.; Obata, K.; Schachner, M. Recovery of emotional behaviour in neural cell adhesion molecule (NCAM) null mutant mice through transgenic expression of NCAM180. Eur. J. Neurosci. 2000, 12, 3291–3306. [Google Scholar] [CrossRef] [PubMed]
- Dityatev, A.; Dityateva, G.; Schachner, M. Synaptic Strength as a Function of Post- versus Presynaptic Expression of the Neural Cell Adhesion Molecule NCAM. Neuron 2000, 26, 207–217. [Google Scholar] [CrossRef] [Green Version]
- Persohn, E.; Schachner, M. Immunohistological Localization of the Neural Adhesion Molecules L1 and N-CAM in the Developing Hippocampus of the Mouse. J. Neurocytol. 1990, 19, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Lüthi, A.; Laurent, J.-P.; Figurovt, A.; Mullert, D.; Schachnert, M. Hippocampal long-term potentiation and neural cell adhesion molecules L1 and NCAM. Nat. Cell Biol. 1994, 372, 777–779. [Google Scholar] [CrossRef]
- Todaro, L.; Puricelli, L.; Gioseffi, H.; Pallotta, M.G.; Lastiri, J.; Joffé, E.B.D.K.; Varela, M.; De Lustig, E.S. Neural cell adhesion molecule in human serum. Increased levels in dementia of the Alzheimer type. Neurobiol. Dis. 2004, 15, 387–393. [Google Scholar] [CrossRef]
- Santuccione, A.; Sytnyk, V.; Leshchyns’Ka, I.; Schachner, M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 2005, 169, 341–354. [Google Scholar] [CrossRef] [Green Version]
- Vawter, M.P. Dysregulation of the neural cell adhesion molecule and neuropsychiatric disorders. Eur. J. Pharmacol. 2000, 405, 385–395. [Google Scholar] [CrossRef]
- Vawter, M.P.; Usen, N.; Thatcher, L.; Ladenheim, B.; Zhang, P.; Vanderputten, D.M.; Conant, K.; Herman, M.M.; Van Kammen, D.P.; Sedvall, G.; et al. Characterization of Human Cleaved N-CAM and Association with Schizophrenia. Exp. Neurol. 2001, 172, 29–46. [Google Scholar] [CrossRef] [Green Version]
- Fujita-Hamabe, W.; Tokuyama, S. The involvement of cleavage of neural cell adhesion molecule in neuronal death under oxidative stress conditions in cultured cortical neurons. Biol. Pharm. Bull. 2012, 35, 624–628. [Google Scholar] [CrossRef] [Green Version]
- Shichi, K.; Fujita-Hamabe, W.; Harada, S.; Mizoguchi, H.; Yamada, K.; Nabeshima, T.; Tokuyama, S. Involvement of Matrix Metalloproteinase-Mediated Proteolysis of Neural Cell Adhesion Molecule in the Development of Cerebral Ischemic Neuronal Damage. J. Pharmacol. Exp. Ther. 2011, 338, 701–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hübschmann, M.V.; Skladchikova, G.; Bock, E.; Berezin, V. Neural Cell Adhesion Molecule Function Is Regulated by Metal-loproteinase- Mediated Ectodomain Release. J. Neurosci. Res. 2005, 80, 826–837. [Google Scholar] [CrossRef] [PubMed]
- Fazeli, M.; Breen, K.C.; Errington, M.; Bliss, T. Increase in extracellular NCAM and amyloid precursor protein following induction of long-term potentiation in the dentate gyrus of anaesthetized rats. Neurosci. Lett. 1994, 169, 77–80. [Google Scholar] [CrossRef]
- Kim, J.S.; Kim, H.Y.; Kim, J.H.; Shin, H.K.; Lee, S.H.; Lee, Y.S.; Son, H. Enhancement of Rat Hippocampal Long-Term Poten-tiation by 17β-Estradiol Involves Mitogen-Activated Protein Kinase-Dependent and -Independent Components. Neurosci. Lett. 2002, 332, 65–69. [Google Scholar] [CrossRef]
- Büttner, B.; Kannicht, C.; Reutter, W.; Horstkorte, R. The neural cell adhesion molecule is associated with major components of the cytoskeleton. Biochem. Biophys. Res. Commun. 2003, 310, 967–971. [Google Scholar] [CrossRef]
- Sullivan, C.S.; Kümper, M.; Temple, B.S.; Maness, P.F. The Neural Cell Adhesion Molecule (NCAM) Promotes Clustering and Activation of EphA3 Receptors in GABAergic Interneurons to Induce Ras Homolog Gene Family, Member A (RhoA)/Rho-associated protein kinase (ROCK)-mediated Growth Cone Collapse. J. Biol. Chem. 2016, 291, 26262–26272. [Google Scholar] [CrossRef] [Green Version]
- Himanen, J.-P.; Nikolov, D.B. Eph receptors and ephrins. Int. J. Biochem. Cell Biol. 2003, 35, 130–134. [Google Scholar] [CrossRef]
- Takeuchi, S.; Katoh, H.; Negishi, M. Eph/ephrin reverse signalling induces axonal retraction through RhoA/ROCK pathway. J. Biochem. 2015, 158, 245–252. [Google Scholar] [CrossRef]
- Klein, R. Excitatory Eph receptors and adhesive ephrin ligands. Curr. Opin. Cell Biol. 2001, 13, 196–203. [Google Scholar] [CrossRef]
- Elowe, S.; Holland, S.J.; Kulkarni, S.; Pawson, T. Downregulation of the Ras–Mitogen-Activated Protein Kinase Pathway by the EphB2 Receptor Tyrosine Kinase Is Required for Ephrin-Induced Neurite Retraction. Mol. Cell. Biol. 2001, 21, 7429–7441. [Google Scholar] [CrossRef] [Green Version]
- Lin, K.-T.; Sloniowski, S.; Ethell, D.W.; Ethell, I.M. Ephrin-B2-induced Cleavage of EphB2 Receptor Is Mediated by Matrix Metalloproteinases to Trigger Cell Repulsion. J. Biol. Chem. 2008, 283, 28969–28979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moeller, M.L.; Shi, Y.; Reichardt, L.F.; Ethell, I.M. EphB Receptors Regulate Dendritic Spine Morphogenesis through the Re-cruitment/Phosphorylation of Focal Adhesion Kinase and RhoA Activation. J. Biol. Chem. 2006, 281, 1587–1598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Pontrello, C.G.; DeFea, K.A.; Reichardt, L.F.; Ethell, I.M. Focal Adhesion Kinase Acts Downstream of EphB Receptors to Maintain Mature Dendritic Spines by Regulating Cofilin Activity. J. Neurosci. 2009, 29, 8129–8142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauterbach, J.; Klein, R. Release of Full-Length EphB2 Receptors from Hippocampal Neurons to Cocultured Glial Cells. J. Neurosci. 2006, 26, 11575–11581. [Google Scholar] [CrossRef] [PubMed]
- Schaupp, A.; Sabet, O.; Dudanova, I.; Ponserre, M.; Bastiaens, P.; Klein, R. The composition of EphB2 clusters determines the strength in the cellular repulsion response. J. Cell Biol. 2014, 204, 409–422. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, M.; Palmer, A.; Köhler, J.; Klein, R. EphB-EphrinB Bi-Directional Endocytosis Terminates Adhesion Allowing Contact Mediated Repulsion. Nat. Cell Biol. 2003, 5, 869–878. [Google Scholar] [CrossRef]
- Gaitanos, T.N.; Koerner, J.; Klein, R. Tiam-Rac Signaling Mediates Trans-Endocytosis of Ephrin Receptor EphB2 and Is Im-portant for Cell Repulsion. J. Cell Biol. 2016, 214, 735. [Google Scholar] [CrossRef]
- Penzes, P.; Beeser, A.; Chernoff, J.; Schiller, M.R.; Eipper, B.A.; Mains, R.E.; Huganir, R.L. Rapid Induction of Dendritic Spine Morphogenesis by Trans-Synaptic EphrinB-EphB Receptor Activation of the Rho-GEF Kalirin. Neuron 2003, 37, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Locke, C.; Machida, K.; Tucker, C.L.; Wu, Y.; Yu, J. Optogenetic activation of EphB2 receptor in dendrites induced actin polymerization by activating Arg kinase. Biol. Open 2017, 6, 1820–1830. [Google Scholar] [CrossRef] [Green Version]
- Hussain, N.K.; Thomas, G.M.; Luo, J.; Huganir, R.L. Regulation of AMPA receptor subunit GluA1 surface expression by PAK3 phosphorylation. Proc. Natl. Acad. Sci. USA 2015, 112, E5883–E5890. [Google Scholar] [CrossRef] [Green Version]
- Irie, F.; Yamaguchi, Y. EphB receptors regulate dendritic spine development via intersectin, Cdc42 and N-WASP. Nat. Neurosci. 2002, 5, 1117–1118. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R.; Gnesutta, N.; Minichiello, L.; White, G.; Roylance, A.J.; Herron, C.E.; Ramsey, M.; Wolfer, D.P.; Cestari, V.; Rossi-Arnaud, C.; et al. A role for the Ras signalling pathway in synaptic transmission and long-term memory. Nat. Cell Biol. 1997, 390, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Heumann, R.; Goemans, C.; Bartsch, D.; Lingenhöhl, K.; Waldmeier, P.C.; Hengerer, B.; Allegrini, P.R.; Schellander, K.; Wagner, E.F.; Arendt, T.; et al. Transgenic Activation of Ras in Neurons Promotes Hypertrophy and Protects from Lesion-Induced Degeneration. J. Cell Biol. 2000, 151, 1537–1548. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Elowe, S.; Nash, P.; Pawson, T. Manipulation of EphB2 Regulatory Motifs and SH2 Binding Sites Switches MAPK Signaling and Biological Activity. J. Biol. Chem. 2003, 278, 6111–6119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClelland, A.C.; Hruska, M.; Coenen, A.J.; Henkemeyer, M.; Dalva, M.B. Trans-synaptic EphB2-ephrin-B3 interaction regulates excitatory synapse density by inhibition of postsynaptic MAPK signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 8830–8835. [Google Scholar] [CrossRef] [Green Version]
- Dail, M.; Richter, M.; Godement, P.; Pasquale, E.B. Eph receptors inactivate R-Ras through different mechanisms to achieve cell repulsion. J. Cell Sci. 2006, 119, 1244–1254. [Google Scholar] [CrossRef] [Green Version]
- Stenmark, P.; Ogg, D.; Flodin, S.; Flores, A.; Kotenyova, T.; Nyman, T.; Nordlund, P.; Kursula, P. The structure of human collapsin response mediator protein 2, a regulator of axonal growth. J. Neurochem. 2006, 101, 906–917. [Google Scholar] [CrossRef]
- Ricard, D.; Stankoff, B.; Bagnard, D.; Aguera, M.; Rogemond, V.; Antoine, J.; Spassky, N.; Zalc, B.; Lubetzki, C.; Belin, M.; et al. Differential Expression of Collapsin Response Mediator Proteins (CRMP/ULIP) in Subsets of Oligodendrocytes in the Postnatal Rodent Brain. Mol. Cell. Neurosci. 2000, 16, 324–337. [Google Scholar] [CrossRef]
- Wang, L.-H.; Strittmatter, S.M. A Family of Rat CRMP Genes Is Differentially Expressed in the Nervous System. J. Neurosci. 1996, 16, 6197–6207. [Google Scholar] [CrossRef]
- Arimura, N.; Ménager, C.; Fukata, Y.; Kaibuchi, K. Role of CRMP-2 in neuronal polarity. J. Neurobiol. 2003, 58, 34–47. [Google Scholar] [CrossRef]
- Inagaki, N.; Chihara, K.; Arimura, N.; Ménager, C.; Kawano, Y.; Matsuo, N.; Nishimura, T.; Amano, M.; Kaibuchi, K. CRMP-2 induces axons in cultured hippocampal neurons. Nat. Neurosci. 2001, 4, 781–782. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.P.; Strittmatter, S.M. Semaphorin-mediated axonal guidance via Rho-related G proteins. Curr. Opin. Cell Biol. 2001, 13, 619–626. [Google Scholar] [CrossRef]
- Niwa, S.; Nakamura, F.; Tomabechi, Y.; Aoki, M.; Shigematsu, H.; Matsumoto, T.; Yamagata, A.; Fukai, S.; Hirokawa, N.; Goshima, Y.; et al. Structural basis for CRMP2-induced axonal microtubule formation. Sci. Rep. 2017, 7, 10681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arimura, N.; Inagaki, N.; Chihara, K.; Ménager, C.; Nakamura, N.; Amano, M.; Iwamatsu, A.; Goshima, Y.; Kaibuchi, K. Phosphorylation of Collapsin Response Mediator Protein-2 by Rho-Kinase: Evidence for Two Separate Signaling Pathways for Growth Cone Collapse. J. Biol. Chem. 2000, 275, 23973–23980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.; Brown, M.; Jacobs, T.; Ferrari, G.; Cann, N.; Teo, M.; Monfries, C.; Lim, L. Collapsin Response Mediator Protein Switches RhoA and Rac1 Morphology in N1E-115 Neuroblastoma Cells and Is Regulated by Rho Kinase. J. Biol. Chem. 2001, 276, 43482–43486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshimura, T.; Arimura, N.; Kawano, Y.; Kawabata, S.; Wang, S.; Kaibuchi, K. Ras regulates neuronal polarity via the PI3-kinase/Akt/GSK-3β/CRMP-2 pathway. Biochem. Biophys. Res. Commun. 2006, 340, 62–68. [Google Scholar] [CrossRef]
- Mimura, F.; Yamagishi, S.; Arimura, N.; Fujitani, M.; Kubo, T.; Kaibuchi, K.; Yamashita, T. Myelin-associated Glycoprotein Inhibits Microtubule Assembly by a Rho-kinase-dependent Mechanism. J. Biol. Chem. 2006, 281, 15970–15979. [Google Scholar] [CrossRef] [Green Version]
- Nan, L.; Yang, L.; Zheng, Y.; He, Y.; Xie, Q.; Chen, Z.; Li, H.; Huang, M. Effects of Gualou Guizhi Decoction Aqueous Extract on Axonal Regeneration in Organotypic Cortical Slice Culture after Oxygen-Glucose Deprivation. Evid. Based Complement. Altern. Med. 2017, 2017, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Petratos, S.; Li, Q.-X.; George, A.J.; Hou, X.; Kerr, M.L.; Unabia, S.E.; Hatzinisiriou, I.; Maksel, D.; Aguilar, M.-I.; Small, D.H. The β-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain 2007, 131, 90–108. [Google Scholar] [CrossRef] [Green Version]
- Pilotte, J.; Kiosses, W.; Chan, S.W.; Makarenkova, H.P.; Dupont-Versteegden, E.; Vanderklish, P.W. Morphoregulatory functions of the RNA-binding motif protein 3 in cell spreading, polarity and migration. Sci. Rep. 2018, 8, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Quarta, S.; Camprubí-Robles, M.; Schweigreiter, R.; Matusica, D.; Haberberger, R.V.; Proia, R.L.; Bandtlow, C.E.; Ferrer-Montiel, A.; Kress, M. Sphingosine-1-Phosphate and the S1P3 Receptor Initiate Neuronal Retraction via RhoA/ROCK Associated with CRMP2 Phosphorylation. Front. Mol. Neurosci. 2017, 10, 317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozés Salvador, V.; Heredia, F.; Berardo, A.; Palandri, A.; Wojnacki, J.; Vivinetto, A.L.; Sheikh, K.A.; Caceres, A.; Lopez, P.H.H. Anti-Glycan Antibodies Halt Axon Regeneration in a Model of Guillain Barrè Syndrome Axonal Neuropathy by Inducing Microtubule Disorganization via RhoA-ROCK-Dependent Inactivation of CRMP-2. Exp. Neurol. 2016, 278, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.-H.; Sun, Z.-L.; Li, H.-J.; Feng, D.-F. Rho GTPase-activating proteins: Regulators of Rho GTPase activity in neuronal development and CNS diseases. Mol. Cell. Neurosci. 2017, 80, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Ahearn, I.M.; Haigis, K.M.; Bar-Sagi, D.; Philips, M.R. Regulating the regulator: Post-translational modification of RAS. Nat. Rev. Mol. Cell Biol. 2011, 13, 39–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, P.J.; Mitin, N.; Keller, P.J.; Chenette, E.J.; Madigan, J.P.; Currin, R.O.; Cox, A.D.; Wilson, O.; Kirschmeier, P.; Der, C.J. Rho Family GTPase Modification and Dependence on CAAX Motif-Signaled Posttranslational Modification. J. Biol. Chem. 2008, 283, 25150–25163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitin, N.; Roberts, P.J.; Chenette, E.J.; Der, C.J. Posttranslational Lipid Modification of Rho Family Small GTPases. Adv. Struct. Saf. Stud. 2011, 827, 87–95. [Google Scholar] [CrossRef]
- Lane, K.T.; Beese, L.S. Thematic Review Series: Lipid Posttranslational Modifications. Structural Biology of Protein Farnesyl-transferase and Geranylgeranyltransferase Type I. J. Lipid Res. 2006, 47, 681–699. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Casey, P.J. Protein prenylation: Unique fats make their mark on biology. Nat. Rev. Mol. Cell Biol. 2016, 17, 110–122. [Google Scholar] [CrossRef]
- Abdrabou, A.; Wang, Z. Post-Translational Modification and Subcellular Distribution of Rac1: An Update. Cells 2018, 7, 263. [Google Scholar] [CrossRef] [Green Version]
- Samuel, F.; Hynds, D.L. RHO GTPase Signaling for Axon Extension: Is Prenylation Important? Mol. Neurobiol. 2010, 42, 133–142. [Google Scholar] [CrossRef]
- Spillane, M.; Gallo, G. Involvement of Rho-family GTPases in axon branching. Small GTPases 2014, 5, e27974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, J.M.; Samuel, F.G.; McConnell, J.A.; Reddy, C.P.; Beck, B.W.; Hynds, D.L. Non-prenylatable, cytosolic Rac1 alters neurite outgrowth while retaining the ability to be activated. Cell. Signal. 2015, 27, 630–637. [Google Scholar] [CrossRef] [PubMed]
- Berzat, A.C.; Brady, N.C.; Fiordalisi, J.J.; Cox, A.D. Using Inhibitors of Prenylation to Block Localization and Transforming Activity. Methods Enzymol. 2006, 407, 575–597. [Google Scholar] [CrossRef] [PubMed]
- Tricarico, P.M.; Romeo, A.; Gratton, R.; Crovella, S.; Celsi, F. Lack of Prenylated Proteins, Autophagy Impairment and Apoptosis in SH-SY5Y Neuronal Cell Model of Mevalonate Kinase Deficiency. Cell. Physiol. Biochem. 2017, 41, 1649–1660. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, H.; Toma-Fukai, S.; Kontani, K.; Katada, T.; Shimizu, T. GEF mechanism revealed by the structure of SmgGDS-558 and farnesylated RhoA complex and its implication for a chaperone mechanism. Proc. Natl. Acad. Sci. USA 2018, 115, 9563–9568. [Google Scholar] [CrossRef] [Green Version]
- Akula, M.K.; Ibrahim, M.X.; Ivarsson, E.G.; Khan, O.M.; Kumar, I.T.; Erlandsson, M.; Karlsson, C.; Xu, X.; Brisslert, M.; Brakebusch, C.; et al. Protein prenylation restrains innate immunity by inhibiting Rac1 effector interactions. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Aslam, M.; Troidl, C.; Tanislav, C.; Rohrbach, S.; Gündüz, D.; Hamm, C.W. Inhibition of Protein Prenylation of GTPases Alters Endothelial Barrier Function. Int. J. Mol. Sci. 2019, 21, 2. [Google Scholar] [CrossRef] [Green Version]
- Kalpachidou, T.; Spiecker, L.; Kress, M.; Quarta, S. Rho GTPases in the Physiology and Pathophysiology of Peripheral Sensory Neurons. Cells 2019, 8, 591. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Chen, Y. Spatiotemporal Regulation of Rho GTPases in Neuronal Migration. Cells 2019, 8, 568. [Google Scholar] [CrossRef] [Green Version]
- Zmurchok, C.; Holmes, W.R. Simple Rho GTPase Dynamics Generate a Complex Regulatory Landscape Associated with Cell Shape. Biophys. J. 2020, 118, 1438–1454. [Google Scholar] [CrossRef]
- Zaręba-Kozioł, M.; Figiel, I.; Bartkowiak-Kaczmarek, A.; Włodarczyk, J. Insights Into Protein. Front. Mol. Neurosci. 2018, 11, 175. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Lérida, I.; Sánchez-Perales, S.; Calvo, M.; Rentero, C.; Zheng, Y.; Enrich, C.; Del Pozo, M.A. A palmitoylation switch mechanism regulates Rac1 function and membrane organization. EMBO J. 2011, 31, 534–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Wan, J.; Arstikaitis, P.; Takahashi, H.; Huang, K.; Bailey, A.O.; Thompson, J.X.; Roth, A.F.; Drisdel, R.C.; Mastro, R.; et al. Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nat. Cell Biol. 2008, 456, 904–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, A.; Chen-Wacker, C.; Wu, Y.-W.; Gorinski, N.; Filippov, M.A.; Pandey, G.; Ponimaskin, E. Dual lipidation of the brain-specific Cdc42 isoform regulates its functional properties. Biochem. J. 2013, 456, 311–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albanesi, J.P.; Barylko, B.; DeMartino, G.N.; Jameson, D.M. Palmitoylated Proteins in Dendritic Spine Remodeling. Front. Synaptic Neurosci. 2020, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Moutin, E.; Nikonenko, I.; Stefanelli, T.; Wirth, A.; Ponimaskin, E.; De Roo, M.; Muller, D. Palmitoylation of cdc42 Promotes Spine Stabilization and Rescues Spine Density Deficit in a Mouse Model of 22q11.2 Deletion Syndrome. Cereb. Cortex 2016, 27, 3618–3629. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.S.; Drake, K.R.; Rogers, C.; Wright, L.; Lippincott-Schwartz, J.; Philips, M.R.; Kenworthy, A.K. Depalmitoylated Ras traffics to and from the Golgi complex via a nonvesicular pathway. J. Cell Biol. 2005, 170, 261–272. [Google Scholar] [CrossRef] [Green Version]
- Rocks, O.; Peyker, A.; Kahms, M.; Verveer, P.J.; Koerner, C.; Lumbierres, M.; Kuhlmann, J.; Waldmann, H.; Wittinghofer, A.; Bastiaens, P.I.H. An Acylation Cycle Regulates Localization and Activity of Palmitoylated Ras Isoforms. Science 2005, 307, 1746–1752. [Google Scholar] [CrossRef]
- Roy, S.; Plowman, S.; Rotblat, B.; Prior, I.A.; Muncke, C.; Grainger, S.; Parton, R.G.; Henis, Y.I.; Kloog, Y.; Hancock, J.F. Indi-vidual Palmitoyl Residues Serve Distinct Roles in H-Ras Trafficking, Microlocalization, and Signaling. Mol. Cell Biol. 2005, 25, 6722–6733. [Google Scholar] [CrossRef] [Green Version]
- Laude, A.J.; Prior, I.A. Palmitoylation and localisation of RAS isoforms are modulated by the hypervariable linker domain. J. Cell Sci. 2008, 121, 421–427. [Google Scholar] [CrossRef] [Green Version]
- Rocks, O.; Gerauer, M.; Vartak, N.; Koch, S.; Huang, Z.-P.; Pechlivanis, M.; Kuhlmann, J.; Brunsveld, L.; Chandra, A.; Ellinger, B.; et al. The Palmitoylation Machinery Is a Spatially Organizing System for Peripheral Membrane Proteins. Cell 2010, 141, 458–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silvius, J.R.; Bhagatji, P.; Leventis, R.; Terrone, D. K-Ras4B and Prenylated Proteins Lacking “Second Signals” Associate Dy-namically with Cellular Membranes. Mol. Biol Cell. 2006, 17, 192–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prior, I.A.; Harding, A.; Yan, J.; Sluimer, J.C.; Parton, R.G.; Hancock, J.F. GTP-dependent segregation of H-ras from lipid rafts is required for biological activity. Nat. Cell Biol. 2001, 3, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Woolfrey, K.M.; Srivastava, D.P. Control of Dendritic Spine Morphological and Functional Plasticity by Small GTPases. Neural Plast. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forget, M.A.; Desrosiers, R.R.; Gingras, D.; Béliveau, R. Phosphorylation States of Cdc42 and RhoA Regulate Their Interactions with Rho GDP Dissociation Inhibitor and Their Extraction from Biological Membranes. Biochem. J. 2002, 361, 243–254. [Google Scholar] [CrossRef]
- Chang, F.; Lemmon, C.A.; Lietha, D.; Eck, M.; Romer, L.H. Tyrosine Phosphorylation of Rac1: A Role in Regulation of Cell Spreading. PLoS ONE 2011, 6, e28587. [Google Scholar] [CrossRef] [Green Version]
- Tu, X.; Yasuda, R.; Colgan, L.A. Rac1 is a downstream effector of PKCα in structural synaptic plasticity. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Álvarez-Moya, B.; López-Alcalá, C.; Drosten, M.; Bachs, O.; Agell, N. K-Ras4B phosphorylation at Ser181 is inhibited by calmodulin and modulates K-Ras activity and function. Oncogene 2010, 29, 5911–5922. [Google Scholar] [CrossRef] [Green Version]
- Mabb, A.M.; Ehlers, M.D. Ubiquitination in Postsynaptic Function and Plasticity. Annu. Rev. Cell Dev. Biol. 2010, 26, 179–210. [Google Scholar] [CrossRef] [Green Version]
- Jura, N.; Scotto-Lavino, E.; Sobczyk, A.; Bar-Sagi, D. Differential Modification of Ras Proteins by Ubiquitination. Mol. Cell 2006, 21, 679–687. [Google Scholar] [CrossRef]
- Bryan, B.; Cai, Y.; Wrighton, K.; Wu, G.; Feng, X.-H.; Liu, M. Ubiquitination of RhoA by Smurf1 promotes neurite outgrowth. FEBS Lett. 2005, 579, 1015–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figiel, I.; Kruk, P.K.; Zaręba-Kozioł, M.; Rybak, P.; Bijata, M.; Wlodarczyk, J.; Dzwonek, J. MMP-9 Signaling Pathways That Engage Rho GTPases in Brain Plasticity. Cells 2021, 10, 166. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010166
Figiel I, Kruk PK, Zaręba-Kozioł M, Rybak P, Bijata M, Wlodarczyk J, Dzwonek J. MMP-9 Signaling Pathways That Engage Rho GTPases in Brain Plasticity. Cells. 2021; 10(1):166. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010166
Chicago/Turabian StyleFigiel, Izabela, Patrycja K. Kruk, Monika Zaręba-Kozioł, Paulina Rybak, Monika Bijata, Jakub Wlodarczyk, and Joanna Dzwonek. 2021. "MMP-9 Signaling Pathways That Engage Rho GTPases in Brain Plasticity" Cells 10, no. 1: 166. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010166