
Why Do We Not Assess Sympathetic Nervous System Activity in Heart Failure Management: Might GRK2 Serve as a New Biomarker?

 , ,
, ,
Abstract
:1. Introduction
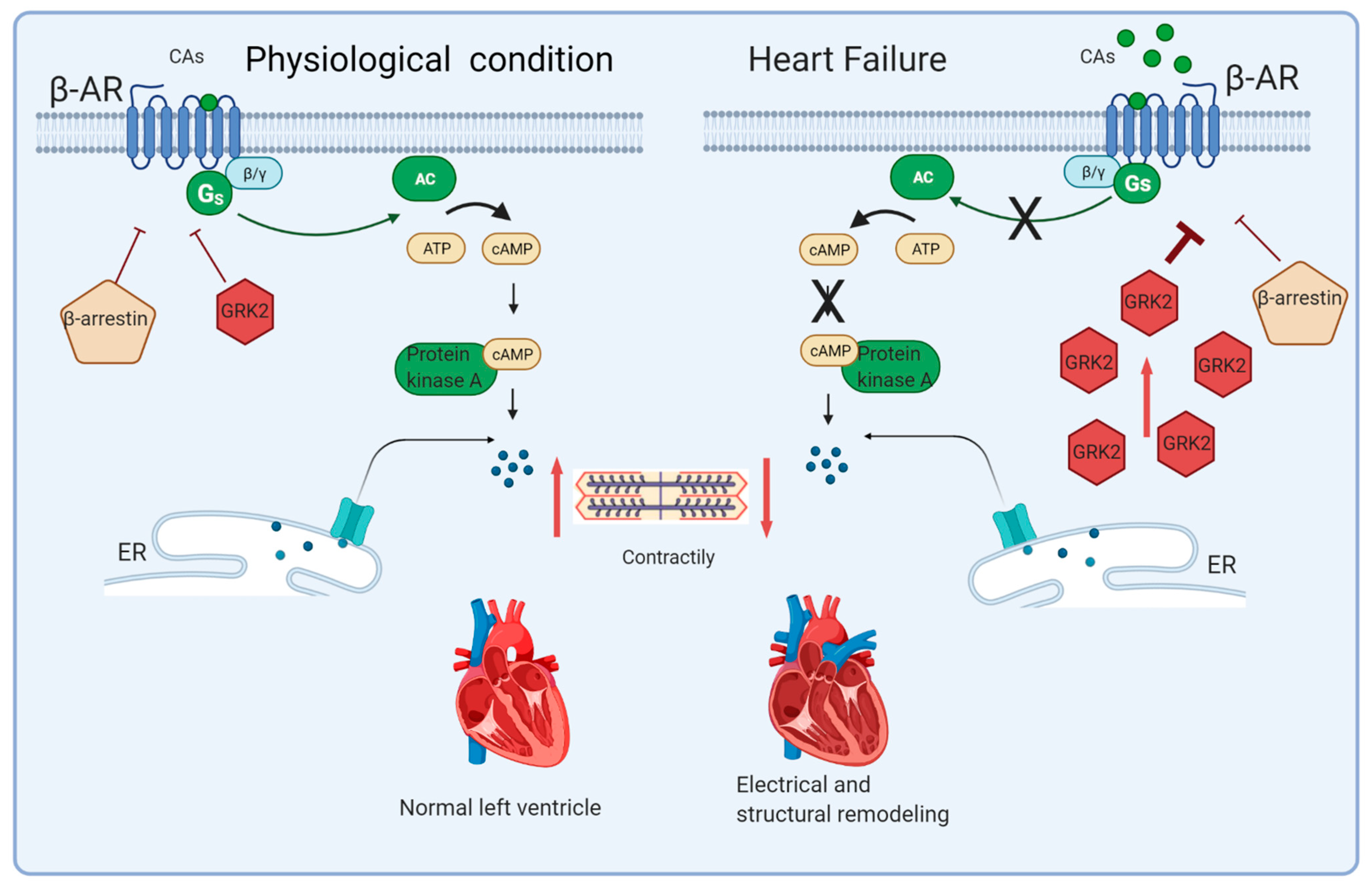
2. Sympathetic Nervous System Hyperactivity in HF Pathophysiology
3. How to Assess Sympathetic Nervous Activity: Methods
4. Biomarkers of HF
Biomarkers of Sympathetic Nervous Activity
5. Lymphocyte GRK2 as Biomarkers of HF
6. Future Perspectives
7. Conclusive Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Savarese, G.; Lund, L.H. Global Public Health Burden of Heart Failure. Card. Fail. Rev. 2017, 3, 7–11. [Google Scholar] [CrossRef]
- Lippi, G.; Sanchis-Gomar, F. Global epidemiology and future trends of heart failure. AME Med. J. 2020, 5, 15. [Google Scholar] [CrossRef]
- Dharmarajan, K.; Rich, M.W. Epidemiology, Pathophysiology, and Prognosis of Heart Failure in Older Adults. Hear. Fail. Clin. 2017, 13, 417–426. [Google Scholar] [CrossRef] [Green Version]
- De Lucia, C.; Eguchi, A.; Koch, W.J. New Insights in Cardiac β-Adrenergic Signaling During Heart Failure and Aging. Front. Pharmacol. 2018, 9, 904. [Google Scholar] [CrossRef] [Green Version]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; (Uk), J.G.F.C.; (Uk), A.J.S.C.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur. J. Hear. Fail. 2016, 18, 891–975. [Google Scholar] [CrossRef]
- Bencivenga, L.; Liccardo, D.; Napolitano, C.; Visaggi, L.; Rengo, G.; Leosco, D. β-Adrenergic receptor signaling and heart failure: From bench to bedside. Heart Fail. Clin. 2019, 15, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Triposkiadis, F.; Karayannis, G.; Giamouzis, G.; Skoularigis, J.; Louridas, G.; Butler, J. The Sympathetic Nervous System in Heart Failure. J. Am. Coll. Cardiol. 2009, 54, 1747–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santulli, G.; Iaccarino, G. Adrenergic signaling in heart failure and cardiovascular aging. Maturitas 2016, 93, 65–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannavo, A.; Komici, K.; Bencivenga, L.; D’Amico, M.L.; Gambino, G.; Liccardo, D.; Ferrara, N.; Rengo, G. GRK2 as a therapeutic target for heart failure. Expert Opin. Ther. Targets 2018, 22, 75–83. [Google Scholar] [CrossRef]
- Florea, V.G.; Cohn, J.N. The Autonomic Nervous System and Heart Failure. Circ. Res. 2014, 114, 1815–1826. [Google Scholar] [CrossRef] [Green Version]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenal adrenoceptors in heart failure: Fine-tuning cardiac stimulation. Trends Mol. Med. 2007, 13, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Moniotte, S.; Kobzik, L.; Feron, O.; Trochu, J.-N.; Gauthier, C.; Balligand, J.-L. Upregulation of β 3 -Adrenoceptors and Altered Contractile Response to Inotropic Amines in Human Failing Myocardium. Circulation 2001, 103, 1649–1655. [Google Scholar] [CrossRef] [Green Version]
- Myagmar, B.-E.; Flynn, J.M.; Cowley, P.M.; Swigart, P.M.; Montgomery, M.D.; Thai, K.; Nair, D.R.; Gupta, R.; Deng, D.X.; Hosoda, C.; et al. Adrenergic Receptors in Individual Ventricular Myocytes. Circ. Res. 2017, 120, 1103–1115. [Google Scholar] [CrossRef] [Green Version]
- Gauthier, C.; Leblais, V.; Kobzik, L.; Trochu, J.N.; Khandoudi, N.; Bril, A.; Balligand, J.-L.; Le Marec, H. The negative inotropic effect of beta3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J. Clin. Investig. 1998, 102, 1377–1384. [Google Scholar] [CrossRef]
- Lefkowitz, R.J. Transduction of Receptor Signals by -Arrestins. Science 2005, 308, 512–517. [Google Scholar] [CrossRef]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic Nervous System in Heart Failure. Circ. Res. 2013, 113, 739–753. [Google Scholar] [CrossRef]
- Woodall, M.C.; Ciccarelli, M.; Woodall, B.P.; Koch, W.J. G protein-coupled receptor kinase 2: A link between myocardial contractile function and cardiac metabolism. Circ. Res. 2014, 114, 1661–1670. [Google Scholar] [CrossRef] [PubMed]
- Woodall, B.P.; Gresham, K.S.; Woodall, M.A.; Valenti, M.-C.; Cannavo, A.; Pfleger, J.; Chuprun, J.K.; Drosatos, K.; Koch, W.J. Alteration of myocardial GRK2 produces a global metabolic phenotype. JCI Insight 2019, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Cannavo, A.; Marzano, F.; Elia, A.; Liccardo, D.; Bencivenga, L.; Gambino, G.; Perna, C.; Rapacciuolo, A.; Cittadini, A.; Ferrara, N.; et al. Aldosterone Jeopardizes Myocardial Insulin and β-Adrenergic Receptor Signaling via G Protein-Coupled Receptor Kinase 2. Front. Pharmacol. 2019, 10, 888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murga, C.; Arcones, A.C.; Cruces-Sande, M.; Briones, A.M.; Salaices, M.; Mayor, F., Jr. G Protein-Coupled Receptor Kinase 2 (GRK2) as a Potential Therapeutic Target in Cardiovascular and Metabolic Diseases. Front. Pharmacol. 2019, 10, 112. [Google Scholar] [CrossRef] [Green Version]
- Thireau, J.; Karam, S.; Roberge, S.; Roussel, J.; Aimond, F.; Cassan, C.; Gac, A.; Babuty, D.; Le Guennec, J.-Y.; Lacampagne, A.; et al. β-Adrenergic blockade combined with subcutaneous B-type natriuretic peptide: A promising approach to reduce ventricular arrhythmia in heart failure? Heart 2014, 100, 833–841. [Google Scholar] [CrossRef] [Green Version]
- Cipolletta, E.; Campanile, A.; Santulli, G.; Sanzari, E.; Leosco, D.; Campiglia, P.; Trimarco, B.; Iaccarino, G. The G protein coupled receptor kinase 2 plays an essential role in beta-adrenergic receptor-induced insulin resistance. Cardiovasc. Res. 2009, 84, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Watari, K.; Nakaya, M.; Kurose, H. Multiple functions of G protein-coupled receptor kinases. J. Mol. Signal. 2014, 9, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orso, F.; Baldasseroni, S.; Maggioni, A.P. Heart Rate in Coronary Syndromes and Heart Failure. Prog. Cardiovasc. Dis. 2009, 52, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Fox, K.; Ford, I.; Steg, P.G.; Tendera, M.; Ferrari, R. Ivabradine for patients with stable coronary artery disease and left-ventricular systolic dysfunction (BEAUTIFUL): A randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 807–816. [Google Scholar] [CrossRef]
- Borovac, J.A.; D’Amario, D.; Bozic, J.; Glavas, D. Sympathetic nervous system activation and heart failure: Current state of evidence and the pathophysiology in the light of novel biomarkers. World J. Cardiol. 2020, 12, 373–408. [Google Scholar] [CrossRef]
- Ewing, D.J.; Martyn, C.N.; Young, R.J.; Clarke, B.F. The Value of Cardiovascular Autonomic Function Tests: 10 Years Experience in Diabetes. Diabetes Care 1985, 8, 491–498. [Google Scholar] [CrossRef]
- Patel, H.; Ozdemir, B.A.; Patel, M.; Xiao, H.B.; Poole-Wilson, P.A.; Rosen, S.D. Impairment of autonomic reactivity is a feature of heart failure whether or not the left ventricular ejection fraction is normal. Int. J. Cardiol. 2011, 151, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, F.; Ginsberg, J.P. An Overview of Heart Rate Variability Metrics and Norms. Front. Public Health 2017, 5, 258. [Google Scholar] [CrossRef] [Green Version]
- Patel, V.N.; Pierce, B.R.; Bodapati, R.K.; Brown, D.L.; Ives, D.G.; Stein, P.K. Association of holter-derived heart rate variability parameters with the development of congestive heart failure in the cardiovascular health study. JACC Hear. Fail. 2017, 5, 423–431. [Google Scholar] [CrossRef]
- Femminella, G.D.; Rengo, G.; Komici, K.; Iacotucci, P.; Petraglia, L.; Pagano, G.; De Lucia, C.; Canonico, V.; Bonaduce, D.; Leosco, D.; et al. Autonomic Dysfunction in Alzheimer’s Disease: Tools for Assessment and Review of the Literature. J. Alzheimer’s Dis. 2014, 42, 369–377. [Google Scholar] [CrossRef]
- Hasking, G.J.; Esler, M.D.; Jennings, G.L.; Burton, D.; Johns, J.A.; Korner, P.I. Norepinephrine spillover to plasma in patients with congestive heart failure: Evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation 1986, 73, 615–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaye, D.M.; Lefkovits, J.; Jennings, G.L.; Bergin, P.; Broughton, A.; Esler, M.D. Adverse consequences of high sympathetic nervous activity in the failing human heart. J. Am. Coll. Cardiol. 1995, 26, 1257–1263. [Google Scholar] [CrossRef] [Green Version]
- Petersson, M.; Friberg, P.; Eisenhofer, G.; Lambert, G.; Rundqvist, B. Long-term outcome in relation to renal sympathetic activity in patients with chronic heart failure. Eur. Hear. J. 2005, 26, 906–913. [Google Scholar] [CrossRef]
- Tygesen, H.; Rundqvist, B.; Waagstein, F.; Wennerblom, B. Heart rate variability measurement correlates with cardiac norepinephrine spillover in congestive heart failure. Am. J. Cardiol. 2001, 87, 1308–1311. [Google Scholar] [CrossRef]
- Grassi, G.; Colombo, M.; Seravalle, G.; Spaziani, D.; Mancia, G. Dissociation Between Muscle and Skin Sympathetic Nerve Activity in Essential Hypertension, Obesity, and Congestive Heart Failure. Hypertension 1998, 31, 64–67. [Google Scholar] [CrossRef] [Green Version]
- Leimbach, W.N.; Wallin, B.G.; Victor, R.G.; Aylward, P.E.; Sundlöf, G.; Mark, A.L. Direct evidence from intraneural recordings for increased central sympathetic outflow in patients with heart failure. Circulation 1986, 73, 913–919. [Google Scholar] [CrossRef] [Green Version]
- Ferguson, D.W.; Berg, W.J.; Sanders, J.S.; Kempf, J.S. Clinical and hemodynamic correlates of sympathetic nerve activity in normal humans and patients with heart failure: Evidence from direct micronenrographic recordings. J. Am. Coll. Cardiol. 1990, 16, 1125–1134. [Google Scholar] [CrossRef] [Green Version]
- Barretto, A.C.; Santos, A.C.; Munhoz, R.; Rondon, M.U.; Franco, F.G.; Trombetta, I.C.; Roveda, F.; De Matos, L.N.; Braga, A.M.; Middlekauff, H.R.; et al. Increased muscle sympathetic nerve activity predicts mortality in heart failure patients. Int. J. Cardiol. 2009, 135, 302–307. [Google Scholar] [CrossRef]
- Jacobson, A.F.; Senior, R.; Cerqueira, M.D.; Wong, N.D.; Thomas, G.S.; Lopez, V.A.; Agostini, D.; Weiland, F.; Chandna, H.; Narula, J. Myocardial Iodine-123 Meta-Iodobenzylguanidine Imaging and Cardiac Events in Heart Failure. J. Am. Coll. Cardiol. 2010, 55, 2212–2221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komici, K.; Bencivenga, L.; Paolillo, S.; Gargiulo, P.; Formisano, R.; Assante, R.; Nappi, C.; Marsico, F.; D’Antonio, A.; De Simini, G.; et al. Impact of body mass index on cardiac adrenergic derangement in heart failure patients: A 123I-mIBG imaging study. Eur. J. Nucl. Med. Mol. Imaging 2019, 47, 1713–1721. [Google Scholar] [CrossRef]
- Narula, J.; Gerson, M.; Thomas, G.S.; Cerqueira, M.D.; Jacobson, A.F. 123I-MIBG Imaging for Prediction of Mortality and Potentially Fatal Events in Heart Failure: The ADMIRE-HFX Study. J. Nucl. Med. 2015, 56, 1011–1018. [Google Scholar] [CrossRef] [Green Version]
- Nakata, T.; Nakajima, K.; Yamashina, S.; Yamada, T.; Momose, M.; Kasama, S.; Matsui, T.; Matsuo, S.; Travin, M.I.; Jacobson, A.F. A Pooled Analysis of Multicenter Cohort Studies of 123I-mIBG Imaging of Sympathetic Innervation for Assessment of Long-Term Prognosis in Heart Failure. JACC: Cardiovasc. Imaging 2013, 6, 772–784. [Google Scholar] [CrossRef] [Green Version]
- Aikawa, T.; Naya, M.; Obara, M.; Oyama-Manabe, N.; Manabe, O.; Magota, K.; Ito, Y.M.; Katoh, C.; Tamaki, N. Regional interaction between myocardial sympathetic denervation, contractile dysfunction, and fibrosis in heart failure with preserved ejection fraction: 11C-hydroxyephedrine PET study. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 1897–1905. [Google Scholar] [CrossRef]
- Nozaki, K.; Hamazaki, N.; Yamamoto, S.; Kamiya, K.; Tanaka, S.; Ichikawa, T.; Nakamura, T.; Yamashita, M.; Maekawa, E.; Matsunaga, A.; et al. Prognostic value of pupil area for all-cause mortality in patients with heart failure. ESC Hear. Fail. 2020, 7, 3067–3074. [Google Scholar] [CrossRef] [PubMed]
- Nadar, S.K.; Shaikh, M.M. Biomarkers in Routine Heart Failure Clinical Care. Card. Fail. Rev. 2019, 5, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, N.E.; Januzzi, J.L. Established and Emerging Roles of Biomarkers in Heart Failure. Circ. Res. 2018, 123, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Altay, H. Biomarkers and Heart Failure. In Biomarker - Indicator of Abnormal Physiological Process; IntechOpen: London, UK, 2018. [Google Scholar]
- Gaggin, H.K.; Januzzi, J.L. Biomarkers and diagnostics in heart failure. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2013, 1832, 2442–2450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, D.C.; Diamandis, E.P.; Januzzi, J.L.; Maisel, A.; Jaffe, A.S.; Clerico, A. Natriuretic Peptides in Heart Failure. Clin. Chem. 2014, 60, 1040–1046. [Google Scholar] [CrossRef] [Green Version]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin–Neprilysin Inhibition versus Enalapril in Heart Failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [Green Version]
- Fonarow, G.C.; Peacock, W.F.; Phillips, C.O.; Givertz, M.M.; Lopatin, M. Admission B-Type Natriuretic Peptide Levels and In-Hospital Mortality in Acute Decompensated Heart Failure. J. Am. Coll. Cardiol. 2007, 49, 1943–1950. [Google Scholar] [CrossRef] [Green Version]
- Doust, J.A.; Pietrzak, E.; Dobson, A.; Glasziou, P. How well does B-type natriuretic peptide predict death and cardiac events in patients with heart failure: Systematic review. BMJ 2005, 330, 625. [Google Scholar] [CrossRef] [Green Version]
- Michtalik, H.J.; Yeh, H.-C.; Campbell, C.Y.; Haq, N.; Park, H.; Clarke, W.; Brotman, D.J. Acute Changes in N-Terminal Pro-B-Type Natriuretic Peptide During Hospitalization and Risk of Readmission and Mortality in Patients With Heart Failure. Am. J. Cardiol. 2011, 107, 1191–1195. [Google Scholar] [CrossRef]
- Stanek, B.; Frey, B.; Hülsmann, M.; Berger, R.; Sturm, B.; Strametz-Juranek, J.; Bergler-Klein, J.; Moser, P.; Bojic, A.; Hartter, E.; et al. Prognostic evaluation of neurohumoral plasma levels before and during beta-blocker therapy in advanced left ventricular dysfunction. J. Am. Coll. Cardiol. 2001, 38, 436–442. [Google Scholar] [CrossRef]
- Yoshimura, M.; Mizuno, Y.; Nakayama, M.; Sakamoto, T.; Sugiyama, S.; Kawano, H.; Soejima, H.; Hirai, N.; Saito, Y.; Nakao, K.; et al. B-type natriuretic peptide as a marker of the effects of enalapril in patients with heart failure. Am. J. Med. 2002, 112, 716–720. [Google Scholar] [CrossRef]
- Berry, C.; Murphy, N.; De Vito, G.; Galloway, S.; Seed, A.; Fisher, C.; Sattar, N.; Vallance, P.; Hillis, W.S.; McMurray, J. Effects of aldosterone receptor blockade in patients with mild-moderate heart failure taking a beta-blocker. Eur. J. Hear. Fail. 2007, 9, 429–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menardi, E.; Vado, A.; Rossetti, G.; Racca, E.; Conte, E.; Deorsola, A.; Bobbio, M.; Feola, M. Cardiac Resynchronization Therapy Modifies the Neurohormonal Profile, Hemodynamic and Functional Capacity in Heart Failure Patients. Arch. Med Res. 2008, 39, 702–708. [Google Scholar] [CrossRef] [PubMed]
- Maisel, A.; Mueller, C.; Nowak, R.; Peacock, W.F.; Landsberg, J.W.; Ponikowski, P.; Mockel, M.; Hogan, C.; Wu, A.H.B.; Richards, M.; et al. Mid-Region Pro-Hormone Markers for Diagnosis and Prognosis in Acute Dyspnea. J. Am. Coll. Cardiol. 2010, 55, 2062–2076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.-N.; Januzzi, J.L. Natriuretic Peptide Testing in Heart Failure. Circulation 2011, 123, 2015–2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayes-Genis, A.; Lloyd-Jones, D.M.; Van Kimmenade, R.R.J.; Lainchbury, J.G.; Richards, A.M.; Ordoñez-Llanos, J.; Santaló, M.; Pinto, Y.M.; Januzzi, J.L. Effect of Body Mass Index on Diagnostic and Prognostic Usefulness of Amino-Terminal Pro–Brain Natriuretic Peptide in Patients With Acute Dyspnea. Arch. Intern. Med. 2007, 167, 400–407. [Google Scholar] [CrossRef] [Green Version]
- Voors, A.; Dorhout, B.; Van Der Meer, P. The potential role of valsartan + AHU377 (LCZ696) in the treatment of heart failure. Expert Opin. Investig. Drugs 2013, 22, 1041–1047. [Google Scholar] [CrossRef]
- Peacock, W.F.; De Marco, T.; Fonarow, G.C.; Diercks, D.B.; Wynne, J.; Apple, F.S.; Wu, A.H. Cardiac Troponin and Outcome in Acute Heart Failure. N. Engl. J. Med. 2008, 358, 2117–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felker, G.M.; Mentz, R.J.; Teerlink, J.R.; Voors, A.A.; Pang, P.S.; Ponikowski, P.; Greenberg, B.H.; Filippatos, G.; Davison, B.A.; Cotter, G.; et al. Serial high sensitivity cardiac troponin T measurement in acute heart failure: Insights from the RELAX-AHF study. Eur. J. Hear. Fail. 2015, 17, 1262–1270. [Google Scholar] [CrossRef]
- Latini, R.; Masson, S.; Anand, I.S.; Missov, E.; Carlson, M.; Vago, T.; Angelici, L.; Barlera, S.; Parrinello, G.; Maggioni, A.P.; et al. Prognostic Value of Very Low Plasma Concentrations of Troponin T in Patients With Stable Chronic Heart Failure. Circulation 2007, 116, 1242–1249. [Google Scholar] [CrossRef] [Green Version]
- McCullough, P.; Olobatoke, A.; Olobatoke, T.E. Galectin-3: A novel blood test for the evaluation and management of patients with heart failure. Rev. Cardiovasc. Med. 2011, 12, 200–210. [Google Scholar]
- Michalski, B.; Trzciński, P.; Kupczyńska, K.; Miśkowiec, D.; Pęczek, ę.; Nawrot, B.; Lipiec, P.; Kasprzak, J.D. The differences in the relationship between diastolic dysfunction, selected biomarkers and collagen turn-over in heart failure patients with preserved and reduced ejection fraction. Cardiol. J. 2017, 24, 35–42. [Google Scholar] [CrossRef]
- Lok, D.J.; Lok, S.I.; De La Porte, P.W.B.-A.; Badings, E.; Lipsic, E.; Van Wijngaarden, J.; De Boer, R.A.; Van Veldhuisen, D.J.; Van Der Meer, P. Galectin-3 is an independent marker for ventricular remodeling and mortality in patients with chronic heart failure. Clin. Res. Cardiol. 2012, 102, 103–110. [Google Scholar] [CrossRef]
- Sharma, U.C.; Pokharel, S.; Van Brakel, T.J.; Van Berlo, J.H.; Cleutjens, J.P.M.; Schroen, B.; Andreé, S.; Crijns, H.J.G.M.; Gabius, H.-J.; Maessen, J.; et al. Galectin-3 Marks Activated Macrophages in Failure-Prone Hypertrophied Hearts and Contributes to Cardiac Dysfunction. Circulation 2004, 110, 3121–3128. [Google Scholar] [CrossRef] [PubMed]
- De Boer, R.A.; Lok, D.J.A.; Jaarsma, T.; Van Der Meer, P.; Voors, A.A.; Hillege, H.L.; Van Veldhuisen, D.J. Predictive value of plasma galectin-3 levels in heart failure with reduced and preserved ejection fraction. Ann. Med. 2010, 43, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Komici, K.; Gnemmi, I.; Bencivenga, L.; Vitale, D.F.; Rengo, G.; Di Stefano, A.; Eleuteri, E. Impact of Galectin-3 Circulating Levels on Frailty in Elderly Patients with Systolic Heart Failure. J. Clin. Med. 2020, 9, 2229. [Google Scholar] [CrossRef]
- Martínez-Martínez, E.; López-Ándres, N.; Jurado-López, R.; Rousseau, E.; Bartolomé, M.V.; Fernández-Celis, A.; Rossignol, P.; Islas, F.; Antequera, A.; Prieto, S.; et al. Galectin-3 Participates in Cardiovascular Remodeling Associated With Obesity. Hypertension 2015, 66, 961–969. [Google Scholar] [CrossRef]
- Piek, A.; Du, W.; De Boer, R.A.; Silljé, H.H.W. Novel heart failure biomarkers: Why do we fail to exploit their potential? Crit. Rev. Clin. Lab. Sci. 2018, 55, 246–263. [Google Scholar] [CrossRef]
- Shah, R.V.; Truong, Q.A.; Gaggin, H.K.; Pfannkuche, J.; Hartmann, O.; Januzzi, J.L. Mid-regional pro-atrial natriuretic peptide and pro-adrenomedullin testing for the diagnostic and prognostic evaluation of patients with acute dyspnoea. Eur. Hear. J. 2012, 33, 2197–2205. [Google Scholar] [CrossRef] [Green Version]
- Sanada, S.; Hakuno, D.; Higgins, L.J.; Schreiter, E.R.; McKenzie, A.N.; Lee, R.T. IL-33 and ST2 comprise a critical biomechanically induced and cardioprotective signaling system. J. Clin. Investig. 2007, 117, 1538–1549. [Google Scholar] [CrossRef] [Green Version]
- Mueller, T.; Dieplinger, B.; Gegenhuber, A.; Poelz, W.; Pacher, R.; Haltmayer, M. Increased Plasma Concentrations of Soluble ST2 are Predictive for 1-Year Mortality in Patients with Acute Destabilized Heart Failure. Clin. Chem. 2008, 54, 752–756. [Google Scholar] [CrossRef] [Green Version]
- Nagy, A.I.; Hage, C.; Merkely, B.; Donal, E.; Daubert, J.-C.; Linde, C.; Lund, L.H.; Manouras, A. Left atrial rather than left ventricular impaired mechanics are associated with the pro-fibrotic ST2 marker and outcomes in heart failure with preserved ejection fraction. J. Intern. Med. 2018, 283, 380–391. [Google Scholar] [CrossRef] [Green Version]
- Januzzi, J.L.; Peacock, W.F.; Maisel, A.S.; Chae, C.U.; Jesse, R.L.; Baggish, A.L.; O’Donoghue, M.; Sakhuja, R.; Chen, A.A.; Van Kimmenade, R.R.; et al. Measurement of the Interleukin Family Member ST2 in Patients With Acute Dyspnea. J. Am. Coll. Cardiol. 2007, 50, 607–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bayes-Genis, A.; Zamora, E.; De Antonio, M.; Galán, A.; Vila, J.; Urrutia, A.; Díez, C.; Coll, R.; Altimir, S.; Lupón, J. Soluble ST2 Serum Concentration and Renal Function in Heart Failure. J. Card. Fail. 2013, 19, 768–775. [Google Scholar] [CrossRef]
- Homsak, E.; Ekart, R. ST2 as a novel prognostic marker in end-stage renal disease patients on hemodiafiltration. Clin. Chim. Acta 2018, 477, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, E.A.; Rozentryt, P.; Witkowska, A.; Nowak, J.; Hartmann, O.; Ponikowska, B.; Borodulin-Nadzieja, L.; Banasiak, W.; Polonski, L.; Filippatos, G.; et al. Iron deficiency: An ominous sign in patients with systolic chronic heart failure. Eur. Hear. J. 2010, 31, 1872–1880. [Google Scholar] [CrossRef]
- Jankowska, E.A.; Tkaczyszyn, M.; Suchocki, T.; Drozd, M.; Von Haehling, S.; Doehner, W.; Banasiak, W.; Filippatos, G.; Anker, S.D.; Ponikowski, P. Effects of intravenous iron therapy in iron-deficient patients with systolic heart failure: A meta-analysis of randomized controlled trials. Eur. J. Hear. Fail. 2016, 18, 786–795. [Google Scholar] [CrossRef]
- De Lucia, C.; Komici, K.; Borghetti, G.; Femminella, G.D.; Bencivenga, L.; Cannavo, A.; Corbi, G.; Ferrara, N.; Houser, S.R.; Koch, W.J.; et al. microRNA in Cardiovascular Aging and Age-Related Cardiovascular Diseases. Front. Med. 2017, 4, 74. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Ma, F.; Zhang, Y.; Wang, C.; Qiu, D.; Zhou, K.; Hua, Y.; Li, Y. miRNAs as biomarkers for diagnosis of heart failure. Medicine 2017, 96, e6825. [Google Scholar] [CrossRef] [PubMed]
- Cohn, J.N.; Levine, T.B.; Olivari, M.T.; Garberg, V.; Lura, D.; Francis, G.S.; Simon, A.B.; Rector, T. Plasma Norepinephrine as a Guide to Prognosis in Patients with Chronic Congestive Heart Failure. N. Engl. J. Med. 1984, 311, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Anand, I.S.; Fisher, L.D.; Chiang, Y.-T.; Latini, R.; Masson, S.; Maggioni, A.P.; Glazer, R.D.; Tognoni, G.; Cohn, J.N. Changes in Brain Natriuretic Peptide and Norepinephrine Over Time and Mortality and Morbidity in the Valsartan Heart Failure Trial (Val-HeFT). Circulation 2003, 107, 1278–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Givertz, M.M.; Braunwald, E. Neurohormones in heart failure: Predicting outcomes, optimizing care. Eur. Hear. J. 2004, 25, 281–282. [Google Scholar] [CrossRef]
- Hjemdahl, P. Plasma catecholamines—Analytical challenges and physiological limitations. Baillière’s Clin. Endocrinol. Metab. 1993, 7, 307–353. [Google Scholar] [CrossRef]
- Ajijola, O.A.; Chatterjee, N.A.; Gonzales, M.J.; Gornbein, J.; Liu, K.; Li, D.; Paterson, D.J.; Shivkumar, K.; Singh, J.P.; Herring, N. Coronary Sinus Neuropeptide Y Levels and Adverse Outcomes in Patients With Stable Chronic Heart Failure. JAMA Cardiol. 2020, 5, 318. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.J.; Cox, H.S.; Lambert, G.W.; Kaye, D.M.; Jennings, G.L.; Meredith, I.T.; Esler, M.D. Region-Specific Neuropeptide Y Overflows at Rest and During Sympathetic Activation in Humans. Hypertension 1997, 29, 137–143. [Google Scholar] [CrossRef]
- Herring, N.; Cranley, J.; Lokale, M.N.; Li, D.; Shanks, J.; Alston, E.N.; Girard, B.M.; Carter, E.; Parsons, R.L.; Habecker, B.A.; et al. The cardiac sympathetic co-transmitter galanin reduces acetylcholine release and vagal bradycardia: Implications for neural control of cardiac excitability. J. Mol. Cell. Cardiol. 2012, 52, 667–676. [Google Scholar] [CrossRef] [Green Version]
- Smith-White, M.; Iismaa, T.P.; Potter, E.K. Galanin and neuropeptide Y reduce cholinergic transmission in the heart of the anaesthetised mouse. Br. J. Pharmacol. 2003, 140, 170–178. [Google Scholar] [CrossRef] [Green Version]
- Gür, D. Özkaramanlı; Savaş, G.; Akyüz, A.; Alpsoy, Şeref Role of sympathetic cotransmitter galanin on autonomic balance in heart failure: An active player or a bystander? Anatol. J. Cardiol. 2017, 18, 281–288. [Google Scholar] [CrossRef]
- Ceconi, C.; Ferrari, R.; Bachetti, T.; Opasich, C.; Volterrani, M.; Colombo, B.; Parrinello, G.; Corti, A. Chromogranin A in heart failure. A novel neurohumoral factor and a predictor for mortality. Eur. Hear. J. 2002, 23, 967–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahata, S.K.; Mahata, M.; Fung, M.M.; O’Connor, D.T. Catestatin: A multifunctional peptide from chromogranin A. Regul. Pept. 2010, 162, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Borovac, J.A.; Glavas, D.; Grabovac, Z.S.; Domic, D.S.; D’Amario, D.; Bozic, J. Catestatin in Acutely Decompensated Heart Failure Patients: Insights from the CATSTAT-HF Study. J. Clin. Med. 2019, 8, 1132. [Google Scholar] [CrossRef] [Green Version]
- Iaccarino, G.; Tomhave, E.D.; Lefkowitz, R.J.; Koch, W.J. Reciprocal In Vivo Regulation of Myocardial G Protein–Coupled Receptor Kinase Expression by β-Adrenergic Receptor Stimulation and Blockade. Circulation 1998, 98, 1783–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, S.; Hein, L.; Wiesmann, F.; Lohse, M.J. Progressive hypertrophy and heart failure in 1-adrenergic receptor transgenic mice. Proc. Natl. Acad. Sci. USA 1999, 96, 7059–7064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liggett, S.B.; Tepe, N.M.; Lorenz, J.N.; Canning, A.M.; Jantz, T.D.; Mitarai, S.; Yatani, A.; Dorn, G.W. Early and Delayed Consequences of 2-Adrenergic Receptor Overexpression in Mouse Hearts: Critical Role for Expression Level. Circulation 2000, 101, 1707–1714. [Google Scholar] [CrossRef] [Green Version]
- Iaccarino, G.; Barbato, E.; Cipolletta, E.; De Amicis, V.; Margulies, K.B.; Leosco, D.; Trimarco, B.; Koch, W.J. Elevated myocardial and lymphocyte GRK2 expression and activity in human heart failure. Eur. Hear. J. 2005, 26, 1752–1758. [Google Scholar] [CrossRef] [Green Version]
- Hata, J.A.; Williams, M.L.; Schroder, J.N.; Lima, B.; Keys, J.R.; Blaxall, B.C.; Petrofski, J.A.; Jakoi, A.; Milano, C.A.; Koch, W.J. Lymphocyte Levels of GRK2 (βARK1) Mirror Changes in the LVAD-Supported Failing Human Heart: Lower GRK2 Associated With Improved β-Adrenergic Signaling After Mechanical Unloading. J. Card. Fail. 2006, 12, 360–368. [Google Scholar] [CrossRef]
- Agüero, J.; Almenar, L.; D’Ocon, P.; Oliver, E.; Monto, F.; Rueda, J.; Vicente, D.; Martínez-Dolz, L.; Salvador, A. Myocardial and Peripheral Lymphocytic Transcriptomic Dissociation of β-adrenoceptors and G Protein–coupled Receptor Kinases in Heart Transplantation. J. Hear. Lung Transplant. 2009, 28, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Rengo, G.; Galasso, G.; Femminella, G.D.; Parisi, V.; Zincarelli, C.; Pagano, G.; De Lucia, C.; Cannavo, A.; Liccardo, D.; Marciano, C.; et al. Reduction of lymphocyte G protein-coupled receptor kinase-2 (GRK2) after exercise training predicts survival in patients with heart failure. Eur. J. Prev. Cardiol. 2014, 21, 4–11. [Google Scholar] [CrossRef]
- Bonita, R.E.; Raake, P.W.; Otis, N.J.; Chuprun, J.K.; Spivack, T.; Dasgupta, A.; Whellan, D.J.; Mather, P.J.; Koch, W.J. Dynamic Changes in Lymphocyte GRK2 Levels in Cardiac Transplant Patients: A Biomarker for Left Ventricular Function. Clin. Transl. Sci. 2010, 3, 14–18. [Google Scholar] [CrossRef]
- Rengo, G.; Pagano, G.; Filardi, P.P.; Femminella, G.D.; Parisi, V.; Cannavo, A.; Liccardo, D.; Komici, K.; Gambino, G.; D’Amico, M.L.; et al. Prognostic Value of Lymphocyte G Protein-Coupled Receptor Kinase-2 Protein Levels in Patients With Heart Failure. Circ. Res. 2016, 118, 1116–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santulli, G.; Campanile, A.; Spinelli, L.; Di Panzillo, E.A.; Ciccarelli, M.; Trimarco, B.; Iaccarino, G. G Protein-Coupled Receptor Kinase 2 in Patients With Acute Myocardial Infarction. Am. J. Cardiol. 2011, 107, 1125–1130. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Biomarkers | Cut Off Values | Production | Increasing in | Hf Phenotype | Role |
|---|---|---|---|---|---|
| BNP | >35 pg/mL | released from myocytes under stress | HF, Aging, LVH, CKD, AS, MI, AF, Obesity | HFrEF > HFpEF | Diagnosis, Prognosis, Follow up |
| NT PRO BNP | >125 pg/mL | fragment of BNP precursor | HF, Aging, LVH, CKD, AS, MI, AF | HFrEF > HFpEF | Diagnosis, Prognosis, Follow up |
| MR PRO ANP | >127 pmol/L | atrial wall as result of stretch | HF, AS, Sepsis, MI, AF, Burns | HFrEF > HFpEF | Diagnosis, Prognosis, Follow up |
| HS-CIN TROPONIN | >34.2 pg/mL | cardiomyocytes injury | HF, MI, Myocarditis, CKD, Sepsis, Hypothyroidism, Trauma | HFrEF > HFpEF | Diagnosis, Prognosis |
| GALECTIN 3 | <17.8 ng/mL | fibroblast proliferation and activation | HF, Aging, DM, CKD, IPF, Obesity, Cirrhosis, Cancer, Inflammatory states | HFpEF > HFrEF | Prognosis |
| MR-PROADM | 0.10–0.64 nmol/L | released in several tissue as result of increased pressure and volume overload | HF, MI, CAD, Hypertension, CKD, Sepsis, Cancer | HFmrEF | Prognosis |
| ST2 | >30 ng/mL | myocardial stretch | HF, CAD, IS | HFpEF > HFrEF | Prognosis |
| NGAL | 50 ng/mL | neutrophils and endothelial cells, involved in response renal injury | HF, RI | HFrEF > HFpEf | Diagnosis, Prognosis |
| IRON DEFICIENCY | Ferritin <15 µg/L | multifactorial condition | HF, IDA, IM, Bleeding | HFrEF > HFpEF | Prognosis |
| NE | >480 pg/dl | Neuroendocrine cells as result of sympathetic overdrive | HF, MI, Hypertension, Pheochromocytoma, Cushing, Stress | HFrEF>HFpEF | Prognosis |
| NPY | >130 pg/mL | Neuroendocrine cells as result of sympathetic overdrive | HF, Obesity, Stress | HFrEF | Prognosis |
| GALANIN | To be determined yet | Neuroendocrine and gastrointestinal cells | HF, Hypertension, Pain, SL, Cancer | HFpEF | To be further elucidated |
| CHROMOGRANIN A /CST | >19.73 ng/mL | Neuroendocrine and myocardial cells | HF, CAD, Sepsis | Independent from LVEF | Prognosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bencivenga, L.; Palaia, M.E.; Sepe, I.; Gambino, G.; Komici, K.; Cannavo, A.; Femminella, G.D.; Rengo, G. Why Do We Not Assess Sympathetic Nervous System Activity in Heart Failure Management: Might GRK2 Serve as a New Biomarker? Cells 2021, 10, 457. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020457
Bencivenga L, Palaia ME, Sepe I, Gambino G, Komici K, Cannavo A, Femminella GD, Rengo G. Why Do We Not Assess Sympathetic Nervous System Activity in Heart Failure Management: Might GRK2 Serve as a New Biomarker? Cells. 2021; 10(2):457. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020457
Chicago/Turabian StyleBencivenga, Leonardo, Maria Emiliana Palaia, Immacolata Sepe, Giuseppina Gambino, Klara Komici, Alessandro Cannavo, Grazia Daniela Femminella, and Giuseppe Rengo. 2021. "Why Do We Not Assess Sympathetic Nervous System Activity in Heart Failure Management: Might GRK2 Serve as a New Biomarker?" Cells 10, no. 2: 457. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020457