New Prognostic Biomarkers in Metastatic Castration-Resistant Prostate Cancer
by
 , and
, and
Vincenza Conteduca
1 ,
,
Alessandra Mosca
2,
Nicole Brighi
1,
Ugo de Giorgi
1 and
Pasquale Rescigno
3,* 1
Department of Medical Oncology, Istituto Scientifico Romagnolo per lo Studio e la Cura dei Tumori (IRST) IRCCS, 47014 Meldola, Italy
2
Multidisciplinary Outpatient Oncology Clinic, Candiolo Cancer Institute, FPO-IRCCS, Candiolo, 10060 Turin, Italy
3
Interdisciplinary Group for Translational Research and Clinical Trials, Urological Cancers (GIRT-Uro), Candiolo Cancer Institute, FPO-IRCCS, Candiolo, 10060 Turin, Italy
*
Author to whom correspondence should be addressed.
Cells 2021, 10(1), 193; https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010193
Submission received: 16 December 2020
/
Revised: 11 January 2021
/
Accepted: 13 January 2021
/
Published: 19 January 2021
(This article belongs to the Special Issue Identification of Prognostic Markers in Cancer Biology)
{kind=link}
{kind=link}
Abstract
:Prostate cancer is one of the most frequent cancers in men and is a common cause of cancer-related death. Despite significant progress in the diagnosis and treatment of this tumor, patients who relapse after radical treatments inevitably develop metastatic disease. Patient stratification is therefore key in this type of cancer, and there is an urgent need for prognostic biomarkers that can define patients’ risk of cancer-related death. In the last 10 years, multiple prognostic factors have been identified and studied. Here, we review the literature available and discuss the most common aberrant genomic pathways in metastatic castration-resistant prostate cancer shown to have a prognostic relevance in this setting.
1. Introduction
Prostate cancer (PC) is the second most common cancer for incidence and the fifth most frequent cause of cancer-related mortality in men worldwide, while in Western countries it represents the second cause of death for cancer in males [1,2]. The incidence of PC has been rising in recent years, due to the broader availability of PSA screening in Western countries’ populations [3].
Prostate cancer is a highly heterogeneous disease, in both clinical and molecular aspects. PC may range from slowly progressing to very aggressive disease, mostly due to the development of resistance to treatment. Although advanced PC typically starts with a strong dependency on androgens, resulting in initial responses to androgen deprivation treatments, most patients progress to castration-resistant disease (CRPC) [4]. CRPC status is defined as the presence of castrate serum testosterone levels (<50 ng/dL or 1.7 nmol/L) plus either biochemical progression (three consecutive rises in prostate-specific antigen (PSA) 1 week apart, resulting in two 50% increases over the nadir, and PSA >2 ng/mL) or radiologic progression (consisting of the appearance of new lesions, such as two or more new bone lesions on bone scan or a soft tissue lesion using the Response Evaluation Criteria in Solid Tumors) [5].
In addition to the different possible clinical presentations, intra-patient tumor heterogeneity and clonal evolution must be taken into account and represent a challenge in the management of patients. Several recurrent molecular pathways have been identified in the metastatic CRPC (mCRPC) molecular landscape, resulting in resistance to treatments and tumor progression, and impacting patients’ survival. These include androgen receptor (AR) aberrations, PTEN loss, DNA repair gene deletions, TP53 mutations, and RB1 loss [6,7].
2. Clinical Studies of Circulating Androgen Receptor (AR) Status in Liquid Biopsy as a Prognostic Biomarker in mCRPC
Androgens are involved in the normal development of the prostate gland, but also in prostate carcinogenesis. In normal prostate tissue, differentiation is promoted, while during the progression of PC, proliferation gradually overcomes differentiation. AR contributes to the control of the balance between cell proliferation and differentiation [8,9,10,11].
Androgen signaling inhibition, either through androgen deprivation or AR activity block, is therefore the mainstay of PC treatment. In the last decade, novel hormonal drugs have been approved in castration-sensitive prostate cancer (CSPC) and/or mCRPC thanks to their mechanisms of action (abiraterone prevents androgen biosynthesis, and enzalutamide, apalutamide and darolutamide inhibit AR translocation to the nucleus) (Figure 1).
Indeed, even when the disease turns castration-resistant, AR remains an important molecular driver [12,13,14,15]. Subsequently, the disease acquires resistance to both androgen deprivation therapies (ADT) and AR-directed treatments through molecular pathways mostly driven by AR aberrations. These may include AR gene mutations, AR splice-variants, an AR gene amplification; also, the presence of AR co-regulators may contribute to resistance to treatments [16,17]. These aberrations are seen to increase during tumor progression: in the setting of castration resistance, they are reported in 10–15% of patients, while they are identified in up to 40% of cases in patients treated with second or further treatment lines for advanced CRPC [18].
The vast majority of patients with AR pathway alterations present AR gene amplifications (or gain) [6]. The efficacy of antagonists in inhibiting a receptor-ligand interaction depends greatly on the concentrations of receptors, agonists, and antagonists. Thus, in the presence of AR gene amplifications, AR antagonists (such as enzalutamide, apalutamide and darolutamide) may be unable to effectively antagonize AR proteins. Moreover, since PC is able to synthetize androgens, even when androgen concentrations are under castrate levels, AR signaling still remains active. Through these mechanisms, AR amplifications and enhanced AR signaling result in resistance to first- and second-generation antiandrogens [19,20].
Other AR alterations such as AR point mutations [16] can also result in treatment resistance. Several point mutations have been described: the most clinically relevant are T878A (formerly F876L and T877A) and W742C, affecting the ligand binding domain and conferring resistance to flutamide and bicalutamide; the point mutation F877L, instead, results in resistance to apalutamide and enzalutamide. Mutations of T878A or L702H have been reported to have a role in abiraterone resistance where AR T878A enables the activation of the receptor by progesterone and L702H the activation by glucocorticoids such as prednisone, which is given together with abiraterone and docetaxel. Other mutations frequently described include V715M, V730M, and H875Y [7,8,21].
Secondary AR alterations, usually emerging after treatment with abiraterone or enzalutamide, are represented by AR splice variants (AR-Vs). AR-V7 is the most frequent variant, conferring a constitutive activation resulting in enhanced transcription, cell proliferation and DNA repair. Thus, AR-Vs are related to treatment resistance and poor outcomes [22,23,24]. However, AR-Vs’ activity is usually seen in the presence of AR amplifications [4,25].
Finally, the presence of AR co-regulators represents another possible mechanism of resistance to treatments. Several co-activators may interact with AR and enhance transcription. Among them, the activity of the members of the p160 steroid receptor coactivator (SRC) family, the histone acetyltransferase CBP/p300, and the pioneer factor forkhead box A1 (FOXA1) have been described to be associated with treatment resistance and worse prognosis [17,26,27].
All these alterations involving the AR pathway can be studied using liquid biopsies. For example, given the heterogeneity of AR-V7 expression at different metastatic sites [28], its study in circulating tumor cells (CTCs) could provide a comprehensive assessment of the AR-V7 status and a monitoring of AR-V7 changes during treatment. The first study by Antonarakis et al. [24] showed that AR-V7 positive CTCs were associated with resistance to second-line AR-directed therapies in mCRPC patients. Of the 62 enrolled patients, 19% of the 31 abiraterone-treated patients and 39% of the 31 enzalutamide-treated patients had AR-V7-positive CTCs, respectively. The AR-V7-positive patients presented significantly worse outcomes, including lower PSA response rates and worse progression-free survival (PFS) and overall survival (OS). In a follow-up study on 202 prospectively enrolled patients, treated with either abiraterone or enzalutamide, AR-V7 positivity was significantly associated with other adverse prognostic factors, including a Gleason score of ≥8, the presence of metastatic disease, and previous treatment with AR-directed therapies and taxanes. On multivariable analysis, the prognostic impact of AR-V7-positivity on PFS and OS was confirmed [29]. The same group also investigated AR-V7 mRNA expression as a predictor of outcomes in mCRPC patients treated with taxanes [30]. The study enrolled 37 patients, of which 46% had AR-V7-positive CTCs. Their outcomes were, however, not dissimilar to those patients with AR-V7-negative CTCs, suggesting that AR-V7 status was not predictive of taxane resistance. On the contrary, the AR-V7-positive patients treated with taxanes had significantly better outcomes than the AR-V7-positive men who had received AR-targeting agents, suggesting that taxane therapy is preferable for AR-V7-positive mCRPC patients. Similar results were observed in a study of 29 mCRPC patients receiving cabazitaxel [31]. Recently, Scher et al. also reported an automated method of detecting intranuclear AR-V7 protein using immunofluorescent staining of CTCs that was also predictive of the clinical response to second generation hormonal treatments, as shown in the tissue-based study of AR-V7 expression [32]. Moreover, nuclear localization of the AR-V7 protein was an independent prognostic factor for survival (HR 2.38; 95% CI, 1.02–5.53; p = 0.045) in taxane-treated patients. Using the same automated immunofluorescent assay of CTC, a multicenter study also confirmed the predictive value of AR-V7 positivity in 142 mCRPC patients, showing a benefit in OS for AR-positive patients treated with chemotherapy compared to those treated with AR-directed agents [33].
Interestingly, in the TAXNERGY study (early switch from first-line docetaxel/prednisone to cabazitaxel/prednisone and the opposite sequence, exploring molecular markers in mCRPC patients), using a novel digital droplet PCR assay, 67% of the 54 docetaxel/ or cabazitaxel treated patients had AR-V7-positive CTCs [34]. These patients presented with lower PSA response rates (78% vs. 58%; p = 0.23) and shorter median PFS (8 vs. 12 months; HR, 0.38; p = 0.01) compared to the AR-V7-negative patients.
Preliminary findings were also reported in 15 AR-V7-positive patients treated with ipilimumab and nivolumab showing lack of efficacy in this subset of patients, with only two patients (13%) achieving a PSA response [35].
Further circulating biomarkers in CRPC are represented by AR gene aberrations that are infrequent in the early stages, but common in the CRPC setting. Therefore, NGS- and PCR-based studies have been particularly focused on AR copy number in advanced disease. Romanel et al. [36] analyzed 274 plasma samples from 97 CRPC patients treated with abiraterone. The authors reported plasma AR gain and point mutations to be mutually exclusive. While AR copy number was not different during treatment, the emergence of T878A or L702H AR amino acid changes was reported in 13% of samples upon progression. There was also an association between plasma AR aberrations and worse OS (HR 7.33, 95% CI 3.51–15.34, p = 1.3 × 10−9) and PFS (HR 3.73, 95%CI 2.17–6.41, p = 5.6 × 10−7). Subsequently, a multicenter biomarker study assessed the role of plasma AR also in chemotherapy-naïve patients [37]. There was a primary cohort of 73 chemotherapy-naïve and 98 post-docetaxel patients treated with enzalutamide or abiraterone and a secondary cohort of 94 chemotherapy-naïve patients treated with enzalutamide in 16 institutions in the PREMIERE trial (NCT02288936). In the primary cohort, 14% pre-chemotherapy and 34% post-docetaxel patients presented AR gain, while 11% post-docetaxel but no chemotherapy-naïve abiraterone-treated patients showed AR point mutations. Chemotherapy-naïve and post-docetaxel AR-gained patients demonstrated a poor survival (OS: HR = 3.98, 95%CI 1.74–9.10, p < 0.001 and HR = 3.81, 95%CI 2.28–6.37, p < 0.001, respectively, and PFS: HR = 2.18, 95%CI 1.08–4.39, p = 0.03, and HR = 1.95, 95%CI 1.23–3.11, p = 0.01, respectively). Patients with AR mutations also showed a significantly worse OS (HR = 3.26, 95%CI 1.47–not reached, p = 0.004). In the validation cohort, 11 (12%) patients had AR gain. AR-gained patients had a reduced biochemical PFS (HR = 4.33, 95%CI 1.94–9.68, p < 0.001), radiographic PFS (HR = 8.06, 95%CI 3.26–19.93, p < 0.001), OS (HR = 11.08, 95%CI 2.16–56.95, p = 0.004).
As previous findings have suggested that the detection of AR-V7 in CTCs could predict a response to AR-signaling inhibitors versus taxane chemotherapy, recently, the role of cell-free AR copy number has also been investigated in CRPC patients treated with taxanes [38,39]. A biomarker study of 163 docetaxel-treated patients assessed the association between plasma AR and outcomes in mCRPC, reporting only a significant worse OS in AR-gained patients (HR = 1.61, 95%CI 1.08–2.39, p = 0.018). Moreover, the same authors incorporated updated data from their prior study [38], and interrogated the interaction between plasma AR and treatment type in the abiraterone/enzalutamide-treated group (73 patients), after and in 115 first-line docetaxel patients. The findings from this study would suggest that AR-normal men could benefit from hormonal treatment in the first-line therapy group, while plasma AR-gained patients had a better response to docetaxel. Similarly, in a subsequent work with mCRPC patients receiving second-line cabazitaxel therapy, a significant treatment interaction between plasma AR and cabazitaxel vs. AR-directed therapies was observed for OS (p = 0.041). An exploratory analysis showed AR-gained patients treated with an initially reduced dose of cabazitaxel presented a meaningfully worse OS/PFS and would probably need a standard initial dose of cabazitaxel. This study suggests that plasma AR may improve clinical decision-making in choosing between adapted and standard regimen of taxanes [39].
Recently, plasma AR gene status has been also explored in a phase 2 clinical trial (NCT03454750) of mCRPC patients treated with 177Lu–PSMA–617 [40]. Early progressive disease was observed in 17 (42.5%) of the 40 patients (12 of 15 (80%) with AR amplification and 5 of 25 (20%) with AR normal (p = 0.0002)). The OR for patients with early disease progression and AR gain was 16.00, 95% CI 3.23–79.27, p = 0.0007. These preliminary data suggest that plasma AR status assessed by ddPCR could be helpful to identify resistance to 177Lu–PSMA–617 in mCRPC patients.
Recent studies determined the impact of AR mutations, e.g., F877L and T878A, responses to novel AR-directed therapies, such as apalutamide [41] and darolutamide [42]. The phase I/II study ARN-509-001 [40] used the sensitive BEAMing assay to detect AR mutations in cell-free DNA in apalutamide-treated nonmetastatic CRPC and mCRPC patients. However, the overall frequency of AR mutations was so low that no conclusions could be drawn.
A recent paper [43] uncovered an orthogonal methylation signature in 25 mCRPC patients treated with abiraterone or enzalutamide. The study showed an association between AR copy number gain and an enrichment for AR binding sequences and hypomethylation of these segments. Moreover, plasma methylome analysis could accurately estimate tumor fraction. All these findings permitted to identify patients with a more aggressive clinical course based on this methylation pattern and so a molecular prognostic stratification of mCRPC individuals.
The role of plasma DNA analysis has been recently investigated in combination with functional imaging and other routinely obtained circulating biomarkers in order to improve prognostication of mCRPC in patients [44]. This work provided innovative insights on utility of integrating functional imaging with plasma DNA analysis including AR status assessment and other noninvasive biomarkers to improve treatment.
3. The Prognostic Role of PTEN Alterations and Its Pathway
Alterations in phosphatase and tensin homolog (PTEN) and activation of the kinases PI3K and AKT, downstream of its axis, are common in primary PC and are enriched in mCRPCs [6,45]. Genomic inactivation of PTEN, mainly through the loss of its locus on 10q23.31, is the most common molecular aberration involving this pathway occurring in approximately 40% of CRPCs [6,7,45]. However, mutations and complex genomic rearrangements can also occur [46], as well as aberrations in PIK3CA, PIK3CB, PIK3R1, PIK3R3 and AKT1 [6]. All these alterations often result in the activation of this pathway and are responsible for cell growth, cell cycle progression, and cell proliferation [47]. The PTEN/PI3K/AKT pathway indirectly also regulates the AR signaling through a negative feedback loop [48,49], and this is particularly relevant for the therapeutic approaches in mCRPC. Immunohistochemistry (IHC) studies have consistently shown PTEN loss as a poor prognostic factor in mCRPC [50,51,52,53].
Ferraldeschi et al. analyzed the PTEN status of 144 patients who had received abiraterone post-docetaxel. In this retrospective study, the loss of PTEN expression was not only associated with shorter median OS (14 vs. 21 mo; hazard ratio [HR]: 1.75; 95% confidence interval [CI], 1.19–2.55; p = 0.004), but also with shorter median duration on abiraterone (24 vs. 28 wk; HR: 1.6; 95% CI, 1.12–2.28; p = 0.009). More importantly, PTEN status was consistent between matched CSPC and CRPC tumor biopsies in nearly 90% of cases [52], meaning that the loss of PTEN is an early event in prostate cancer tumorigenesis. Similarly, PI3K and AKT alterations were identified in 11% of 418 prostate cancer tissue samples, of these, 26/418 (6%) were pathogenic pathway activating mutations. These mutations were associated with a shorter OS (2.8 vs. 4.3 years; HR: 2.73; p < 0.001) and duration of abiraterone/enzalutamide (5.9 vs. 10.0 mo; p < 0.001) [54].
Altogether, these data highlight the relevance of the aberrations in the PTEN pathway as prognostic factor in mCRPC. However, more recently, these alterations have been studied as biomarkers of response to the combination of new generation hormonal treatments and AKT inhibitors such as ipatasertib and capivasertib [55,56].
4. DNA Repair Defects as Prognostic Biomarkers in mCRPC
Genomic instability is a common denominator of many different cancers. This instability derives from the high rate of cell division that is responsible for the fast accumulation of genomic aberrations [57]. Therefore, defects in the DNA damage repair (DDR) play a key role in the promotion of cancer growth [58]. Most of the endogenous or exogenous mutagens can cause DNA single-strand breaks (SSBs), although double-strand breaks (DSBs) are more lethal to cells. Thus, most DDR-directed therapies target the repair mechanisms associated with DSBs, increasing replication stress; or inhibit cell cycle checkpoints that facilitate DNA repair. More specifically, defects in (or inhibition of) the high-fidelity DDR system, such as homologous recombination (HR), increase genomic instability, since cells will try to rely on compensatory mechanisms of repair that are often error-prone in order to survive [59].
Breast cancer-associated gene2 (BRCA2), a key member of the HR and Fanconi anemia complex, is the most commonly mutated DDR gene in prostate cancer. In the metastatic setting, germline BRCA2 (gBRCA) mutations have a prevalence of 3–6%, while somatic mutations and homozygous deletions account for ~20% of metastatic CRPC (mCRPC) cases [6,7,19,60]. On the other hand, alterations in other HR members, BRCA1, PALB2 and RAD51, are present in <1% of mCRPC. The prognostic role of germline and somatic mutations in HR genes, and specifically of BRCA2 alterations, has been investigated in detail in recent years. BRCA2 mutation carriers have a 5-year prostate cancer-specific survival (CSS) rate of ~50%, progressing rapidly from localized PC to mCRPC [61,62,63,64]. Similarly, in the PROREPAIR-B study, which prospectively followed a cohort of 419 gDDR-defective mCRPC patients in order to evaluate the impact of these alterations on outcomes, gBRCA2 patients were found to have a shorter CSS compared to non-carriers (17.4 months vs. 33.2 months, p = 0.027) [65]. A recent meta-analysis of 10 studies including 525 BRCA2 mutation-carriers and 8,463 non-carriers confirmed that carrying a BRCA2 alteration correlates with a reduced CSS and OS (Hazard Ratios (HRs) 2.53 vs. 2.21 respectively, p < 0.001). The results also demonstrated that BRCA2-mutation carriers were characterized by a higher Gleason Score (GS) (>7), TNM stage (>T3, N1, M1) at diagnosis [66]. The predictive role of these mutations as biomarkers of response to standard treatments for mCRPC remains, however, controversial [67,68,69]; nevertheless, it is undoubted that BRCA2 alterations predict response to PARP inhibitors in mCRPC [70,71].
ATM (ataxia telangiectasia, mutated) is a member of the PI3 family of serine-threonine kinases and functions as a DNA damage sensor [72]. Therefore, it is involved in DNA repair, triggering the action of others DDR proteins [73]. Its alterations are found in nearly 10% of mCRPC patients [6,60], but its loss does not determine the same mutational signature as BRCA1 or BRCA2 biallelic loss [74,75]. Its prognostic value is unclear, but ATM loss does not seem to affect mCRPC patients’ outcome [76].
CDK12 is reportedly involved in regulating expression of several HR genes [77]; somatic defects in CDK12 are associated with a genome-wide focal tandem duplication (FTD) signature [78]. Multiple retrospective studies have reported a worse survival for CK12 mutant mCRPC, which are also characterized by a high Gleason score (>8) and a tumor immune infiltrate, potentially immunosuppressive, comprised of CD4+FOXP3- cells [79]. The FTD phenotype has also been correlated with increased neo-antigen burden in those tumors, positioning CDK12 among the putative biomarkers of response to novel immunotherapies in mCRPC [74].
The mismatch repair (MMR) system is a post-replicative, high-fidelity, single-strand repair mechanism that recognizes and reverses DNA base mismatches and insertion/deletion loops, compromised MMR results in microsatellite instability, and a hypermutator phenotype that has been associated with chemotherapy resistance but immunotherapy sensitivity [80]. The prevalence of MMR aberrations ranges between 3% and 12%, depending on the assay and population selected [6,81,82], and could be underestimated since alterations in MSH2 and MSH6, the two main players of the MMR system, involve noncoding regions detectable only by whole-genome studies. MMR-defective status (dMMR) was associated with poor OS compared to MMR-proficient tumors (3.8 vs. 7.0 years from start of luteinizing hormone–releasing hormone; p = 0.005), increased T-cell infiltration and elevated PD-L1 protein expression in a cohort of 124 mCRPC biopsies from a single institution [83]. Transcriptome analysis has also revealed that cancers with an MMR mutational signature are characterized by increased expression of inferred immune cells, immune checkpoints, and T-cell-associated transcripts [83].
Considering their localization on chromosome 13, BRCA2 losses often co-occur with the retinoblastoma (RB1) ones [84]. RB1 is a tumor suppressor that also plays a relevant role in mCRPC. Comprehensive genomic, transcriptomic and histologic analysis of 429 patients with mCRPC linked with longitudinal clinical outcomes identified RB1 genomic alteration as a potent predictor of poor outcome especially when associated with tumor protein p53 (TP53) alterations [85]. TP53 plays a role in cell cycle and genomic stability. It is able to activate DNA repair proteins upon DNA damage, arresting cell growth at the G1/S regulation point. In case of irreparable damage, TP53 is able to induce apoptosis and cell death [86] and represent with PTEN one of the most common alteration in mCRPC [6]. The prognostic role of RB1 and TP53 aberrations was also confirmed by another large retrospective study in which 470 treatment-naïve prostate cancer diagnostic biopsies and 61 matched mCRPC biopsies were analyzed using targeted and low-pass whole-genome sequencing. Moreover, this study demonstrated that RB1 losses along with TP53 and AR aberrations were enriched in later stages [87].
5. Use of Molecular Biomarkers in Localized Prostate Cancer
We explored the relevance of prognostic biomarkers in mCRPC; however, in the last few years, some studies have proposed stratifying patients with localized PC based on molecular biomarkers. Traditionally, Gleason score, clinical staging and PSA at diagnosis have helped classifying localized PCs as being at low, intermediate, and high risk of relapse [88]. Recently several tissue-based multigene expression (Decipher, Oncotype Dx Prostate, Polaris) and a protein-based (ProMark) classifiers have been proposed to identify patients with biologically significant disease [89,90,91,92].
Using RNA analysis on formalin fixed paraffin embedded clinical samples, studying biological pathways with a known role in prostate tumorigenesis (i.e., proliferation, cell cycle, etc.), these classifiers (Decipher, Oncotype Dx Prostate, Polaris) offer scores that, integrating other clinical–pathological features, might help identifying patients with localized disease but with worse outcomes. Potentially, these tools could help clinicians in deciding which patients would benefit from surveillance vs. active treatment, or a salvage approach after a radical treatment [89,90,91,92].
However, tissue-based molecular testing is dependent on tissue preservation and influenced by the heterogeneity of the disease [93]. Moreover, the high costs and the lack of prospective validation in clinical trials represent the main limitations to their use in the daily clinical practice. Therefore, based on these considerations, the American society of clinical oncology (ASCO) has not recommended tissue-based molecular biomarkers for routine use [94].
6. Conclusions
Here, we presented the most common genomic pathway aberrations in prostate cancer that have an impact on patient outcomes. All of these studies and data are relevant to understanding the importance of genomic studies in the metastatic setting in order to better evaluate patient prognosis. Since some of these biomarkers can be used as either therapeutic targets or as biomarkers of response to novel agents and standard treatments in mCRPC, they also have emerging potential in targeted therapy and therapy response prediction (Figure 2).
Funding
Prostate Cancer Foundation: PCF YIA 2019.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
Pasquale Rescigno’s work is funded by the Prostate Cancer Foundation through a PCFYI award. Research at the Candiolo Cancer Institute is also supported by the Italian Minister of Health through the ’Ricerca Corrente 2020’ national grant.
Conflicts of Interest
Vincenza Conteduca has received speaker honoraria or travel support from Astellas, Janssen-Cilag and Sanofi-Aventis, and has received a consulting fee from Bayer. Nicole Brighi has received travel support from Ipsen, Novartis and Pfizer. Ugo De Giorgi has served as consultant/advisory board member for Astellas, Bayer, BMS, Ipsen, Janssen, Merck, Pfizer and Sanofi; has received travel support from BMS, Ipsen, Janssen and Pfizer; and has received research funding from AstraZeneca, Roche and Sanofi (Inst). The other The authors have no conflicts to declare.
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, M.; Karim-Kos, H.E.; Coebergh, J.W.; Byrnes, G.; Antilla, A.; Ferlay, J.; Renehan, A.G.; Forman, D.; Soerjomataram, I. Recent trends in incidence of five common cancers in 26 European countries since 1988: Analysis of the European Cancer Observatory. Eur. J. Cancer 2015, 51, 1164–1187. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Attard, G.; Balk, S.P.; Burnstein, K.; Cato, L.; Cherkasov, A.; De Bono, J.S.; Dong, Y.; Gao, A.C.; Gleave, M.; et al. Role of Androgen Receptor Variants in Prostate Cancer: Report from the 2017 Mission Androgen Receptor Variants Meeting. Eur. Urol. 2018, 73, 715–723. [Google Scholar] [CrossRef]
- Cornford, P.; Bellmunt, J.; Bolla, M.; Briers, E.; De Santis, M.; Gross, T.; Henry, A.M.; Joniau, S.; Lam, T.B.; Mason, M.D.; et al. EAU-ESTRO-SIOG Guidelines on Prostate Cancer. Part II: Treatment of Relapsing, Metastatic, and Castration-Resistant Prostate Cancer. Eur. Urol. 2017, 71, 630–642. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Waltering, K.K.; Urbanucci, A.; Visakorpi, T. Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol. Cell. Endocrinol. 2012, 360, 38–43. [Google Scholar] [CrossRef]
- Hendriksen, P.J.; Dits, N.F.; Kokame, K.; Veldhoven, A.; van Weerden, W.M.; Bangma, C.H.; Trapman, J.; Jenster, G. Evolution of the androgen receptor pathway during progression of prostate cancer. Cancer Res. 2006, 66, 5012–5020. [Google Scholar] [CrossRef] [Green Version]
- Conteduca, V.; Gurioli, G.; Brighi, N.; Lolli, C.; Schepisi, G.; Casadei, C.; Burgio, S.L.; Gargiulo, S.; Ravaglia, G.; Rossi, L.; et al. Plasma Androgen Receptor in Prostate Cancer. Cancers 2019, 11, 1719. [Google Scholar] [CrossRef] [Green Version]
- Helsen, C.; Van den Broeck, T.; Voet, A.; Prekovic, S.; Van Poppel, H.; Joniau, S.; Claessens, F. Androgen receptor antagonists for prostate cancer therapy. Endocr. Relat. Cancer 2014, 21, T105–T118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.E.; Sternberg, C.N.; Miller, K.; de Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Smith, M.R.; de Bono, J.S.; Molina, A.; Logothetis, C.J.; de Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karantanos, T.; Evans, C.P.; Tombal, B.; Thompson, T.C.; Montironi, R.; Isaacs, W.B. Understanding the mechanisms of androgen deprivation resistance in prostate cancer at the molecular level. Eur. Urol. 2015, 67, 470–479. [Google Scholar] [CrossRef] [Green Version]
- Culig, Z. Molecular mechanisms of enzalutamide resistance in prostate cancer. Curr. Mol. Biol. Rep. 2017, 3, 230–235. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial interindividual and limited intraindividual genomic diversity among tumors from men with metastatic prostate cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef]
- Beltran, H.; Yelensky, R.; Frampton, G.M.; Park, K.; Downing, S.R.; MacDonald, T.Y.; Jarosz, M.; Lipson, D.; Tagawa, S.T.; Nanus, D.M.; et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur. Urol. 2013, 63, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Veldscholte, J.; Berrevoets, C.A.; Ris-Stalpers, C.; Kuiper, G.G.; Jenster, G.; Trapman, J.; Brinkmann, A.O.; Mulder, E. The androgen receptor in LNCaP cells contains a mutation in the ligand binding domain which affects steroid binding characteristics and response to antiandrogens. J. Steroid Biochem. Mol. Biol. 1992, 41, 665–669. [Google Scholar] [CrossRef]
- Joseph, J.D.; Lu, N.; Qian, J.; Sensintaffar, J.; Shao, G.; Brigham, D.; Moon, M.; Maneval, E.C.; Chen, I.; Darimont, B.; et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer Discov. 2013, 3, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Morrissey, C.; Sun, S.; Ketchandji, M.; Nelson, P.S.; True, L.D.; Vakar-Lopez, F.; Vessella, R.L.; Plymate, S.R. Androgen receptor variants occur frequently in castration resistant prostate cancer metastases. PLoS ONE 2011, 6, e27970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, S.; Sprenger, C.C.; Vessella, R.L.; Haugk, K.; Soriano, K.; Mostaghel, E.A.; Page, S.T.; Coleman, I.M.; Nguyen, H.M.; Sun, H.; et al. Castration resistance in human prostate cancer is conferred by a frequently occurring androgen receptor splice variant. J. Clin. Invest. 2010, 120, 2715–2730.23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Isaacs, W.B.; Luo, J. A snapshot of the expression signature of androgen receptor splicing variants and their distinctive transcriptional activities. Prostate 2011, 71, 1656–1667. [Google Scholar] [CrossRef] [Green Version]
- Agoulnik, I.U.; Vaid, A.; Bingman, W.E.; Erdeme, H.; Frolov, A.; Smith, C.L.; Ayala, G.; Ittmann, M.M.; Weigel, N.L. Role of SRC-1 in the promotion of prostate cancer cell growth and tumor progression. Cancer Res. 2005, 65, 7959–7967. [Google Scholar] [CrossRef] [Green Version]
- Agoulnik, I.U.; Vaid, A.; Nakka, M.; Alvarado, M.; Bingman, W.E., 3rd; Erdem, H.; Frolov, A.; Smith, C.L.; Ayala, G.E.; Ittmann, M.M.; et al. Androgens modulate expression of transcription intermediary factor 2, an androgen receptor coactivator whose expression level correlates with early biochemical recurrence in prostate cancer. Cancer Res. 2006, 66, 10594–10602. [Google Scholar] [CrossRef] [Green Version]
- Sharp, A.; Coleman, I.; Yuan, W.; Sprenger, C.; Dolling, D.; Rodrigues, D.N.; Russo, J.W.; Figueiredo, I.; Bertan, C.; Seed, G.; et al. Androgen receptor splice variant-7 expression emerges with castration resistance in prostate cancer. J. Clin. Invest. 2019, 129, 192–208. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Zhu, Y.; Silberstein, J.L.; Taylor, M.N.; Maughan, B.L.; Denmeade, S.R.; et al. Clinical significance of androgen receptor splice variant-7 mRNA detection in circulating tumor cells of men with metastatic castration-resistant prostate cancer treated with first- and second- line abiraterone and enzalutamide. J. Clin. Oncol. 2017, 35, 2149–2156. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Wang, H.; Chen, Y.; Nakazawa, M.; Nadal, R.; Paller, C.J.; Denmeade, S.R.; Carducci, M.A.; et al. Androgen receptor splice variant 7 and efficacy of taxane chemotherapy in patients with metastatic castration-resistant prostate cancer. JAMA Oncol. 2015, 1, 582–591. [Google Scholar] [CrossRef] [Green Version]
- Onstenk, W.; Sieuwerts, A.M.; Kraan, J.; Van, M.; Nieuweboer, A.J.; Mathijssen, R.H.; Hamberg, P.; Meulenbeld, H.J.; De Laere, B.; Dirix, L.Y.; et al. Efficacy of cabazitaxel in castration resistant prostate cancer is independent of the presence of AR-V7 in circulating tumor cells. Eur. Urol. 2015, 68, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Lu, D.; Schreiber, N.A.; Louw, J.; Graf, R.P.; Vargas, H.A.; Johnson, A.; Jendrisak, A.; Bambury, R.; Danila, D.; et al. Association of AR-V7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration resistant prostate cancer. JAMA Oncol. 2016, 2, 1441–1449. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Graf, R.P.; Schreiber, N.A.; Jayaram, A.; Winquist, E.; McLaughlin, B.; Lu, D.; Fleisher, M.; Orr, S.; Lowes, L.; et al. Assessment of the validity of nuclear localized androgen receptor splice variant 7 in circulating tumor cells as a predictive biomarker for castration-resistant prostate cancer. JAMA Oncol. 2018, 4, 1179–1186. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, S.T.; Antonarakis, E.S.; Gjyrezi, A.; Galletti, G.; Kim, S.; Worroll, D.; Stewart, J.; Zaher, A.; Szatrowski, T.P.; Ballman, K.V.; et al. Expression of AR-V7 and ARv(567es) in circulating tumor cells correlates with outcomes to taxane therapy in men with metastatic prostate cancer treated in TAXYNERGY. Clin. Cancer Res. 2019, 25, 1880–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boudadi, K.; Suzman, D.L.; Anagnostou, V.; Fu, W.; Luber, B.; Wang, H.; Niknafs, N.; White, J.R.; Silberstein, J.L.; Sullivan, R.; et al. Ipilimumab plus nivolumab and DNA-repair defects in AR-V7-expressing metastatic prostate cancer. Oncotarget 2018, 9, 28561–28571. [Google Scholar] [CrossRef] [Green Version]
- Romanel, A.; Tandefelt, D.G.; Conteduca, V.; Jayaram, A.; Casiraghi, N.; Wetterskog, D.; Salvi, S.; Amadori, D.; Zafeiriou, Z.; Rescigno, P.; et al. Plasma AR and abiraterone-resistant prostate cancer. Sci. Transl. Med. 2015, 7, 312re10. [Google Scholar] [CrossRef] [Green Version]
- Conteduca, V.; Wetterskog, D.; Sharabiani, M.T.A.; Grande, E.; Fernandez-Perez, M.P.; Jayaram, A.; Salvi, S.; Castellano, D.; Romanel, A.; Lolli, C.; et al. Androgen receptor gene status in plasma DNA associates with worse outcome on enzalutamide or abiraterone for castration-resistant prostate cancer: A multi-institution correlative biomarker study. Ann. Oncol. 2017, 28, 1508–1516. [Google Scholar] [CrossRef]
- Conteduca, V.; Jayaram, A.; Romero-Laorden, N.; Wetterskog, D.; Salvi, S.; Gurioli, G.; Scarpi, E.; Castro, E.; Marin-Aguilera, M.; Lolli, C.; et al. Plasma Androgen Receptor and Docetaxel for Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2019, 75, 368–373. [Google Scholar] [CrossRef] [Green Version]
- Conteduca, V.; Castro, E.; Wetterskog, D.; Scarpi, E.; Jayaram, A.; Romero-Laorden, N.; Olmos, D.; Gurioli, G.; Lolli, C.; Sáez, M.I.; et al. Plasma AR status and cabazitaxel in heavily treated metastatic castration-resistant prostate cancer. Eur. J. Cancer 2019, 116, 158–168. [Google Scholar] [CrossRef]
- De Giorgi, U.; Severi, S.; Sarnelli, A.; Sansovini, M.; Monti, M.; Gurioli, G.; Nicolini, S.; Scarpi, E.; Casadei, C.; Conteduca, V.; et al. Circulating androgen receptor (AR) gene amplification and resistance to 177Lu-PSMA-617 in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): Results of a phase II clinical trial. J. Clin. Oncol. 2019, 37, 3020. [Google Scholar] [CrossRef]
- Rathkopf, D.E.; Smith, M.R.; Ryan, C.J.; Berry, W.R.; Shore, N.D.; Liu, G.; Higano, C.S.; Alumkal, J.J.; Hauke, R.; Tutrone, R.F.; et al. Androgen receptor mutations in patients with castration-resistant prostate cancer treated with apalutamide. Ann. Oncol. 2017, 28, 2264–2271. [Google Scholar] [CrossRef] [PubMed]
- Borgmann, H.; Lallous, N.; Ozistanbullu, D.; Beraldi, E.; Paul, N.; Dalal, K.; Fazli, L.; Haferkamp, A.; Lejeune, P.; Cherkasov, A.; et al. Moving Towards Precision Urologic Oncology: Targeting Enzalutamide-resistant Prostate Cancer and Mutated Forms of the Androgen Receptor Using the Novel Inhibitor Darolutamide (ODM-201). Eur. Urol. 2018, 73, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Cremaschi, P.; Wetterskog, D.; Conteduca, V.; Franceschini, G.M.; Kleftogiannis, D.; Jayaram, A.; Sandhu, S.; Wong, S.Q.; Benelli, M.; et al. Genome-wide plasma DNA methylation features of metastatic prostate cancer. J. Clin. Invest. 2020, 130, 1991–2000. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Scarpi, E.; Matteucci, F.; Caroli, P.; Ravaglia, G.; Fantini, L.; Gurioli, G.; Schepisi, G.; Wetterskog, D.; Menna, C.; et al. Multimodal approach to outcome prediction in metastatic castration-resistant prostate cancer by integrating functional imaging and plasma DNA analysis. JCO Precis. Oncol. 2019, 3, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2019, 51, 1194. [Google Scholar] [CrossRef]
- Reid, A.H.; Attard, G.; Brewer, D.; Miranda, S.; Riisnaes, R.; Clark, J.; Hylands, L.; Merson, S.; Vergis, R.; Jameson, C.; et al. Novel, gross chromosomal alterations involving PTEN cooperate with allelic loss in prostate cancer. Mod. Pathol. 2012, 25, 902–910. [Google Scholar] [CrossRef] [Green Version]
- Sarker, D.; Reid, A.H.; Yap, T.A.; de Bono, J.S. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin. Cancer Res. 2009, 15, 4799–4805. [Google Scholar] [CrossRef] [Green Version]
- Munkley, J.; Livermore, K.E.; McClurg, U.L.; Kalna, G.; Knight, B.; McCullagh, P.; McGrath, J.; Crundwell, M.; Leung, H.Y.; Robson, C.N.; et al. The PI3K regulatory subunit gene PIK3R1 is under direct control of androgens and repressed in prostate cancer cells. Oncoscience 2015, 2, 755–764. [Google Scholar] [CrossRef]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [Green Version]
- Reid, A.H.; Attard, G.; Ambroisine, L.; Fisher, G.; Kovacs, G.; Brewer, D.; Clark, J.; Flohr, P.; Edwards, S.; Berney, D.M.; et al. Molecular characterisation of ERG, ETV1 and PTEN gene loci identifies patients at low and high risk of death from prostate cancer. Br. J. Cancer 2010, 102, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Leinonen, K.A.; Saramäki, O.R.; Furusato, B.; Kimura, T.; Takahashi, H.; Egawa, S.; Suzuki, H.; Keiger, K.; Ho Hahm, S.; Isaacs, W.B.; et al. Loss of PTEN is associated with aggressive behavior in ERG-positive prostate cancer. Cancer Epidemiol. Biomarkers Prev. 2013, 22, 2333–2344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferraldeschi, R.; Nava Rodrigues, D.; Riisnaes, R.; Miranda, S.; Figueiredo, I.; Rescigno, P.; Ravi, P.; Pezaro, C.; Omlin, A.; Lorente, D.; et al. PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur. Urol. 2015, 67, 795–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rescigno, P.; Lorente, D.; Dolling, D.; Ferraldeschi, R.; Rodrigues, D.N.; Riisnaes, R.; Miranda, S.; Bianchini, D.; Zafeiriou, Z.; Sideris, S.; et al. Docetaxel Treatment in PTEN- and ERG-aberrant Metastatic Prostate Cancers. Eur. Urol. Oncol. 2018, 1, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, P.; Rediti, M.; Dolling, D.; Rodrigues, D.N.; Bianchini, D.; Riisnaes, R.; Messina, C.; Barrero, M.; Petremolo, A.; Sharp, A.; et al. PI3K/AKT pathway deleterious mutations in lethal prostate cancer. Ann. Oncol. 2018, 29, viii293. [Google Scholar] [CrossRef]
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arranz Arija, J.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef] [Green Version]
- Kolinsky, M.P.; Rescigno, P.; Bianchini, D.; Zafeiriou, Z.; Mehra, N.; Mateo, J.; Michalarea, V.; Riisnaes, R.; Crespo, M.; Figueiredo, I.; et al. A phase I dose-escalation study of enzalutamide in combination with the AKT inhibitor AZD5363 (capivasertib) in patients with metastatic castration-resistant prostate cancer. Ann. Oncol. 2020, 31, 619–625. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, L.R.; Chen, H.; Collins, A.R.; Connell, M.; Damia, G.; Dasgupta, S.; Malhotra, M.; Meeker, A.K.; Amedei, A.; Amin, A.; et al. Genomic instability in human cancer: Molecular insights and opportunities for therapeutic attack and prevention through diet and nutrition. Semin. Cancer Biol. 2015, 35, S5–S24. [Google Scholar] [CrossRef]
- Annala, M.; Vandekerkhove, G.; Khalaf, D.; Taavitsainen, S.; Beja, K.; Warner, E.W.; Sunderland, K.; Kollmannsberger, C.; Eigl, B.J.; Finch, D.; et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov. 2018, 8, 444–457. [Google Scholar] [CrossRef] [Green Version]
- Castro, E.; Goh, C.; Leongamornlert, D.; Saunders, E.; Tymrakiewicz, M.; Dadaev, T.; Govindasami, K.; Guy, M.; Ellis, S.; Frost, D.; et al. Effect of BRCA mutations on metastatic relapse and cause-specific survival after radical treatment for localized prostate cancer. Eur. Urol. 2015, 68, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Kote-Jarai, Z.; Leongamornlert, D.; Saunders, E.; Tymrakiewicz, M.; Castro, E.; Mahmud, N.; Guy, M.; Edwards, S.; O’Brien, L.; Sawyer, E.; et al. BRCA2 is a moderate penetrance gene contributing to young-onset prostate cancer: Implications for genetic testing in prostate cancer patients. Br. J. Cancer 2011, 105, 1230–1244. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.A.; Fraser, M.; Livingstone, J.; Espiritu, S.M.; Thorne, H.; Huang, V.; Lo, W.; Shiah, Y.J.; Yamaguchi, T.N.; Sliwinski, A.; et al. Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nat. Commun. 2017, 8, 13671. [Google Scholar] [CrossRef] [PubMed]
- Na, R.; Zheng, S.L.; Han, M.; Yu, H.; Jiang, D.; Shah, S.; Ewing, C.M.; Zhang, L.; Novakovic, K.; Petkewicz, J.; et al. Germline mutations in ATM and BRCA1/2 distinguish risk for lethal and indolent prostate cancer and are associated with early age at death. Eur. Urol. 2017, 71, 740–747. [Google Scholar] [CrossRef] [Green Version]
- Castro, E.; Romero-Laorden, N.; Del Pozo, A.; Lozano, R.; Medina, A.; Puente, J.; Piulats, J.M.; Lorente, D.; Saez, M.I.; Morales-Barrera, R.; et al. PROREPAIR-B: A Prospective Cohort Study of the Impact of Germline DNA Repair Mutations on the Outcomes of Patients With Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2019, 37, 490–503. [Google Scholar] [CrossRef]
- Cui, M.; Gao, X.S.; Gu, X.; Guo, W.; Li, X.; Ma, M.; Qin, S.; Qi, X.; Xie, M.; Peng, C.; et al. BRCA2 mutations should be screened early and routinely as markers of poor prognosis: Evidence from 8,988 patients with prostate cancer. Oncotarget 2017, 8, 40222–40232. [Google Scholar] [CrossRef] [Green Version]
- Antonarakis, E.S.; Lu, C.; Luber, B.; Liang, C.; Wang, H.; Chen, Y.; Silberstein, J.L.; Piana, D.; Lai, Z.; Chen, Y.; et al. Germline DNA-repair genemutations and outcomes in men with metastatic castration-resistantprostate cancer receiving first-line abiraterone and enzalutamide. Eur. Urol. 2018, 74, 218–225. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Hussain, M.; Daignault-Newton, S.; Twardowski, P.W.; Albany, C.; Stein, M.N.; Kunju, L.P.; Siddiqui, J.; Wu, Y.M.; Robinson, D.; Lonigro, R.J.; et al. Targeting androgen receptor and DNA repair in metastatic castration-resistant prostate cancer: Results from NCI 9012. J. Clin. Oncol. 2018, 36, 991–999. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet. Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Carmichael, J.; Maza, M.L.D.F.; Rescigno, P.; Chandran, K.; de Bono, J.S. Targeting defective DNA repair in prostate cancer. Curr. Opin. Oncol. 2020, 32, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.M.; Cieslik, M.; Lonigro, R.J.; Vats, P.; Reimers, M.A.; Cao, X.; Ning, Y.; Wang, L.; Kunju, L.P.; de Sarkar, N.; et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell 2018, 173, 1770–1782. [Google Scholar] [CrossRef] [Green Version]
- Viswanathan, S.R.; Ha, G.; Hoff, A.M.; Wala, J.A.; Carrot-Zhang, J.; Whelan, C.W.; Haradhvala, N.J.; Freeman, S.S.; Reed, S.C.; Rhoades, J.; et al. Structural alterations driving castration-resistant prostate cancer revealed by linked-read genome sequencing. Cell 2018, 174, 433–447. [Google Scholar] [CrossRef] [Green Version]
- Kaur, H.; Salles, D.C.; Murali, S.; Hicks, J.L.; Nguyen, M.; Pritchard, C.C.; De Marzo, A.M.; Lanchbury, J.S.; Trock, B.J.; Isaacs, W.B.; et al. Genomic and Clinicopathologic Characterization of ATM-deficient Prostate Cancer. Clin. Cancer Res. 2020, 26, 4869–4881. [Google Scholar] [CrossRef]
- Blazek, D.; Kohoutek, J.; Bartholomeeusen, K.; Johansen, E.; Hulinkova, P.; Luo, Z.; Cimermancic, P.; Ule, J.; Peterlin, B.M. The Cyclin K/Cdk12complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes. Dev. 2011, 25, 2158–2172. [Google Scholar] [CrossRef] [Green Version]
- Nik-Zainal, S.; Davies, H.; Staaf, J.; Ramakrishna, M.; Glodzik, D.; Zou, X.; Martincorena, I.; Alexandrov, L.B.; Martin, S.; Wedge, D.C.; et al. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 2016, 534, 47–54. [Google Scholar] [CrossRef]
- Rescigno, P.; Gurel, B.; Pereira, R.; Crespo, M.; Rekowski, J.; Rediti, M.; Barrero, M.; Mateo, J.; Bianchini, D.; Messina, C.; et al. Characterizing CDK12-Mutated Prostate Cancers. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Nilbert, M.; Planck, M.; Fernebro, E.; Borg, A.; Johnson, A. Microsatellite instability is rare in rectal carcinomas and signifies hereditary cancer. Eur. J. Cancer 1999, 35, 942–945. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Morrissey, C.; Kumar, A.; Zhang, X.; Smith, C.; Coleman, I.; Salipante, S.J.; Milbank, J.; Yu, M.; Grady, W.M.; et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat. Commun. 2014, 5, 4988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedes, L.; Antonarakis, E.S.; Schweizer, M.T.; Mirkheshti, N.; Almutairi, F.; Park, J.C.; Glavaris, S.; Hicks, J.; Eisenberger, M.A.; De Marzo, A.M.; et al. MSH2 Loss in Primary Prostate Cancer. Clin. Cancer Res. 2017, 23, 6863–6874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, D.N.; Rescigno, P.; Liu, D.; Yuan, W.; Carreira, S.; Lambros, M.B.; Seed, G.; Mateo, J.; Riisnaes, R.; Mullane, S.; et al. Immunogenomic analyses associate immunological alterations with mismatch repair defects in prostate cancer. J. Clin. Invest. 2018, 128, 4441–4453. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, G.; Armenia, J.; Mazzu, Y.Z.; Nandakumar, S.; Stopsack, K.H.; Atiq, M.O.; Komura, K.; Jehane, L.; Hirani, R.; Chadalavada, K.; et al. Significance of BRCA2 and RB1 Co-loss in Aggressive Prostate Cancer Progression. Clin. Cancer Res. 2020, 26, 2047–2064. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, S.; Phillips, A.C.; Clark, P.A.; Stott, F.; Peters, G.; Ludwig, R.L.; Vousden, K.H. p14ARF links the tumour suppressors RB and p53. Nature 1998, 395, 124–125. [Google Scholar] [CrossRef]
- Mateo, J.; Seed, G.; Bertan, C.; Rescigno, P.; Dolling, D.; Figueiredo, I.; Miranda, S.; Nava Rodrigues, D.; Gurel, B.; Clarke, M.; et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J. Clin. Invest. 2020, 130, 1743–1751. [Google Scholar] [CrossRef]
- Stephenson, A.J.; Scardino, P.T.; Eastham, J.A.; Bianco, F.J., Jr.; Dotan, Z.A.; Fearn, P.A.; Kattan, M.W. Preoperative nomogram predicting the 10-year probability of prostate cancer recurrence after radical prostatectomy. J. Natl. Cancer Inst. 2006, 98, 715–717. [Google Scholar] [CrossRef] [Green Version]
- Van Den Eeden, S.K.; Lu, R.; Zhang, N.; Quesenberry, C.P., Jr.; Shan, J.; Han, J.S.; Tsiatis, A.C.; Leimpeter, A.D.; Lawrence, H.J.; Febbo, P.G.; et al. A Biopsy-based 17-gene Genomic Prostate Score as a Predictor of Metastases and Prostate Cancer Death in Surgically Treated Men with Clinically Localized Disease. Eur. Urol. 2018, 73, 129–138. [Google Scholar] [CrossRef]
- Cullen, J.; Rosner, I.L.; Brand, T.C.; Zhang, N.; Tsiatis, A.C.; Moncur, J.; Ali, A.; Chen, Y.; Knezevic, D.; Maddala, T.; et al. A Biopsy-based 17-gene Genomic Prostate Score Predicts Recurrence After Radical Prostatectomy and Adverse Surgical Pathology in a Racially Diverse Population of Men with Clinically Low- and Intermediate-risk Prostate Cancer. Eur. Urol. 2015, 68, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Spratt, D.E.; Zhang, J.; Santiago-Jiménez, M.; Dess, R.T.; Davis, J.W.; Den, R.B.; Dicker, A.P.; Kane, C.J.; Pollack, A.; Stoyanova, R.; et al. Development and Validation of a Novel Integrated Clinical-Genomic Risk Group Classification for Localized Prostate Cancer. J. Clin. Oncol. 2018, 36, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.C.; Tosoian, J.J.; Qi, J.; Kaye, D.; Johnson, A.; Linsell, S.; Montie, J.E.; Ghani, K.R.; Miller, D.C.; Wojno, K.; et al. Clinical Utility of Gene Expression Classifiers in Men With Newly Diagnosed Prostate Cancer. JCO Precis. Oncol. 2018, 2, PO.18.00163. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Wang, J.; Lampert, E.; Schlanger, S.; DePriest, A.D.; Hu, Q.; Gomez, E.C.; Murakam, M.; Glenn, S.T.; Conroy, J.; et al. Intratumoral and Intertumoral Genomic Heterogeneity of Multifocal Localized Prostate Cancer Impacts Molecular Classifications and Genomic Prognosticators. Eur. Urol. 2017, 71, 183–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eggener, S.E.; Rumble, R.B.; Armstrong, A.J.; Morgan, T.M.; Crispino, T.; Cornford, P.; van der Kwast, T.; Grignon, D.J.; Rai, A.J.; Agarwal, N.; et al. Molecular Biomarkers in Localized Prostate Cancer: ASCO Guideline. J. Clin. Oncol. 2020, 38, 1474–1494. [Google Scholar] [CrossRef] [PubMed]
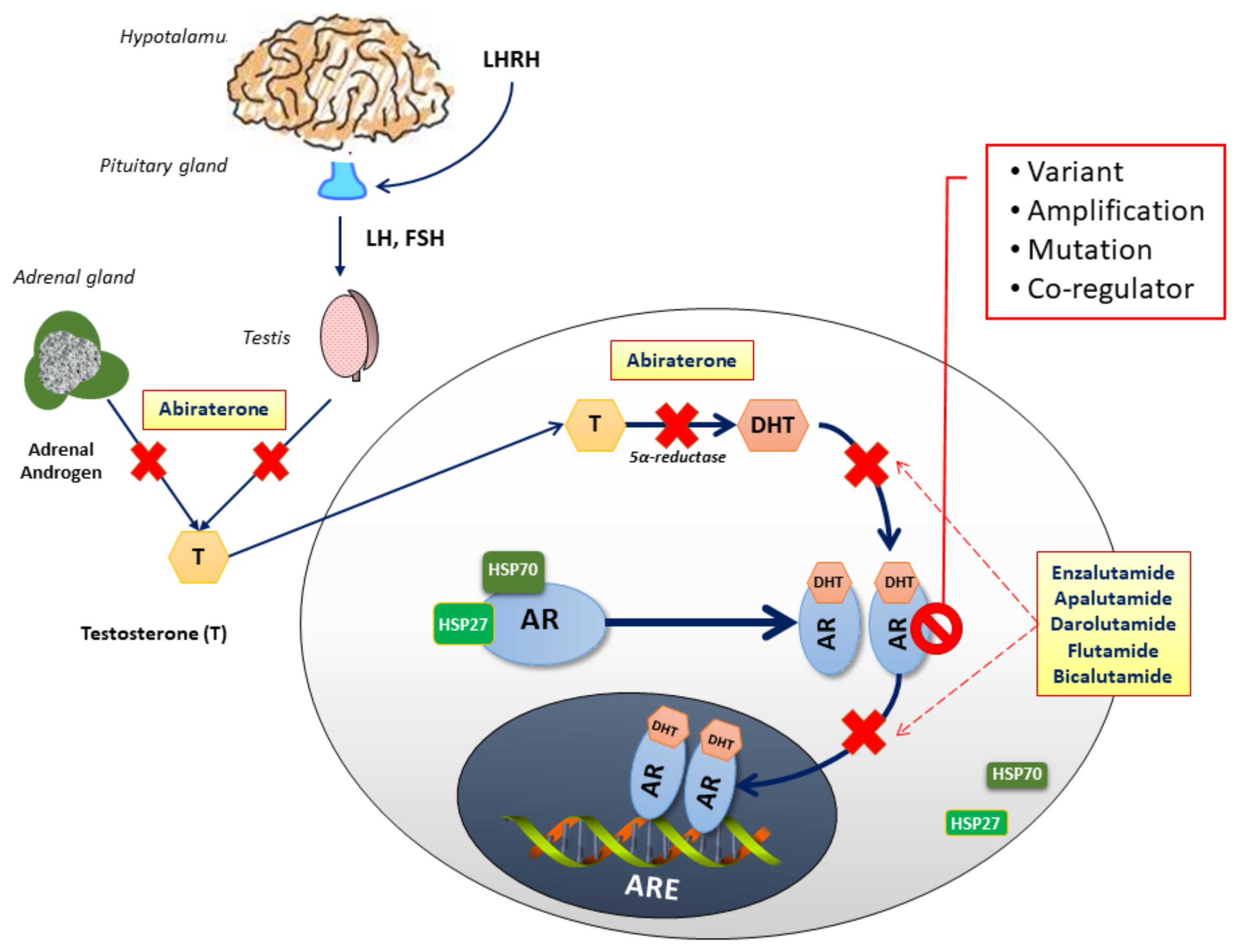
Figure 1.
Androgen-dependent signaling through the androgen receptor (AR). After its production in the testes ad the adrenal glands, testosterone (T) is converted by 5α-reductase to dihydrotestosterone (DHT), its active metabolite. Generally, androgens bind to AR, dissociating chaperone proteins, including members of the heat shock protein family (HSP27 and HSP70). Upon homodimerization, ligand-bound AR dimers translocate to the nucleus where they bind to androgen response elements (ARE), acting as transcription factors to downstream targets. AR-directed drugs are illustrated at their points of pathway alteration: Abiraterone disrupts the androgen biosynthesis inhibiting the 17α-hydroxylase/C17,20-lyase (CYP17), an enzime expressed in testicular, prostate, and adrenal tissue; Flutamide, and Bicalutamide, reversibly, and Enzalutamide, Apalutamide, Darolutamide, irreversibly, prevent testosterone binding to the AR, incapacitating its translocation to the nucleus.
Figure 1.
Androgen-dependent signaling through the androgen receptor (AR). After its production in the testes ad the adrenal glands, testosterone (T) is converted by 5α-reductase to dihydrotestosterone (DHT), its active metabolite. Generally, androgens bind to AR, dissociating chaperone proteins, including members of the heat shock protein family (HSP27 and HSP70). Upon homodimerization, ligand-bound AR dimers translocate to the nucleus where they bind to androgen response elements (ARE), acting as transcription factors to downstream targets. AR-directed drugs are illustrated at their points of pathway alteration: Abiraterone disrupts the androgen biosynthesis inhibiting the 17α-hydroxylase/C17,20-lyase (CYP17), an enzime expressed in testicular, prostate, and adrenal tissue; Flutamide, and Bicalutamide, reversibly, and Enzalutamide, Apalutamide, Darolutamide, irreversibly, prevent testosterone binding to the AR, incapacitating its translocation to the nucleus.

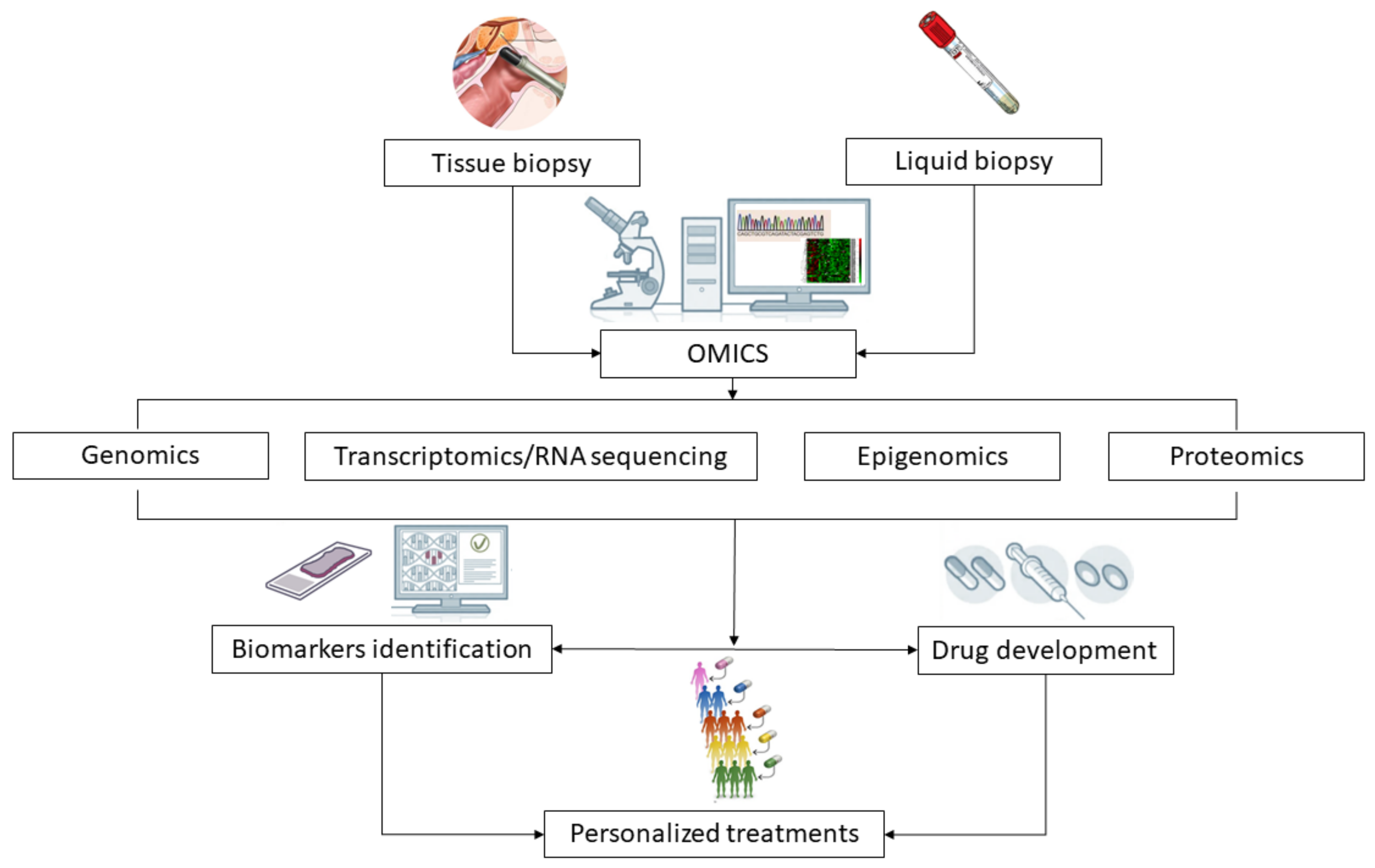
Figure 2.
Clinical significance of omics-driven approaches applied to tissue and liquid biopsy analysis in prostate cancer research. The analysis of clinical samples (tissue biopsies, plasma, urine) through the use of novel techniques (such as genomics, transcriptomics, epigenomics and proteomics) allows the identification of prognostic and predictive biomarkers and may guide the development of drugs targeting molecular pathways altered in cancer cells. These advances will contribute to improve patients and treatments selection and to guide personalized interventions for the management of prostate cancer.
Figure 2.
Clinical significance of omics-driven approaches applied to tissue and liquid biopsy analysis in prostate cancer research. The analysis of clinical samples (tissue biopsies, plasma, urine) through the use of novel techniques (such as genomics, transcriptomics, epigenomics and proteomics) allows the identification of prognostic and predictive biomarkers and may guide the development of drugs targeting molecular pathways altered in cancer cells. These advances will contribute to improve patients and treatments selection and to guide personalized interventions for the management of prostate cancer.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Conteduca, V.; Mosca, A.; Brighi, N.; de Giorgi, U.; Rescigno, P. New Prognostic Biomarkers in Metastatic Castration-Resistant Prostate Cancer. Cells 2021, 10, 193. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010193
AMA Style
Conteduca V, Mosca A, Brighi N, de Giorgi U, Rescigno P. New Prognostic Biomarkers in Metastatic Castration-Resistant Prostate Cancer. Cells. 2021; 10(1):193. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010193
Chicago/Turabian StyleConteduca, Vincenza, Alessandra Mosca, Nicole Brighi, Ugo de Giorgi, and Pasquale Rescigno. 2021. "New Prognostic Biomarkers in Metastatic Castration-Resistant Prostate Cancer" Cells 10, no. 1: 193. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010193
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.