
Cerebral Insulin Bolus Revokes the Changes in Hepatic Lipid Metabolism Induced by Chronic Central Leptin Infusion

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Design
2.2.1. Central Leptin and Insulin Infusion
2.2.2. Peripheral Leptin and Central Insulin Administration
2.3. Serum and Tissue Non-Esterified Fatty Acids (NEFA) and Triglyceride Levels
2.4. Enzyme-Linked Immunosorbent Assays (ELISAs)
2.4.1. Phosphorylation of Insulin Receptor (IR)
2.4.2. Phosphorylation of Insulin-Like Growth Factor I Receptor (IGF-IR)
2.4.3. Serum Growth Hormone (GH) Levels
2.5. Enzyme Activity Assays
2.5.1. Glucose-6-Phosphate Dehydrogenase
2.5.2. Malic Enzyme (ME)
2.6. Extraction of RNA and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Analysis
2.7. Western Blotting
2.8. Multiplexed Bead Immunoassay
2.9. Statistical Analysis
3. Results
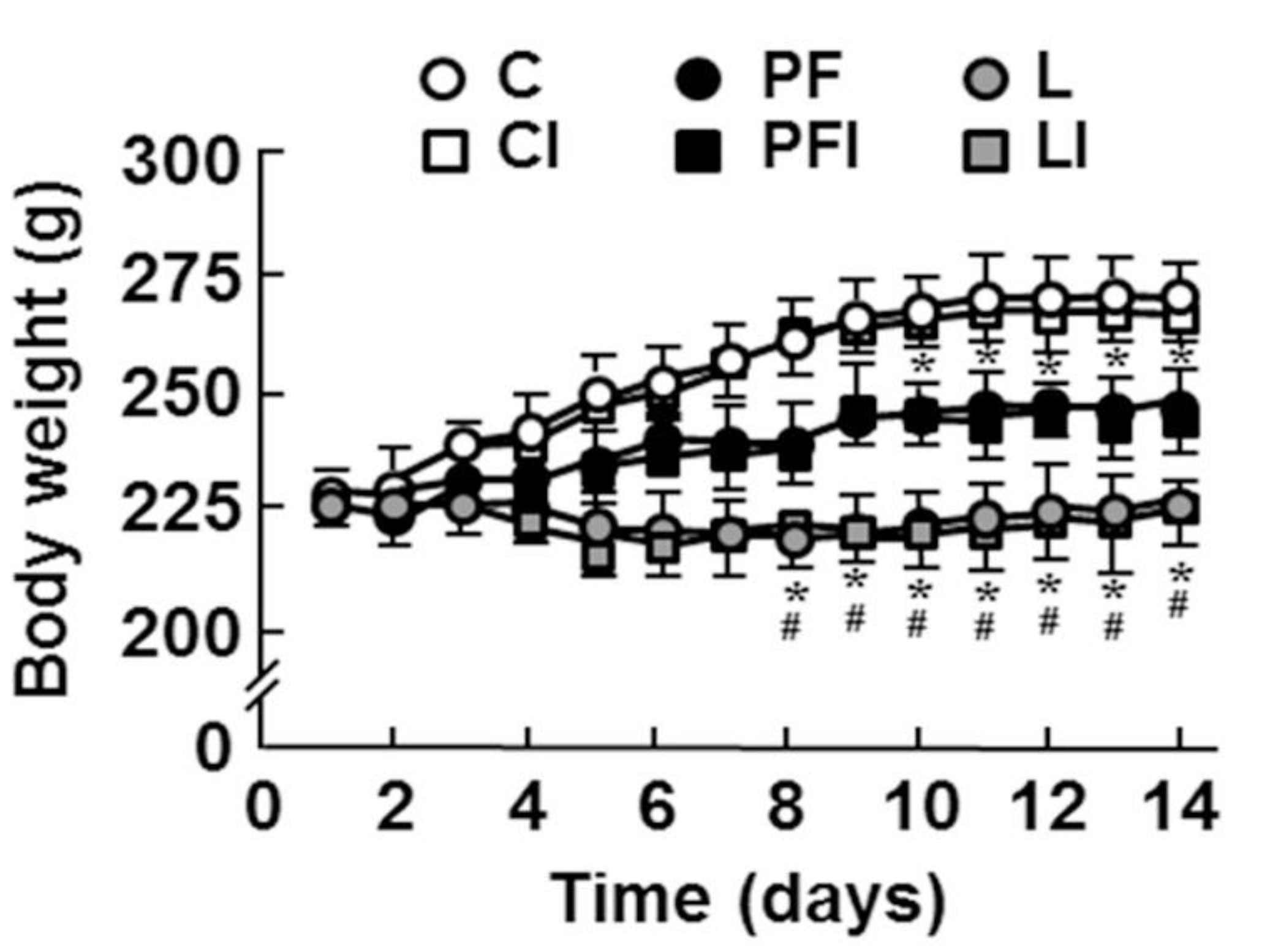
3.1. General Characteristics of Experimental Groups
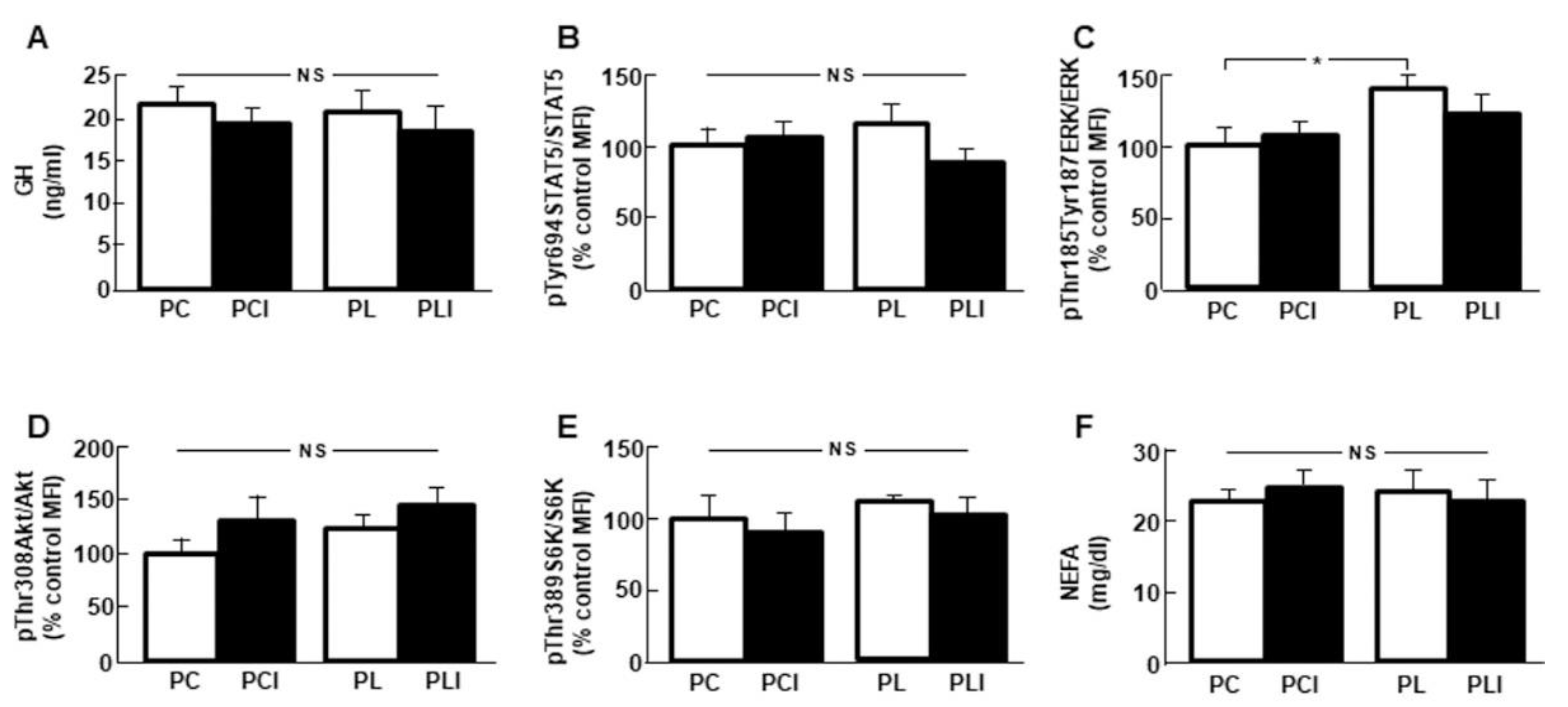
3.2. Combined Treatment Increases Serum GH Levels
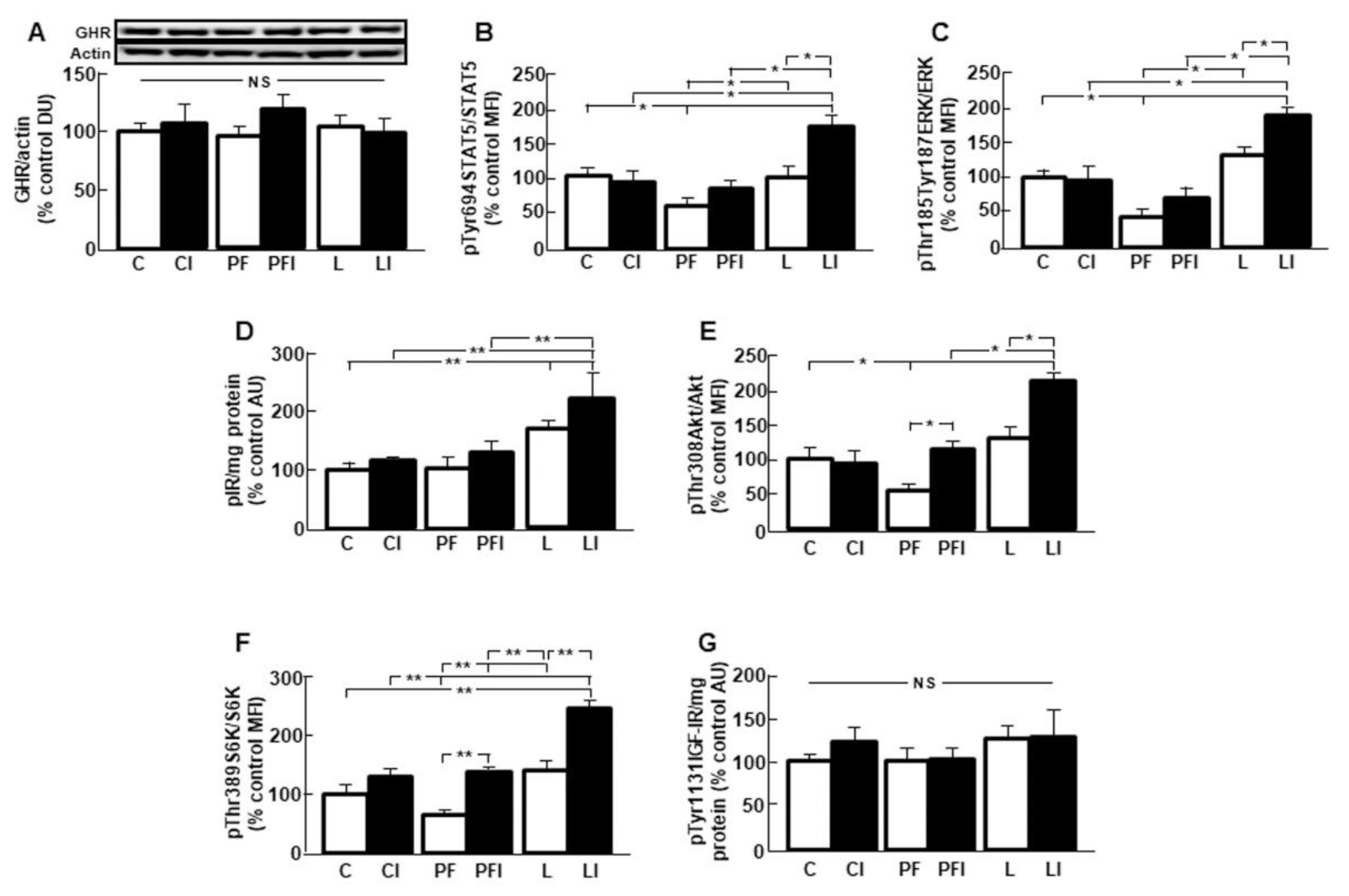
3.3. Hepatic GH Signaling is Increased after Insulin Infusion in Leptin-Treated Rats
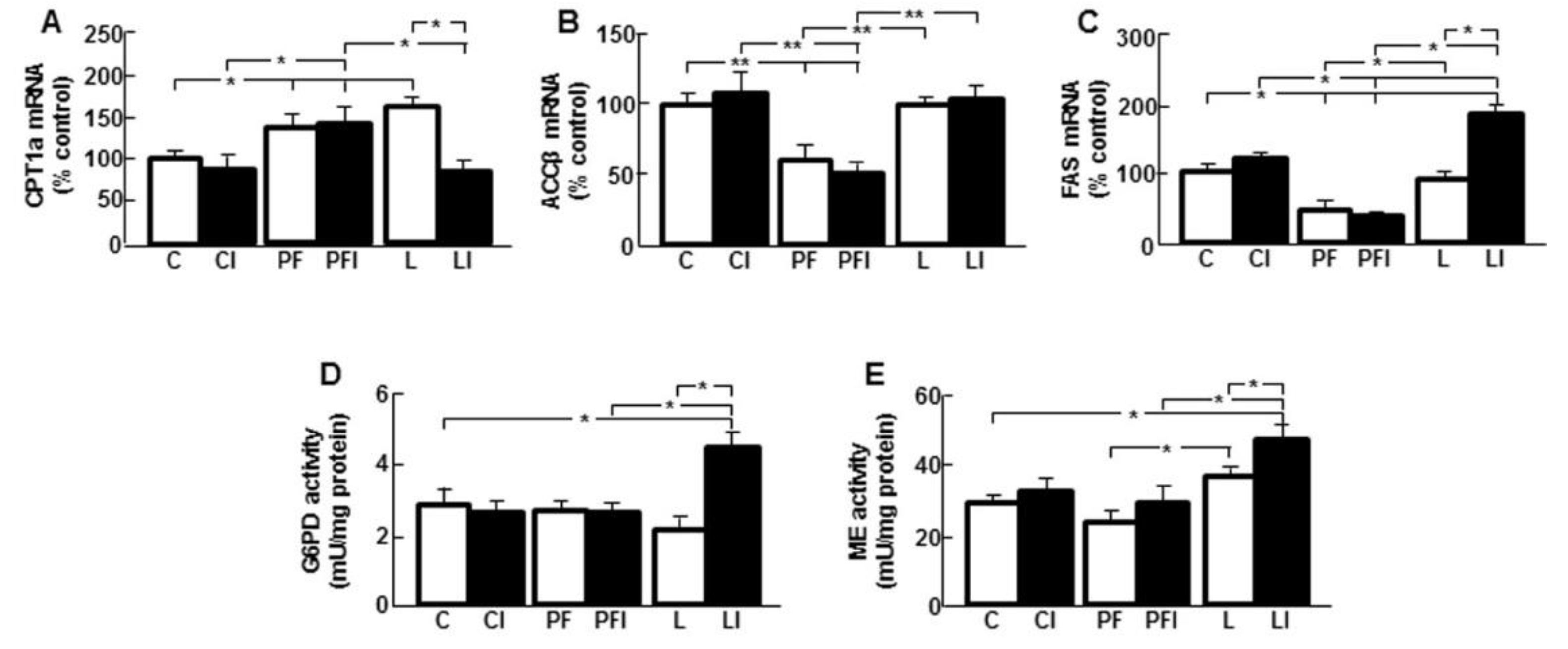
3.4. Leptin Modifies the Effect of Insulin on Markers of Hepatic Lipid Metabolism
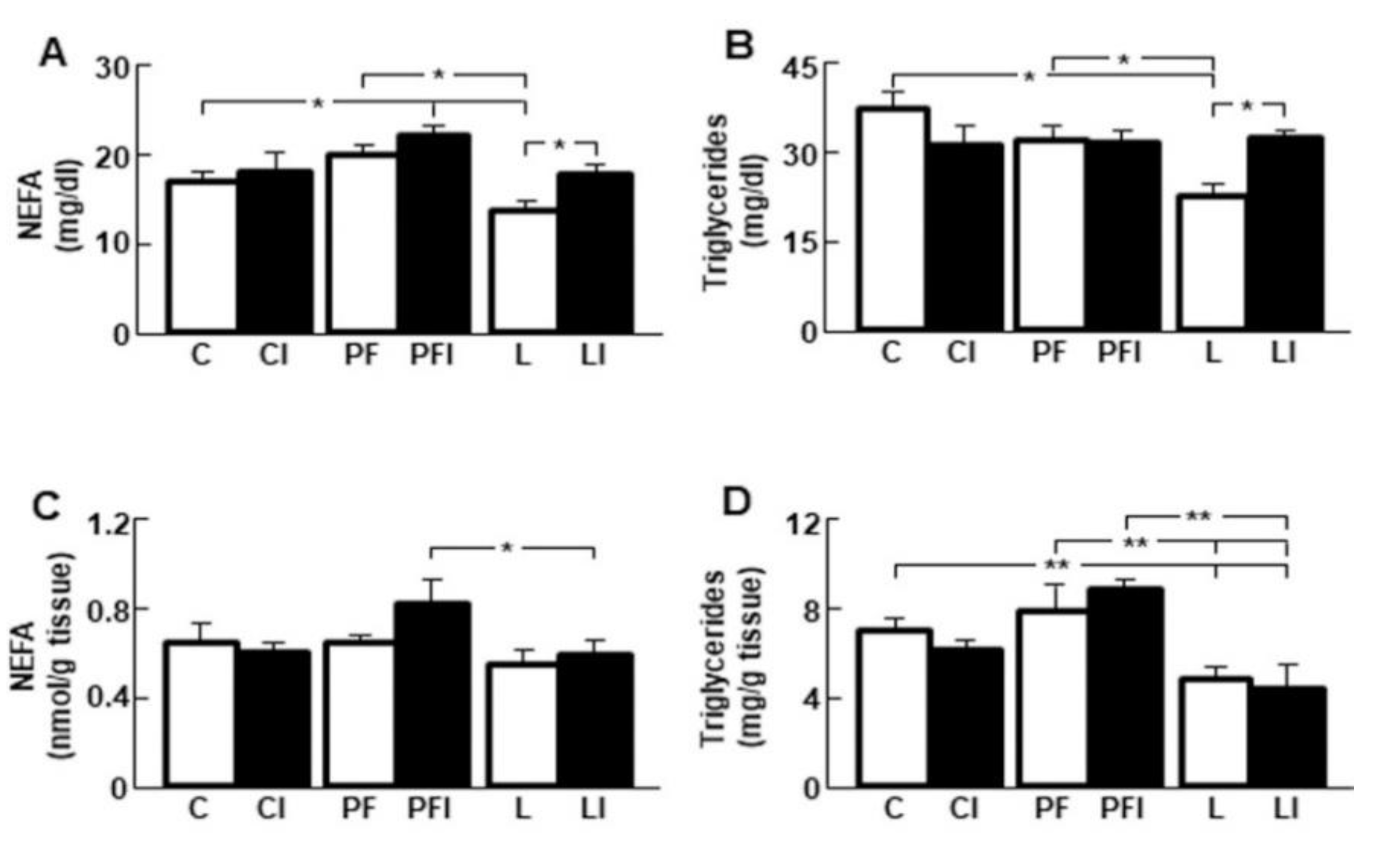
3.5. Insulin Normalizes the Reduction in Serum Lipid Levels Induced by Leptin
3.6. Effect of Leptin and Insulin Infusion in Subcutaneous Fat
3.7. Effect of Peripheral Leptin Infusion
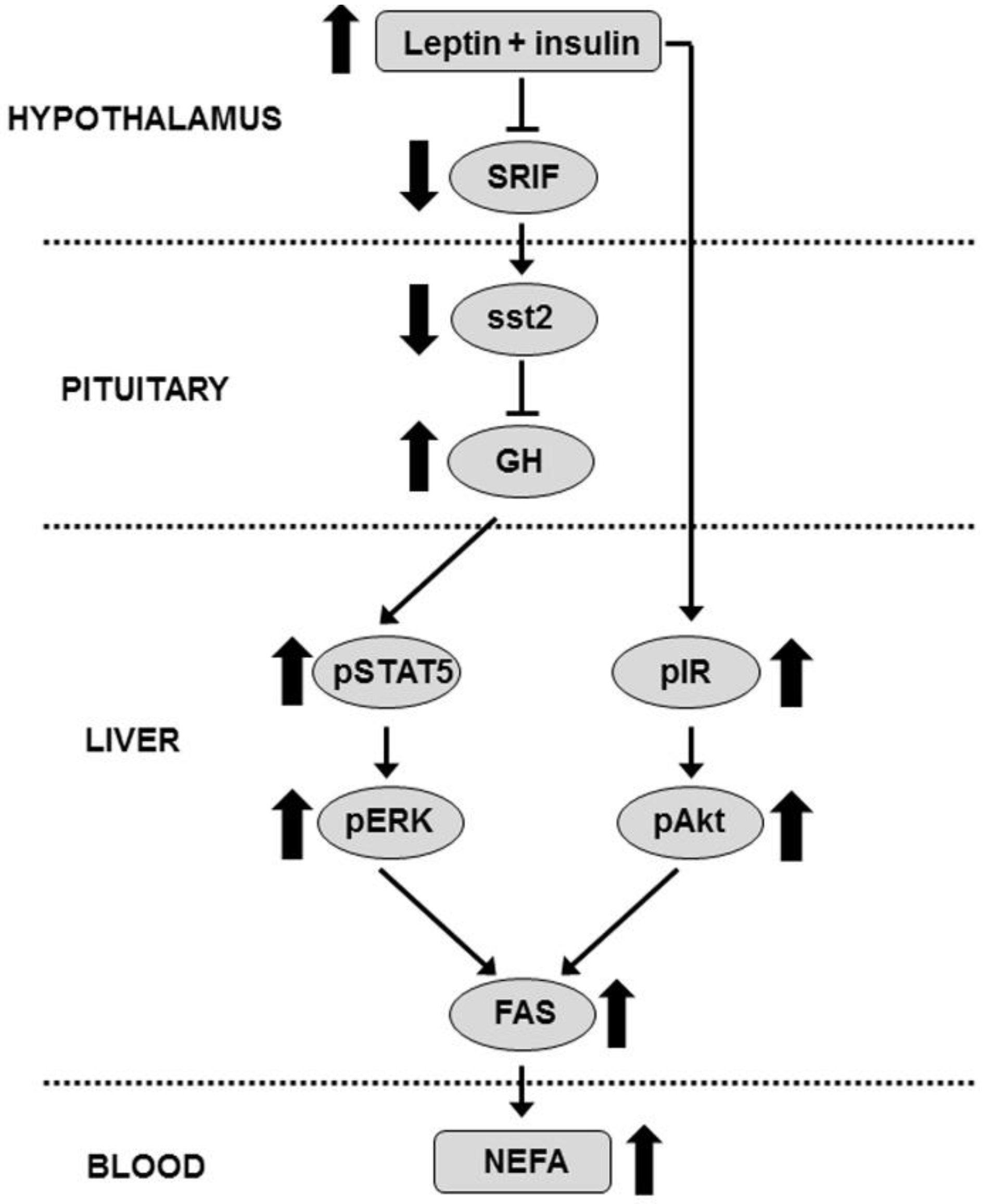
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zadeh, R.K.; Yu, S.; Jiang, Y.; Laque, A.; Schwartzenburg, C.; Morrison, C.D.; Derbenev, A.V.; Zsombok, A.; Münzberg, H. Leptin receptor neurons in the dorsomedial hypothalamus are key regulators of energy expenditure and body weight, but not food intake. Mol. Metab. 2014, 3, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Figlewicz, D.P.; Benoit, S.C. Insulin, leptin, and food reward: Update Am. J. Physiol. Integr. Comp. Physiol. 2009, 296, R9–R19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, E.A.; Reis, D.; Romanatto, T.; Maimoni, D.; Ferreira, E.A.; Santos, G.A.; Torsoni, A.S.; Velloso, L.A.; Torsoni, M.A. Central leptin action improves skeletal muscle AKT, AMPK, and PGC1α activation by hypothalamic PI3K-dependent mechanism. Mol. Cell. Endocrinol. 2010, 314, 62–69. [Google Scholar] [CrossRef]
- Song, Y.F.; Wu, K.; Tan, X.Y.; Zhang, L.H.; Zhuo, M.Q.; Pan, Y.X.; Chen, Q.L. Effects of recombinant human leptin administration on hepatic lipid metabolism in yellow catfish Pelteobagrus fulvidraco: In vivo and in vitro studies. Gen. Comp. Endocrinol. 2015, 212, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Geisler, E.C.; Renquist, B.J. Hepatic lipid accumulation: Cause and consequence of dysregulated glucoregulatory hormones. J. Endocrinol. 2017, 234, R1–R21. [Google Scholar] [CrossRef]
- Goodridge, A.G.; Crish, J.F.; Hillgartner, F.B.; Wilson, S.B. Nutritional and Hormonal Regulation of the Gene for Avian Malic Enzyme. J. Nutr. 1989, 119, 299–308. [Google Scholar] [CrossRef]
- Taniguchi, M.; Mori, N.; Iramina, C.; Yasutake, A. Elevation of Glucose 6-Phosphate Dehydrogenase Activity Induced by Amplified Insulin Response in Low Glutathione Levels in Rat Liver. Sci. World J. 2016, 2016, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velasco, P.; Sieiro, A.M.; Ibarguren, I.; Martinez, R.J.I.; Barcia, R. The modulation of the oxidative phase of the pentose phosphate pathway in mouse liver. Int. J. Biochem. Cell Biol. 1995, 27, 1015–1019. [Google Scholar] [CrossRef]
- Kullmann, S.; Valenta, V.; Wagner, R.; Tschritter, O.; Machann, J.; Häring, H.U.; Preissl, H.; Fritsche, A.; Heni, M. Brain insulin sensitivity is linked to adiposity and body fat distribution. Nat. Commun. 2020, 11, 1841. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, A. Renaming NAFLD to MAFLD: Could the LDE System Assist in This Transition? J. Clin. Med. 2021, 10, 492. [Google Scholar] [CrossRef]
- Tannenbaum, G.S.; Gurd, W.; Lapointe, M. Leptin Is a Potent Stimulator of Spontaneous Pulsatile Growth Hormone (GH) Secretion and the GH Response to GH-Releasing Hormone*. Endocrinology 1998, 139, 3871–3875. [Google Scholar] [CrossRef]
- Watanobe, H.; Habu, S. Leptin Regulates Growth Hormone-Releasing Factor, Somatostatin, and α-Melanocyte-Stimulating Hormone But Not Neuropeptide Y Release in Rat HypothalamusIn Vivo: Relation with Growth Hormone Secretion. J. Neurosci. 2002, 22, 6265–6271. [Google Scholar] [CrossRef] [Green Version]
- Carvalheira, J.B.; Siloto, R.M.; Ignacchitti, I.; Brenelli, S.L.; Carvalho, C.R.; Leite, A.; Velloso, L.A.; Gontijo, J.A.; Saad, M.J. Insulin modulates leptin-induced STAT3 activation in rat hypothalamus. FEBS Lett. 2001, 500, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Kerrigan, J.R.; Martha, P.M.; Krieg, R.J.; Rogol, A.D.; Evans, W.S. Somatostatin Inhibition of Growth Hormone Secretion by Somatotropes from Male, Female, and Androgen Receptor-Deficient Rats: Evidence for Differing Sensitivities*. Endocrinology 1989, 125, 3078–3083. [Google Scholar] [CrossRef]
- Sugihara, H.; Minami, S.; Okada, K.; Kamegai, J.; Hasegawa, O.; Wakabayashi, I. Somatostatin reduces transcription of the growth hormone gene in rats. Endocrinology 1993, 132, 1225–1229. [Google Scholar] [CrossRef] [PubMed]
- Acunzo, J.; Thirion, S.; Roche, C.; Saveanu, A.; Gunz, G.; Germanetti, A.L.; Couderc, B.; Cohen, R.; Figarella-Branger, D.; Dufour, H.; et al. Somatostatin Receptor sst2 Decreases Cell Viability and Hormonal Hypersecretion and Reverses Octreotide Resistance of Human Pituitary Adenomas. Cancer Res. 2008, 68, 10163–10170. [Google Scholar] [CrossRef] [Green Version]
- Quintela, M.; Señaris, R.; Heiman, M.L.; Casanueva, F.F.; Diéguez, C. Leptin Inhibits in Vitro Hypothalamic Somatostatin Secretion and Somatostatin mRNA Levels. Endocrinology 1997, 138, 5641–5644. [Google Scholar] [CrossRef]
- Stepanyan, Z.; Kocharyan, A.; Behrens, M.; Koebnick, C.; Pyrski, M.; Meyerhof, W. Somatostatin, a negative-regulator of central leptin action in the rat hypothalamus. J. Neurochem. 2006, 100, 468–478. [Google Scholar] [CrossRef]
- Matsumoto, R.; Fukuoka, H.; Iguchi, G.; Nishizawa, H.; Bando, H.; Suda, K.; Takahashi, M.; Takahashi, Y. Long-term effects of growth hormone replacement therapy on liver function in adult patients with growth hormone deficiency. Growth Horm. IGF Res. 2014, 24, 174–179. [Google Scholar] [CrossRef]
- Liebe, R.; Esposito, I.; Bock, H.H.; Dahl, S.V.; Stindt, J.; Baumann, U.; Luedde, T.; Keitel, V. Diagnosis and management of secondary causes of steatohepatitis. J. Hepatol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Regaiey, A.K.A.; Masternak, M.M.; Bonkowski, M.; Sun, L.; Bartke, A. Long-Lived Growth Hormone Receptor Knockout Mice: Interaction of Reduced Insulin-Like Growth Factor I/Insulin Signaling and Caloric Restriction. Endocrinology 2005, 146, 851–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kling, P.; Jönsson, E.; Nilsen, T.O.; Einarsdottir, I.E.; Rønnestad, I.; Stefansson, S.O.; Björnsson, B.T. The role of growth hormone in growth, lipid homeostasis, energy utilization and partitioning in rainbow trout: Interactions with leptin, ghrelin and insulin-like growth factor I. Gen. Comp. Endocrinol. 2012, 175, 153–162. [Google Scholar] [CrossRef]
- Evijayakumar, A.; Eyakar, S.; Eleroith, D. The Intricate Role of Growth Hormone in Metabolism. Front. Endocrinol. 2011, 2, 32. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Messina, J.L. Chapter 6 Crosstalk between Growth Hormone and Insulin Signaling. Vitam. Horm. 2009, 80, 125–153. [Google Scholar] [CrossRef]
- Deane, E.E.; Woo, N.Y.S. Upregulation of the Somatotropic Axis Is Correlated with Increased G6PDH Expression in Black Sea Bream Adapted to Iso-osmotic Salinity. Ann. N. Y. Acad. Sci. 2005, 1040, 293–296. [Google Scholar] [CrossRef]
- Gao, Y.; Su, P.; Wang, C.; Zhu, K.; Chen, X.; Liu, S.; He, J. The Role of PTEN in Chronic Growth Hormone-Induced Hepatic Insulin Resistance. PLoS ONE 2013, 8, 8105. [Google Scholar] [CrossRef] [Green Version]
- Ramos, B.E.; Canelles, S.; Rodríguez, A.; Ambrosi, G.J.; Frago, L.M.; Chowen, J.A.; Frühbeck, G.; Argente, J.; Barrios, V. Chronic central leptin infusion modulates the glycemia response to insulin administration in male rats through regulation of hepatic glucose metabolism. Mol. Cell. Endocrinol. 2015, 415, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Stanley, G.S. A simple method for the isolation and purification of total lipids from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Geer, B.; Krochko, D.; Oliver, M.; Walker, V.; Williamson, J. A comparative study of the NADP-malic enzymes from Drosophila and chick liver. Comp. Biochem. Physiol. Part B Comp. Biochem. 1980, 65, 25–34. [Google Scholar] [CrossRef]
- Chomczynski, P. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques 1993, 15, 532–534. [Google Scholar]
- Khan, S.S.; Smith, M.S.; Reda, D.; Suffredini, A.F.; McCoy, J.P. Multiplex bead array assays for detection of soluble cytokines: Comparisons of sensitivity and quantitative values among kits from multiple manufacturers. Cytometry 2004, 61, 35–39. [Google Scholar] [CrossRef]
- Shlomo, B.A.; Pichurin, O.; Khalafi, R.; Zhou, C.; Chesnokova, V.; Ren, S.G.; Liu, N.A.; Melmed, S. Constitutive Somatostatin Receptor Subtype 2 Activity Attenuates GH Synthesis. Endocrinology 2013, 154, 2399–2409. [Google Scholar] [CrossRef]
- Parmar, R.; Chan, W.S.; Dashkevicz, M.; Hayes, E.; Rohrer, S.; Smith, R.; Schaeffer, J.; Blake, A. Nonpeptidyl Somatostatin Agonists Demonstrate That sst2 and sst5 Inhibit Stimulated Growth Hormone Secretion from Rat Anterior Pituitary Cells. Biochem. Biophys. Res. Commun. 1999, 263, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Saleri, R.; Giustina, A.; Tamanini, C.; Valle, D.; Burattin, A.; Wehrenberg, W.B.; Baratta, M. Leptin Stimulates Growth Hormone Secretion via a Direct Pituitary Effect Combined with a Decreased Somatostatin Tone in a Median Eminence-Pituitary Perifusion Study. Neuroendocrinology 2004, 79, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Wein, S.; Ukropec, J.; Gašperíková, D.; Klimeš, I.; Sebokova, E. Concerted Action of Leptin in Regulation of Fatty Acid Oxidation in Skeletal Muscle and Liver. Exp. Clin. Endocrinol. Diabetes 2007, 115, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; A Viscarra, J.; Kim, S.J.; Sul, H.S. Transcriptional regulation of hepatic lipogenesis. Nat. Rev. Mol. Cell Biol. 2015, 16, 678–689. [Google Scholar] [CrossRef] [Green Version]
- Cortés, V.A.; Cautivo, K.M.; Rong, S.; Garg, A.; Horton, J.D.; Agarwal, A.K. Leptin ameliorates insulin resistance and hepatic steatosis in Agpat2 lipodystrophic mice independent of hepatocyte leptin receptors. J. Lipid Res. 2014, 55, 276–288. [Google Scholar] [CrossRef] [Green Version]
- Sachithanandan, N.; Fam, B.C.; Fynch, S.; Dzamko, N.; Watt, M.J.; Wormald, S.; Honeyman, J.; Galic, S.; Proietto, J.; Andrikopoulos, S.; et al. Liver-specific suppressor of cytokine signaling-3 deletion in mice enhances hepatic insulin sensitivity and lipogenesis resulting in fatty liver and obesity. Hepatology 2010, 52, 1632–1642. [Google Scholar] [CrossRef]
- Dong, Q.; Majumdar, G.; Meally, O.R.N.; Cole, R.N.; Elam, M.B.; Raghow, R. Insulin-induced de novo lipid synthesis occurs mainly via mTOR-dependent regulation of proteostasis of SREBP-1c. Mol. Cell. Biochem. 2019, 463, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Sakaguchi, M.; Kleinridders, A.; Pino, G.G.D.; Dreyfuss, J.M.; Neill, O.B.T.; Ramirez, A.K.; Pan, H.; Winnay, J.N.; Boucher, J.; et al. Domain-dependent effects of insulin and IGF-1 receptors on signalling and gene expression. Nat. Commun. 2017, 8, 4892. [Google Scholar] [CrossRef]
- Vijayakumar, A.; Novosyadlyy, R.; Wu, Y.; Yakar, S.; Leroith, D. Biological effects of growth hormone on carbohydrate and lipid metabolism. Growth Horm. IGF Res. 2010, 20, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Frick, F.; Lindén, D.; Améen, C.; Edén, S.; Mode, A.; Oscarsson, J. Interaction between growth hormone and insulin in the regulation of lipoprotein metabolism in the rat. Am. J. Physiol. Metab. 2002, 283, E1023–E1031. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Keeton, A.B.; Franklin, J.L.; Li, X.; Venable, D.Y.; Frank, S.J.; Messina, J.L. Insulin Enhances Growth Hormone Induction of the MEK/ERK Signaling Pathway. J. Biol. Chem. 2006, 281, 982–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bian, Y.; Yu, Y.; Wang, S.; Li, L. Up-regulation of fatty acid synthase induced by EGFR/ERK activation promotes tumor growth in pancreatic cancer. Biochem. Biophys. Res. Commun. 2015, 463, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Martinez, C.S.; Piazza, V.G.; Díaz, M.E.; Boparai, R.K.; Arum, O.; Ramirez, M.C.; González, L.; Villalobos, B.D.; Bartke, A.; Turyn, D.; et al. GH/STAT5 signaling during the growth period in livers of mice overexpressing GH. J. Mol. Endocrinol. 2015, 54, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.Y.; Zhai, G.; Gong, Y.L.; Su, J.Z.; Peng, X.Y.; Shang, G.H.; Han, N.; Jin, J.Y.; Liu, H.K.; Du, Z.Y.; et al. Different physiological roles of insulin receptors in mediating nutrient metabolism in zebrafish. Am. J. Physiol. Metab. 2018, 315, E38–E51. [Google Scholar] [CrossRef] [PubMed]
- Elias, A.E.; Kun, B.; Sabula, I.M.C.; Mello, G.G.; Zablah, A.C.; Kreiling, J.A. The mir-465 family is upregulated with age and attenuates growth hormone signaling in mouse liver. Aging Cell 2019, 18, 2892. [Google Scholar] [CrossRef]
- Huong, D.T.T.; Ide, T. Dietary lipoic acid-dependent changes in the activity and mRNA levels of hepatic lipogenic enzymes in rats. Br. J. Nutr. 2008, 100, 79–87. [Google Scholar] [CrossRef] [Green Version]
- Stanton, R.C. Glucose-6-phosphate dehydrogenase, NADPH, and cell survival. IUBMB Life 2012, 64, 362–369. [Google Scholar] [CrossRef] [Green Version]
- Reed, B.Y.; Veech, R.L. The effects of chronic administration of T4, growth hormone and epidermal growth factor on hepatic lipogenic enzymes in hypophysectomised rats. Biochem. Biophys. Res. Commun. 1986, 141, 78–83. [Google Scholar] [CrossRef]
- Peng, K.; Mo, Z.; Tian, G. Serum Lipid Abnormalities and Nonalcoholic Fatty Liver Disease in Adult Males. Am. J. Med. Sci. 2017, 353, 236–241. [Google Scholar] [CrossRef]
- Lee, H.J.; Jung, H.; Cho, H.; Lee, K.; Hwang, K.T. Black Raspberry Seed Oil Improves Lipid Metabolism by Inhibiting Lipogenesis and Promoting Fatty-Acid Oxidation in High-Fat Diet-Induced Obese Mice and db/db Mice. Lipids 2018, 53, 491–504. [Google Scholar] [CrossRef]
- Ramos, B.E.; Canelles, S.; Rodríguez, A.; Frago, L.M.; Ambrosi, G.J.; Chowen, J.A.; Frühbeck, G.; Argente, J.; Barrios, V. The increase in fiber size in male rat gastrocnemius after chronic central leptin infusion is related to activation of insulin signaling. Mol. Cell. Endocrinol. 2018, 470, 48–59. [Google Scholar] [CrossRef]
- Voshol, P.J.; Haemmerle, G.; Ouwens, D.M.; Zimmermann, R.; Zechner, R.; Teusink, B.; Maassen, J.A.; Havekes, L.M.; Romijn, J.A. Increased Hepatic Insulin Sensitivity Together with Decreased Hepatic Triglyceride Stores in Hormone-Sensitive Lipase-Deficient Mice. Endocrinology 2003, 144, 3456–3462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sperling, M.A. Traditional and novel aspects of the metabolic actions of growth hormone. Growth Horm. IGF Res. 2016, 28, 69–75. [Google Scholar] [CrossRef]
- Fan, Y.; Menon, R.K.; Cohen, P.; Hwang, D.; Clemens, T.; DiGirolamo, D.J.; Kopchick, J.J.; Le Roith, D.; Trucco, M.; Sperling, M.A. Liver-specific Deletion of the Growth Hormone Receptor Reveals Essential Role of Growth Hormone Signaling in Hepatic Lipid Metabolism. J. Biol. Chem. 2009, 284, 19937–19944. [Google Scholar] [CrossRef] [Green Version]
- Lonardo, A.; Mantovani, A.; Lugari, S.; Targher, G. NAFLD in Some Common Endocrine Diseases: Prevalence, Pathophysiology, and Principles of Diagnosis and Management. Int. J. Mol. Sci. 2019, 20, 2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, C.D.; A Daniel, J.; Holmberg, B.J.; Djiane, J.; Raver, N.; Gertler, A.; Keisler, D.H. Central infusion of leptin into well-fed and undernourished ewe lambs: Effects on feed intake and serum concentrations of growth hormone and luteinizing hormone. J. Endocrinol. 2001, 168, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Kurose, Y.; Takahashi, H.; Asakuma, S.; Azuma, Y.; Kobayashi, S. The differences in feeding-inhibitory responses to peripheral and central leptin between non-lactating and lactating rats. J. Endocrinol. 2010, 207, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Ravussin, Y.; A Leduc, C.; Watanabe, K.H.; Mueller, B.; A Skowronski, A.; Rosenbaum, M.; Leibel, R.L. Effects of chronic leptin infusion on subsequent body weight and composition in mice: Can body weight set point be reset? Mol. Metab. 2014, 3, 432–440. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.G.; Tang, G.B.; Wang, D.H.; Speakman, J.R. Effects of leptin infusion during peak lactation on food intake, body composition, litter growth, and maternal neuroendocrine status in female Brandt’s voles (Lasiopodomys brandtii). Am. J. Physiol. Integr. Comp. Physiol. 2011, 300, R447–R459. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | C | CI | PF | PFI | L | LI |
|---|---|---|---|---|---|---|
| Body weight (g) | 267 ± 8 | 261 ± 7 | 253 ± 6 * | 250 ± 5 * | 228 ± 3 *# | 227 ± 5 *# |
| Body weight gain (g) | 43.4 ± 3.2 | 43.0 ± 3.6 | 25.1 ± 2.2 ** | 25.3 ± 2.0 ** | 2.8 ± 1.5 **## | 2.9 ± 1.4 **## |
| Gastrocnemius (%) | 0.87 ± 0.05 | 0.90 ± 0.06 | 0.75 ± 0.03 | 0.75 ± 0.04 | 1.12 ± 0.07 # | 1.14 ± 0.08 # |
| Soleus (%) | 0.07 ± 0.01 | 0.06 ± 0.01 | 0.07 ± 0.01 | 0.07 ± 0.01 | 0.08 ± 0.02 | 0.09 ± 0.02 |
| Liver weight (%) | 3.53 ± 0.19 | 3.54 ± 0.18 | 3.58 ± 0.24 | 3.59 ± 0.22 | 3.80 ± 0.32 | 3.83 ± 0.26 |
| Epididymal fat (%) | 1.39 ± 0.06 | 1.43 ± 0.10 | 1.03 ± 0.09 ** | 1.07 ± 0.08 ** | 0.65 ± 0.10 **## | 0.66 ± 0.13 **## |
| Inguinal fat (%) | 1.06 ± 0.03 | 1.08 ± 0.05 | 0.88 ± 0.05 * | 0.87 ± 0.04 * | 0.66 ± 0.06 *# | 0.65 ± 0.07 *# |
| Mesenteric fat (%) | 1.12 ± 0.14 | 1.15 ± 0.13 | 0.71 ± 0.06 * | 0.74 ± 0.08 * | 0.67 ± 0.08 * | 0.65 ± 0.10 * |
| Retroperitoneal fat (%) | 0.37 ± 0.04 | 0.37 ± 0.05 | 0.35 ± 0.03 | 0.34 ± 0.04 | 0.28 ± 0.02 * | 0.29 ± 0.02 * |
| iBAT (%) | 0.26 ± 0.03 | 0.26 ± 0.02 | 0.17 ± 0.02 * | 0.18 ± 0.03 * | 0.24 ± 0.03 | 0.25 ± 0.04 |
| Parameter | C | CI | PF | PFI | L | LI |
|---|---|---|---|---|---|---|
| SRIF mRNA (% control) | 100.0 ± 7.7 | 110.5 ± 8.4 | 132.5 ± 9.6 * | 169.2 ± 8.5 * | 46.0 ± 3.3 * | 21.5 ± 1.7 *# |
| sst2 mRNA (% control) | 100.0 ± 9.0 | 19.7 ± 5.6 * | 45.9 ± 11.0 * | 42.9 ± 12.8 * | 42.5 ± 6.4 * | 20.3 ± 3.6 *# |
| GH mRNA (% control) | 100.0 ± 10.6 | 64.0 ± 7.8 * | 60.0 ± 6.6 * | 80.5 ± 21.1 | 85.9 ± 16.9 | 141.6 ± 11.3 *# |
| GH (ng/mL) | 16.8 ± 2.4 | 22.4 ± 2.5 | 13.7 ± 2.0 * | 16.2 ± 3.0 | 32.9 ± 4.6 | 45.9 ± 5.8 *# |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrios, V.; López-Villar, E.; Frago, L.M.; Canelles, S.; Díaz-González, F.; Burgos-Ramos, E.; Frühbeck, G.; Chowen, J.A.; Argente, J. Cerebral Insulin Bolus Revokes the Changes in Hepatic Lipid Metabolism Induced by Chronic Central Leptin Infusion. Cells 2021, 10, 581. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10030581
Barrios V, López-Villar E, Frago LM, Canelles S, Díaz-González F, Burgos-Ramos E, Frühbeck G, Chowen JA, Argente J. Cerebral Insulin Bolus Revokes the Changes in Hepatic Lipid Metabolism Induced by Chronic Central Leptin Infusion. Cells. 2021; 10(3):581. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10030581
Chicago/Turabian StyleBarrios, Vicente, Elena López-Villar, Laura M. Frago, Sandra Canelles, Francisca Díaz-González, Emma Burgos-Ramos, Gema Frühbeck, Julie A. Chowen, and Jesús Argente. 2021. "Cerebral Insulin Bolus Revokes the Changes in Hepatic Lipid Metabolism Induced by Chronic Central Leptin Infusion" Cells 10, no. 3: 581. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10030581