
Maternal Exercise Mediates Hepatic Metabolic Programming via Activation of AMPK-PGC1α Axis in the Offspring of Obese Mothers

 , , , , , , , add
Show full author list
, , , , , , , add
Show full author list
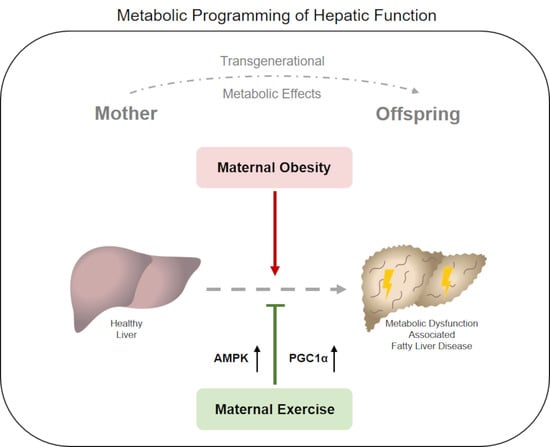
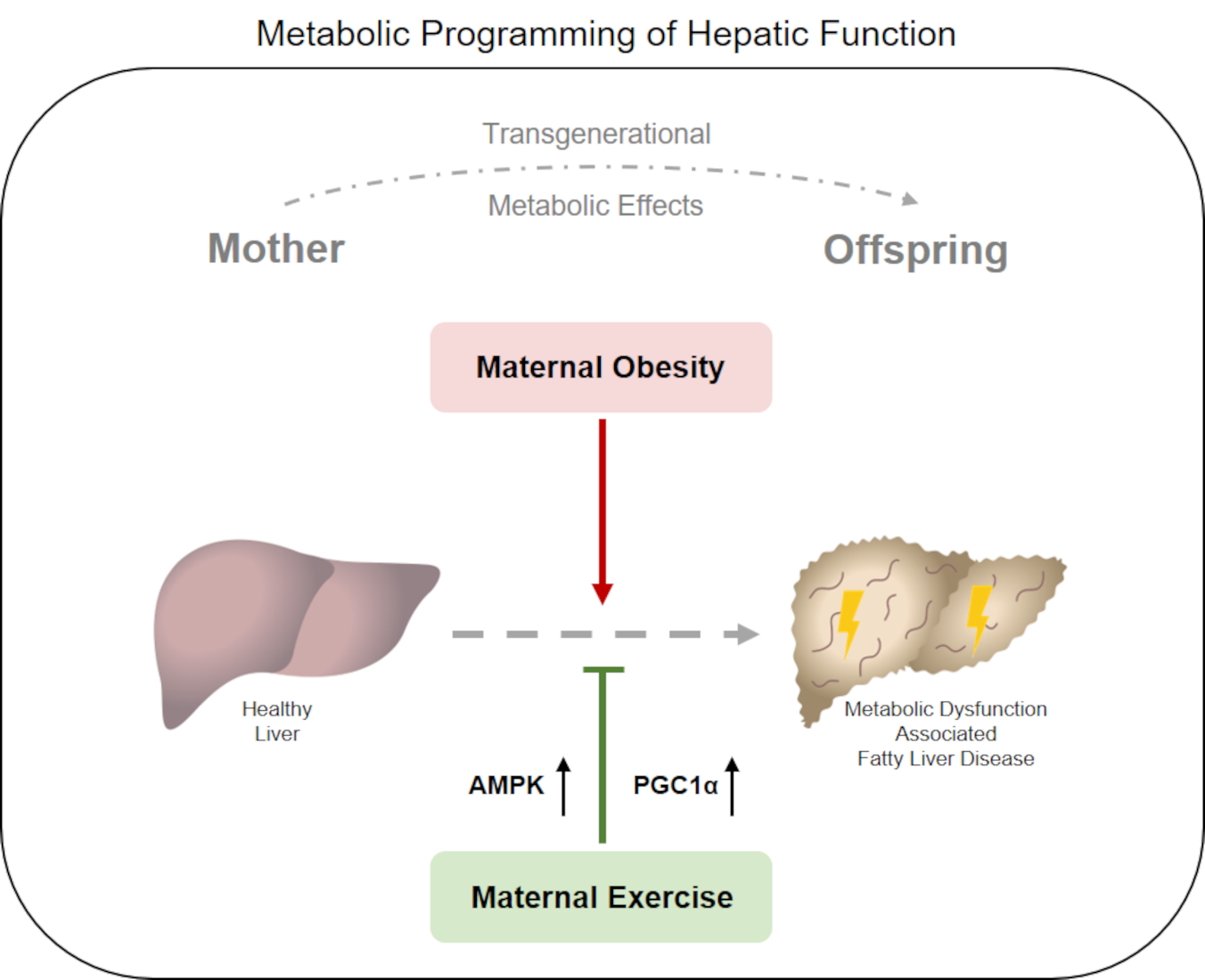
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
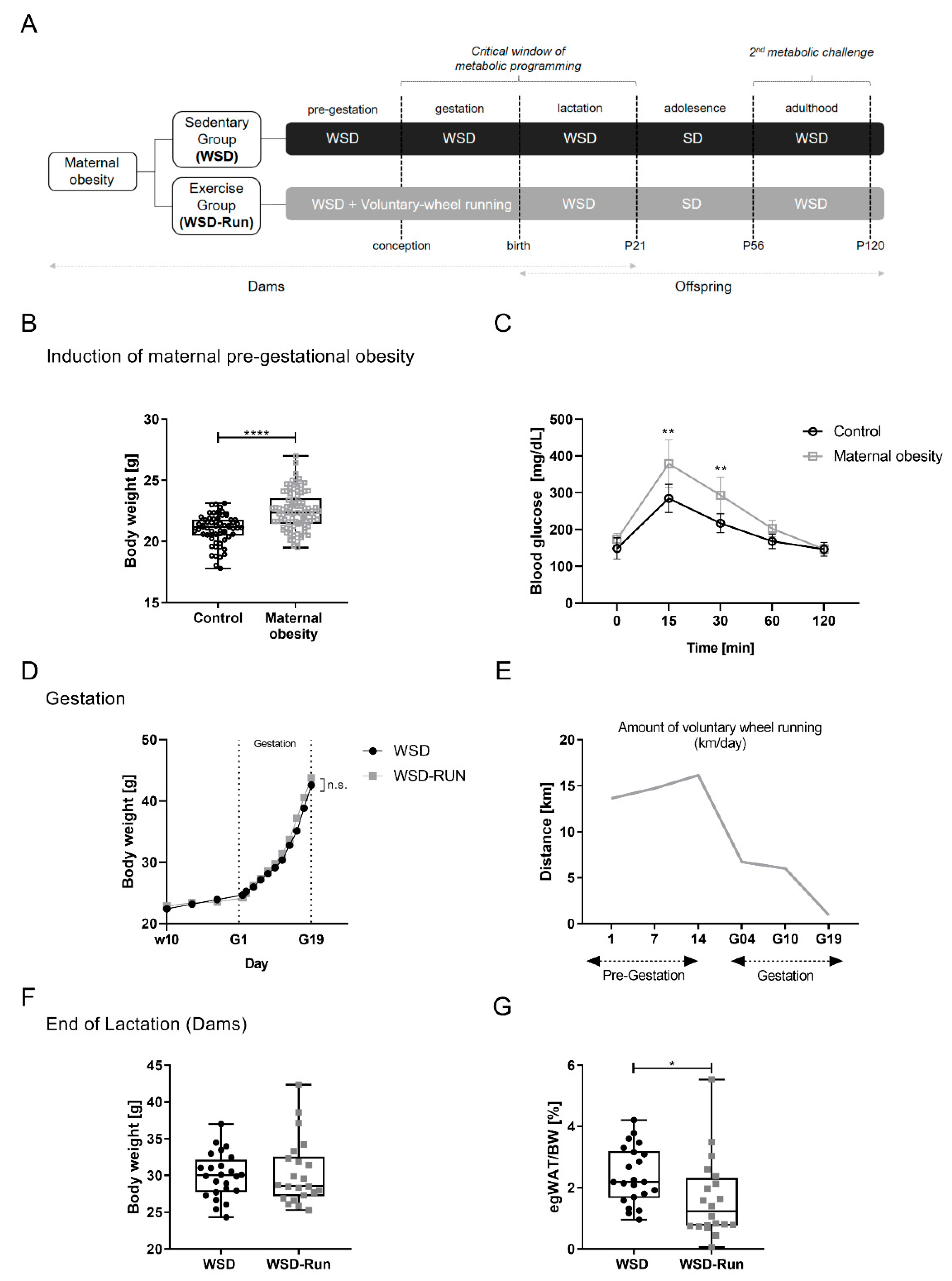
2.1. Animal Model
2.2. Analytical Procedures and Biochemical Measurements
2.3. Intraperitoneal Glucose Tolerance Test (ipGTT) and Insulin Tolerance Test (ipITT)
2.4. Biomarker Analyses
2.5. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.6. Protein Isolation
2.7. Immunoblotting
2.8. Histological Analysis of the Liver Tissue
2.9. Statistical Analysis
3. Results
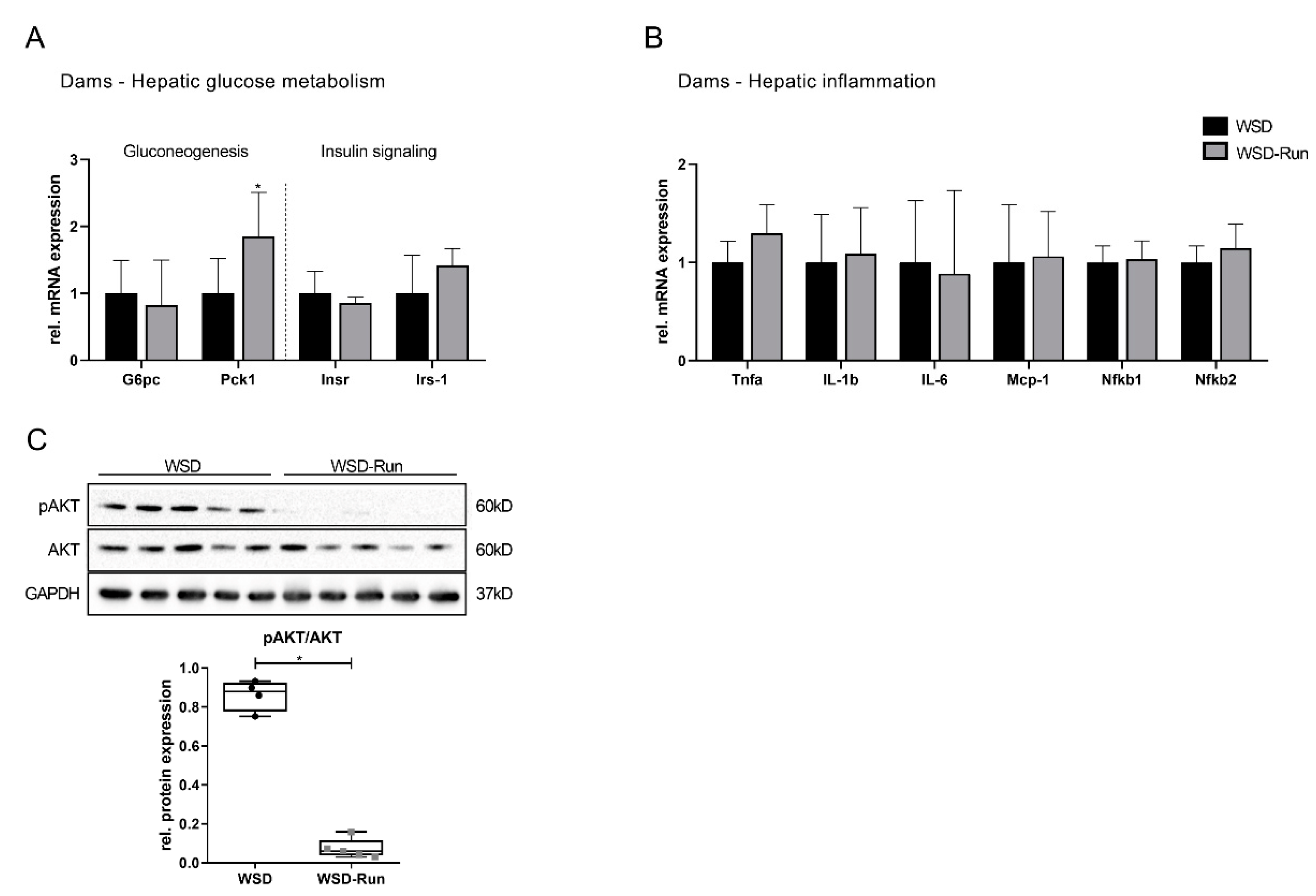
3.1. ME before and during Pregnancy Induces an Altered Body Composition in Obese Dams with a Reduced Amount of Epigonadal Fat Mass

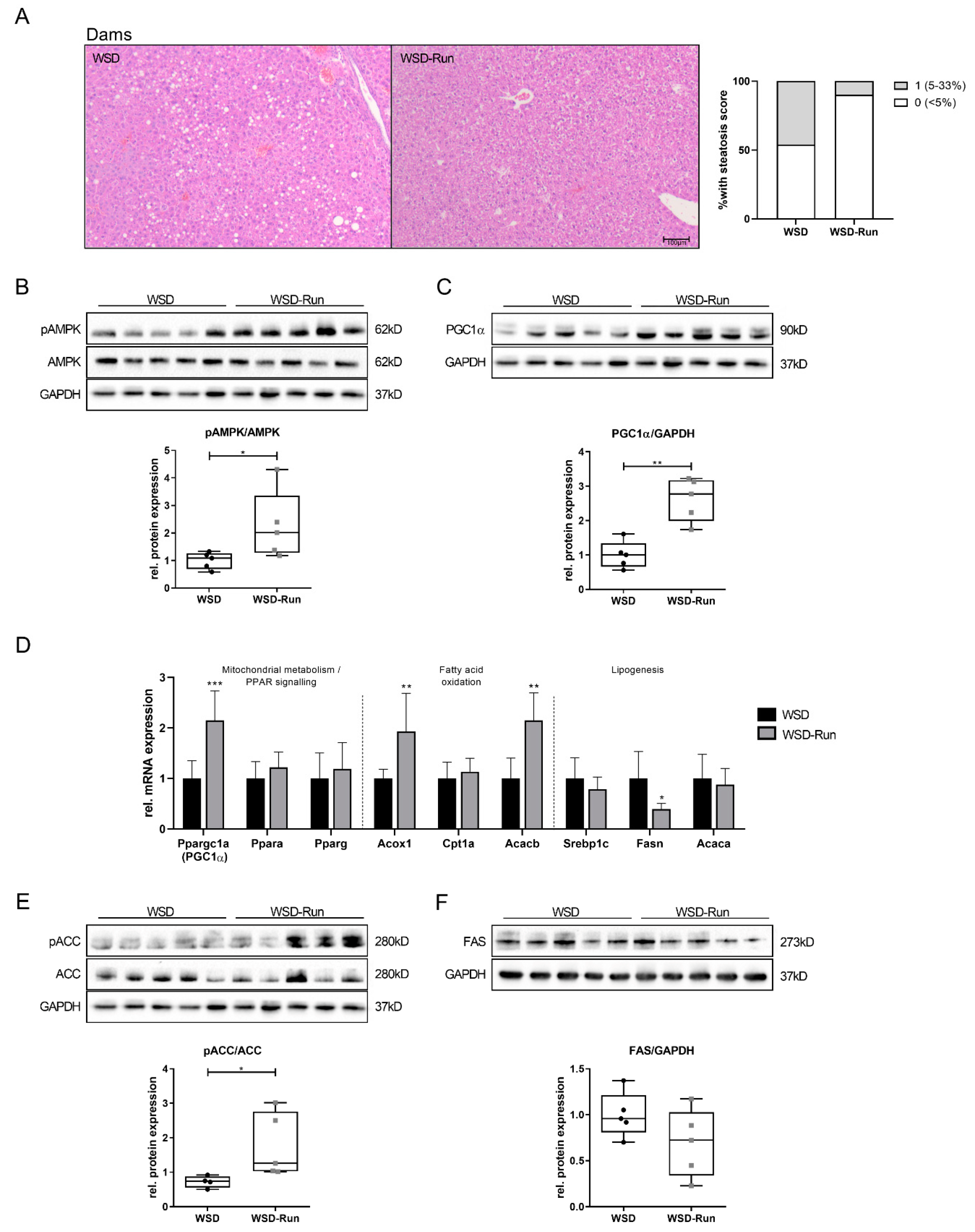
3.2. ME Prevents Obese Dams from WSD-Induced Hepatic Steatosis
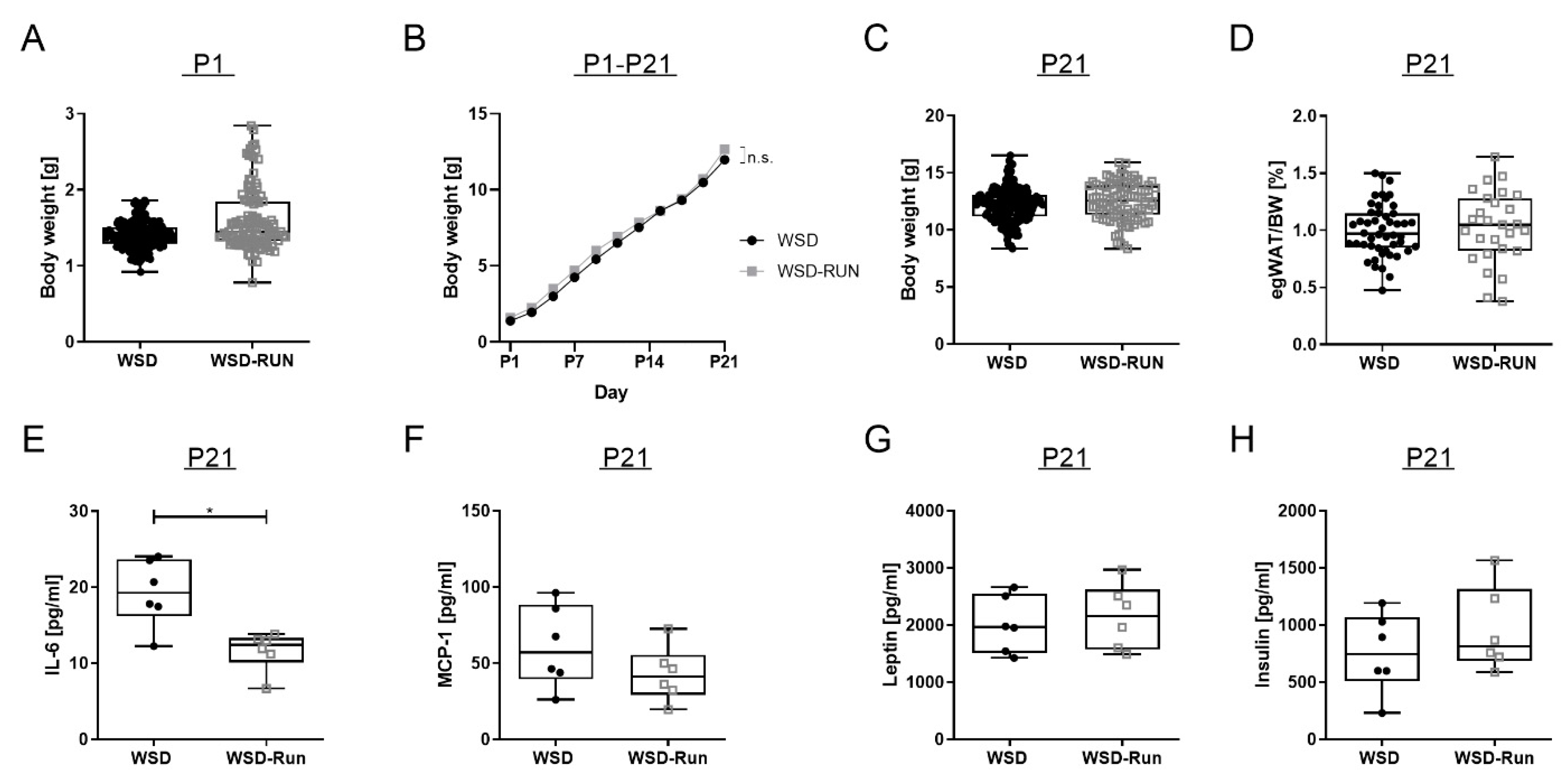
3.3. ME Exerts No Influence on Offspring’s Body Weight and Body Composition at P21
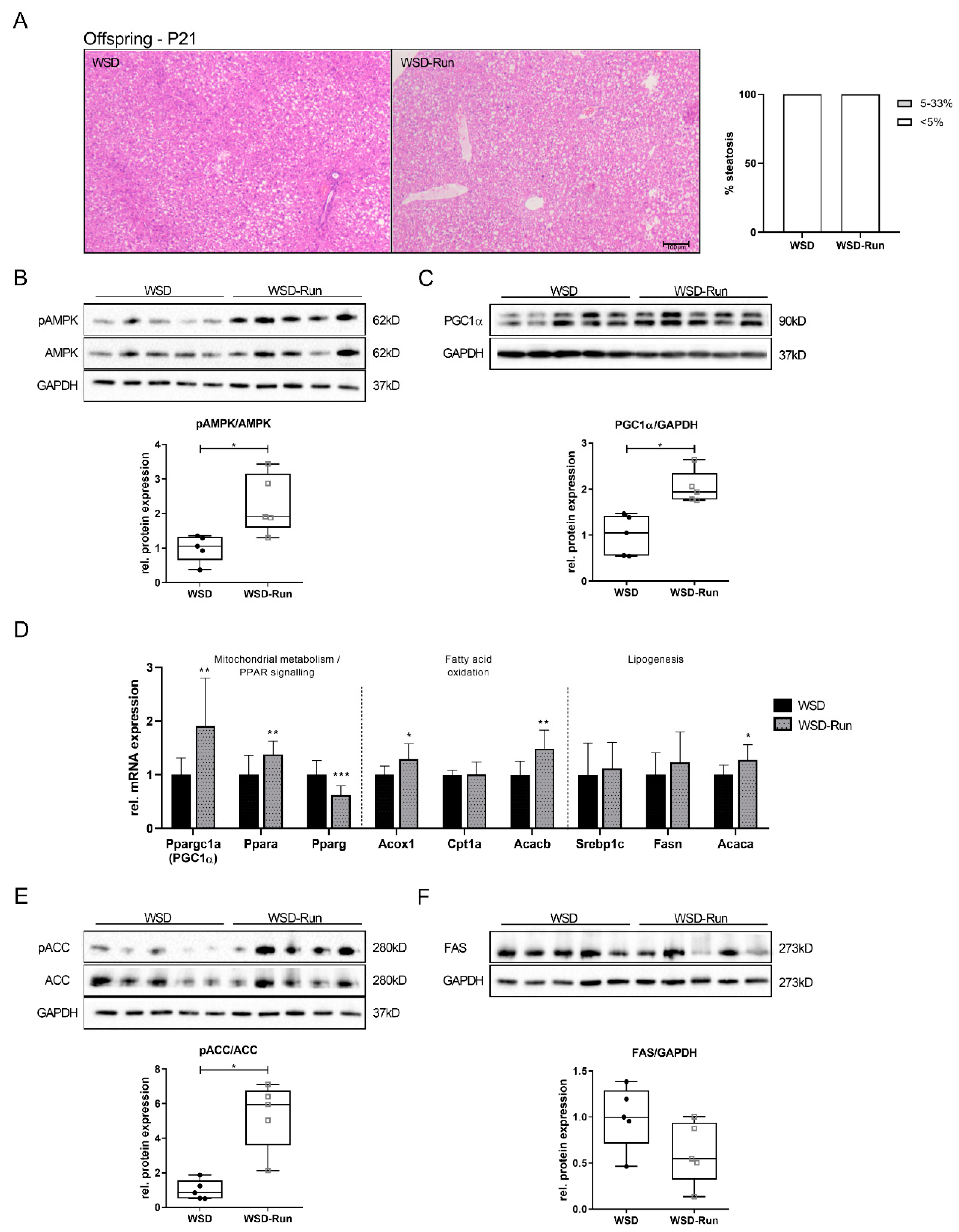
3.4. Offspring of Exercised Obese Dams Exhibit an Increased Hepatic AMPK-PGC1α Axis in Early Life
3.5. Offspring of Exercised Obese Dams Were Protected from WSD Induced Hepatic Steatosis and Increase in Adipose Tissue Mass in Later Life
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Catalano, P.M.; Shankar, K. Obesity and pregnancy: Mechanisms of short term and long term adverse consequences for mother and child. BMJ 2017, 356, j1. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, K.; Reynolds, R.; Prescott, S.; Nyirenda, M.; Jaddoe, V.; Eriksson, J.; Broekman, B. Influence of maternal obesity on the long-term health of offspring. Lancet Diabetes Endocrinol. 2017, 5, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Ogden, C.; Carroll, M.D.; Kit, B.; Flegal, K. Prevalence of obesity and trends in body mass index among US children and adolescents, 1999-2010. JAMA 2012, 307, 483–490. [Google Scholar] [CrossRef] [Green Version]
- Devlieger, R.; Benhalima, K.; Damm, P.; Van Assche, A.; Mathieu, C.; Mahmood, T.; Dunne, F.; Bogaerts, A. Maternal obesity in Europe: Where do we stand and how to move forward?: A scientific paper commissioned by the European Board and College of Obstetrics and Gynaecology (EBCOG). Eur. J. Obs. Gynecol. Reprod. Biol. 2016, 201, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Poston, L.; Caleyachetty, R.; Cnattingius, S.; Corvalán, C.; Uauy, R.; Herring, S.; Gillman, M. Preconceptional and maternal obesity: Epidemiology and health consequences. Lancet Diabetes Endocrinol. 2016, 4, 1025–1036. [Google Scholar] [CrossRef]
- Lisonkova, S.; Muraca, G.M.; Potts, J.; Liauw, J.; Chan, W.-S.; Skoll, A.; Lim, K.I. Association Between Prepregnancy Body Mass Index and Severe Maternal Morbidity. JAMA 2017, 318, 1777–1786. [Google Scholar] [CrossRef] [PubMed]
- Catalano, P.; DeMouzon, S.H. Maternal obesity and metabolic risk to the offspring: Why lifestyle interventions may have not achieved the desired outcomes. Int. J. Obes. 2015, 39, 642–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voerman, E.; Santos, S.; Patro Golab, B.; Amiano, P.; Ballester, F.; Barros, H.; Bergström, A.; Charles, M.-A.; Chatzi, L.; Chevrier, C.; et al. Maternal body mass index, gestational weight gain, and the risk of overweight and obesity across childhood: An individual participant data meta-analysis. PLoS Med. 2019, 16, e1002744. [Google Scholar] [CrossRef]
- Maffeis, C.; Morandi, A. Effect of Maternal Obesity on Foetal Growth and Metabolic Health of the Offspring. Obes. Facts 2017, 10, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Lahti-Pulkkinen, M.; Bhattacharya, S.; Wild, S.H.; Lindsay, R.S.; Räikkönen, K.; Norman, J.E.; Bhattacharya, S.; Reynolds, R.M. Consequences of being overweight or obese during pregnancy on diabetes in the offspring: A record linkage study in Aberdeen, Scotland. Diabetologia 2019, 62, 1412–1419. [Google Scholar] [CrossRef] [Green Version]
- Azzu, V.; Vacca, M.; Virtue, S.; Allison, M.; Vidal-Puig, A. Adipose Tissue-Liver Cross Talk in the Control of Whole-Body Metabolism: Implications in Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 158, 1899–1912. [Google Scholar] [CrossRef]
- Priest, C.; Tontonoz, P. Inter-organ cross-talk in metabolic syndrome. Nat. Metab. 2019, 1, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Byrne, C.D.; Targher, G. What’s new in NAFLD pathogenesis, biomarkers and treatment? Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 70–71. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.; Sarin, S.; Anstee, Q.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.; Schattenberg, J.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Foretz, M.; Violett, B. Regulation of hepatic metabolism by AMPK. J. Hepatol. 2011, 54, 827–829. [Google Scholar] [CrossRef]
- Viollet, B.; Guigas, B.; Leclerc, J.; Hébrard, S.; Lantier, L.; Mounier, M.; Andreelli, F.; Foretz, M. AMP-activated protein kinase in the regulation of hepatic energy metabolism: From physiology to therapeutic perspectives. Acta Physiol. 2009, 196, 81–98. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Even, P.; Viollet, B. AMPK Activation Reduces Hepatic Lipid Content by Increasing Fat Oxidation In Vivo. Int. J. Mol. Sci. 2018, 19, 2826. [Google Scholar] [CrossRef] [Green Version]
- Jeon, S.-M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol. Metab. 2017, 28, 545–560. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef]
- Smith, B.K.; Marcinko, K.; Desjardins, E.M.; Lally, J.S.; Ford, R.J.; Steinberg, G.R. Treatment of nonalcoholic fatty liver disease: Role of AMPK. Am. J. Physiol. Endocrinol. Meta.b 2016, 311, E730–E740. [Google Scholar] [CrossRef] [Green Version]
- Christoforou, E.R.; Sferruzzi-Perri, A.N. Molecular mechanisms governing offspring metabolic programming in rodent models of in utero stress. Cell Mol. Life Sci. 2020, 77, 4861–4898. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, D.J.; Reynolds, R.M.; Hardy, D.B. Developmental origins of health and disease: Current knowledge and potential mechanisms. Nutr. Rev. 2017, 75, 951–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barker, D.J.; Hales, C.N.; Fall, C.H.; Osmond, C.; Phipps, K.; Clark, P.M. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): Relation to reduced fetal growth. Diabetologia 1993, 36, 62–67. [Google Scholar] [CrossRef] [Green Version]
- Kilkenny, C.; Browne, W.; Cuthill, I.; Emerson, M.; Altmann, D. Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. PLoS Biol. 2010, 8, e1000412. [Google Scholar] [CrossRef]
- Bae-Gartz, I.; Kasper, P.; Großmann, N.; Breuer, S.; Janoschek, R.; Kretschmer, T.; Appel, S.; Schmitz, L.; Vohlen, C.; Quaas, A.; et al. Maternal exercise conveys protection against NAFLD in the offspring via hepatic metabolic programming. Sci. Rep. 2020, 10, 15424. [Google Scholar] [CrossRef]
- Wasinski, F.; Bacurau, R.; Estrela, G.; Klempin, F.; Arakaki, A.; Batista, R.; Mafra, F.; Do Nascimento, L.; Hiyane, M.; Velloso, L.; et al. Exercise during pregnancy protects adult mouse offspring from diet-induced obesity. Nutr. Metab. 2015, 12, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Carter, L.; Lewis, K.; Wilkerson, D.; Tobia, C.; Ngo Tenlep, S.; Shridas, P.; Garcia-Cazarin, M.; Wolff, G.; Andrade, F.; Charnigo, R.; et al. Perinatal exercise improves glucose homeostasis in adult offspring. Am. J. Physiol. Endocrinol. Metab. 2012, 303, 1061–1068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, J.; Baer, L.; Stanford, K. Maternal exercise improves the metabolic health of adult offspring. Trends Endocrinol. Metab. 2018, 29, 164–177. [Google Scholar] [CrossRef]
- Rother, E.; Kuschewski, R.; Alcazar, M.; Oberthuer, A.; Bae-Gartz, I.; Vohlen, C.; Roth, B.; Dötsch, J. Hypothalamic JNK1 and IKKβ activation and impaired early postnatal glucose metabolism after maternal perinatal high-fat feeding. Endocrinology 2012, 153, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, L.; Kuglin, R.; Bae-Gartz, I.; Janoschek, R.; Appel, S.; Mesaros, A.; Jakovcevski, I.; Vohlen, C.; Handwerk, M.; Ensenauer, R.; et al. Hippocampal insulin resistance links maternal obesity with impaired neuronal plasticity in adult offspring. Psychoneuroendocrinology 2018, 89, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Bae-Gartz, I.; Janoschek, R.; Kloppe, C.; Vohlen, C.; Roels, F.; Oberthür, A.; Alejandre Alcazar, M.; Lippach, G.; Muether, P.; Dinger, K.; et al. Running exercise in obese pregnancies prevents IL-6 trans-signaling in male offspring. Med. Sci. Sport. Exerc. 2016, 48, 829–838. [Google Scholar] [CrossRef]
- Kasper, P.; Vohlen, C.; Dinger, K.; Mohr, J.; Hucklenbruch-Rother, E.; Janoschek, R.; Köth, J.; Matthes, J.; Appel, S.; Dötsch, J.; et al. Renal Metabolic Programming Is Linked to the Dynamic Regulation of a Leptin-Klf15 Axis and Akt/AMPKα Signaling in Male Offspring of Obese Dams. Endocrinology 2017, 158, 3399–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrari, N.; Bae-Gartz, I.; Bauer, C.; Janoschek, R.; Koxholt, I.; Mahabir, E.; Appel, S.; Alejandre Alcazar, M.; Grossmann, N.; Vohlen, C.; et al. Exercise during pregnancy and its impact on mothers and offspring in humans and mice. J. Dev. Orig. Health Dis. 2018, 9, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Brunt, E.; Janney, C.; Di Bisceglie, A.; Neuschwander-Tetri, B.; Bacon, B. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474. [Google Scholar] [CrossRef]
- Kristiansen, M.; Veidal, S.; Rigbolt, K.; Tølbøl, K.; Roth, J.; Jelsing, J.; Vrang, N. Obese diet-induced mouse models of nonalcoholic steatohepatitis-tracking disease by liver biopsy. World J. Hepatol. 2016, 8, 673–684. [Google Scholar] [CrossRef]
- Kleiner, D.; Brunt, E.; Van Natta, A.; Behling, C.; Contos, M.; Cummings, O.; Ferrell, L.; Liu, Y.; Torbenso, M.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Kersten, S. Integrated physiology and systems biology of PPARα. Mol. Metab. 2014, 3, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Francque, S.; Szabo, G.; Abdelmalek, M.F.; Byrne, C.D.; Cusi, K.; Dufour, J.-F.; Roden, M.; Sacks, F.; Tacke, F. Nonalcoholic steatohepatitis: The role of peroxisome proliferator-activated receptors. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 24–39. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Matsusue, K.; Kashireddy, P.; Cao, W.-Q.; Yeldandi, V.; Yeldandi, A.V.; Rao, M.S.; Gonzalez, F.J.; Reddy, J.K. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression. J. Biol. Chem. 2003, 278, 498–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [Green Version]
- Piccinin, E.; Villani, G.; Moschetta, A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: The role of PGC1 coactivators. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 160–174. [Google Scholar] [CrossRef]
- Morris, E.; Meers, G.; Booth, F.; Fritsche, K.; Hardin, C.; Thyfault, J.; Ibdah, J. PGC-1α overexpression results in increased hepatic fatty acid oxidation with reduced triacylglycerol accumulation and secretion. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G979–G992. [Google Scholar] [CrossRef] [Green Version]
- Woods, A.; Williams, J.; Muckett, P.; Mayer, F.; Liljevald, M.; Bohlooly-Y, M.; Carling, D. Liver-Specific activation of AMPK prevents steatosis on a high-fructose diet. Cell Rep. 2017, 18, 3043–3051. [Google Scholar] [CrossRef] [Green Version]
- Garcia, D.; Hellberg, K.; Chaix, A.; Wallace, M.; Herzig, S.; Badur, M.G.; Lin, T.; Shokhirev, M.N.; Pinto, A.F.M.; Ross, D.S.; et al. Genetic Liver-Specific AMPK Activation Protects against Diet-Induced Obesity and NAFLD. Cell Rep. 2019, 26, 192–208.e6. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.-F.; Tian, M.-X.; Sun, R.-Q.; Zhang, M.-L.; Zhou, L.-S.; Jin, L.; Chen, L.-L.; Zhou, W.-J.; Duan, K.-L.; Chen, Y.-J.; et al. SIRT5 inhibits peroxisomal ACOX1 to prevent oxidative damage and is downregulated in liver cancer. EMBO Rep. 2018, 19, e45124. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161. [Google Scholar] [CrossRef]
- Vincent, M.F.; Erion, M.D.; Gruber, H.E.; Van den Berghe, G. Hypoglycaemic effect of AICAriboside in mice. Diabetologia 1996, 39, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Myers, R.; Li, Y.; Chen, Y.; Shen, X.; Fenyk-Melody, J.; Wu, M.; Ventre, J.; Doebber, T.; Fujii, N.; et al. Role of AMP-activated protein kinase in mechanism of metformin action. J. Clin. Invest. 2001, 108, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Cokorinos, E.C.; Delmore, J.; Reyes, A.R.; Albuquerque, B.; Kjøbsted, R.; Jørgensen, N.O.; Tran, J.-L.; Jatkar, A.; Cialdea, K.; Esquejo, R.M.; et al. Activation of Skeletal Muscle AMPK Promotes Glucose Disposal and Glucose Lowering in Non-human Primates and Mice. Cell Metab. 2017, 25, 1147–1159. [Google Scholar] [CrossRef]
- Esquejo, R.M.; Salatto, C.T.; Delmore, J.; Albuquerque, B.; Reyes, A.; Shi, Y.; Moccia, R.; Cokorinos, E.; Peloquin, M.; Monetti, M.; et al. Activation of Liver AMPK with PF-06409577 Corrects NAFLD and Lowers Cholesterol in Rodent and Primate Preclinical Models. EBioMedicine 2018, 31, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J. Clin. Invest. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fullerton, M.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.; O’Neill, H.; Ford, R.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [Green Version]
- Selen, E.S.; Choi, J.; Wolfgang, M.J. Discordant hepatic fatty acid oxidation and triglyceride hydrolysis leads to liver disease. JCI Insight 2021, 6, e135626. [Google Scholar] [CrossRef]
- Staehr, P.; Hother-Nielsen, O.; Landau, B.R.; Chandramouli, V.; Holst, J.J.; Beck-Nielsen, H. Effects of free fatty acids per se on glucose production, gluconeogenesis, and glycogenolysis. Diabetes 2003, 52, 260–267. [Google Scholar] [CrossRef] [Green Version]
- Wesolowski, S.; El Kasmi, K.; Jonscher, K.; Friedman, J. Developmental origins of NAFLD: A womb with a clue. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 81–96. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Reynolds, C.; Segovia, S.; Gray, C.; Vickers, M. Developmental programming of nonalcoholic fatty liver disease: The effect of early life nutrition on susceptibility and disease severity in later life. Biomed. Res. Int. 2015, 437107, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dearden, L.; Ozanne, S. Early life origins of metabolic disease: Developmental programming of hypothalamic pathways controlling energy homeostasis. Front. Neuroendocr. 2015, 39, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Twinn, D.; Ozanne, S. Early life nutrition and metabolic programming. Ann. N. Y. Acad. Sci. 2010, 1212, 78–96. [Google Scholar] [CrossRef] [PubMed]
- Wasinski, F.; Estrela, G.; Arakaki, A.; Bader, M.; Alenina, N.; Klempin, F.; Araujo, R. Maternal forced swimming reduces cell proliferation in the postnatal dentate gyrus of mouse offspring. Front. Neurosci. 2016, 10, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Raipuria, M.; Bahari, H.; Morris, M. Effects of Maternal Diet and Exercise during Pregnancy on Glucose Metabolism in Skeletal Muscle and Fat of Weanling Rats. PLoS ONE 2015, 10, e0120980. [Google Scholar] [CrossRef] [Green Version]
- Goedeke, L.; Bates, J.; Vatner, D.F.; Perry, R.J.; Wang, T.; Ramirez, R.; Li, L.; Ellis, M.W.; Zhang, D.; Wong, K.E.; et al. Acetyl-CoA Carboxylase Inhibition Reverses NAFLD and Hepatic Insulin Resistance but Promotes Hypertriglyceridemia in Rodents. Hepatology 2018, 68, 2197–2211. [Google Scholar] [CrossRef] [Green Version]
- Matsusue, K.; Haluzik, M.; Lambert, G.; Yim, S.-H.; Gavrilova, O.; Ward, J.M.; Brewer, B.J.; Reitman, M.L.; Gonzalez, F.J. Liver-specific disruption of PPARgamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Invest. 2003, 111, 737–747. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, A.; Zygmunt, M.; Clapp, J.F. 3rd Running throughout pregnancy: Effect on placental villous vascular volume and cell proliferation. Placenta 2004, 25, 694–698. [Google Scholar] [CrossRef]
- Thompson, M.D. Developmental Programming of NAFLD by Parental Obesity. Hepatol. Commun. 2020, 4, 1392–1403. [Google Scholar] [CrossRef]
- Aharoni-Simon, M.; Hann-Obercyger, M.; Pen, S.; Madar, Z.; Tirosh, O. Fatty liver is associated with impaired activity of PPARγ-coactivator 1α (PGC1α) and mitochondrial biogenesis in mice. Lab. Invest. 2011, 91, 1018–1028. [Google Scholar] [CrossRef] [Green Version]
- Laker, R.; Lillard, T.; Okutsu, M.; Zhang, M.; Hoehn, K.; Connelly, J.; Yan, Z. Exercise prevents maternal high-fat diet-induced hypermethylation of the Pgc-1α gene and age-dependent metabolic dysfunction in the offspring. Diabetes 2014, 63, 1605–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metere, A.; Graves, C.E. Factors Influencing Epigenetic Mechanisms: Is There A Role for Bariatric Surgery? High. Throughput 2020, 9, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wankhade, U.D.; Zhong, Y.; Kang, P.; Alfaro, M.; Chintapalli, S.V.; Thakali, K.M.; Shankar, K. Enhanced offspring predisposition to steatohepatitis with maternal high-fat diet is associated with epigenetic and microbiome alterations. PLoS ONE 2017, 12, e0175675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soderborg, T.K.; Borengasser, S.J.; Barbour, L.A.; Friedman, J.E. Microbial transmission from mothers with obesity or diabetes to infants: An innovative opportunity to interrupt a vicious cycle. Diabetologia 2016, 59, 895–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soderborg, T.K.; Clark, S.E.; Mulligan, C.E.; Janssen, R.C.; Babcock, L.; Ir, D.; Young, B.; Krebs, N.; Lemas, D.J.; Johnson, L.K.; et al. The gut microbiota in infants of obese mothers increases inflammation and susceptibility to NAFLD. Nat. Commun. 2018, 9, 4462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wankhade, U.D.; Zhong, Y.; Kang, P.; Alfaro, M.; Chintapalli, S.V.; Piccolo, B.D.; Mercer, K.E.; Andres, A.; Thakali, K.M.; Shankar, K. Maternal High-Fat Diet Programs Offspring Liver Steatosis in a Sexually Dimorphic Manner in Association with Changes in Gut Microbial Ecology in Mice. Sci. Rep. 2018, 8, 16502. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Xiao, X.; Li, M.; Zhang, Q.; Yu, M.; Zheng, J.; Deng, M. Maternal Exercise Improves High-Fat Diet-Induced Metabolic Abnormalities and Gut Microbiota Profiles in Mouse Dams and Offspring. Front. Cell Infect. Microbiol. 2020, 10, 292. [Google Scholar] [CrossRef]
- Sun, X.; Zhu, M.-J. AMP-activated protein kinase: A therapeutic target in intestinal diseases. Open Biol. 2017, 7, 170104. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Qin, L.; Hu, H.; Wang, Z. Alteration of the Gut Microbiota and Its Effect on AMPK/NADPH Oxidase Signaling Pathway in 2K1C Rats. Biomed. Res. Int. 2019, 2019, 8250619. [Google Scholar] [CrossRef] [Green Version]
- Kruse, M.; Seki, Y.; Vuguin, P.M.; Du, X.Q.; Fiallo, A.; Glenn, A.S.; Singer, S.; Breuhahn, K.; Katz, E.B.; Charron, M.J. High-fat intake during pregnancy and lactation exacerbates high-fat diet-induced complications in male offspring in mice. Endocrinology 2013, 154, 3565–3576. [Google Scholar] [CrossRef]
- Bruce, K.D.; Cagampang, F.R.; Argenton, M.; Zhang, J.; Ethirajan, P.L.; Burdge, G.C.; Bateman, A.C.; Clough, G.F.; Poston, L.; Hanson, M.A.; et al. Maternal high-fat feeding primes steatohepatitis in adult mice offspring, involving mitochondrial dysfunction and altered lipogenesis gene expression. Hepatology 2009, 50, 1796–1808. [Google Scholar] [CrossRef]
- McCurdy, C.E.; Bishop, J.M.; Williams, S.M.; Grayson, B.E.; Smith, M.S.; Friedman, J.E.; Grove, K.L. Maternal high-fat diet triggers lipotoxicity in the fetal livers of nonhuman primates. J. Clin. Invest. 2009, 119, 323–335. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Peng, H.; Xu, H.; Li, J.; Golovko, M.; Cheng, H.; Lynch, E.C.; Liu, L.; McCauley, N.; Kennedy, L.; et al. Maternal diet intervention before pregnancy primes offspring lipid metabolism in liver. Lab. Invest. 2020, 100, 553–569. [Google Scholar] [CrossRef]
- Hawley, S.A.; Ford, R.J.; Smith, B.K.; Gowans, G.J.; Mancini, S.J.; Pitt, R.D.; Day, E.A.; Salt, I.P.; Steinberg, G.R.; Hardie, D.G. The Na+/Glucose Cotransporter Inhibitor Canagliflozin Activates AMPK by Inhibiting Mitochondrial Function and Increasing Cellular AMP Levels. Diabetes 2016, 65, 2784–2794. [Google Scholar] [CrossRef] [Green Version]
- Steneberg, P.; Lindahl, E.; Dahl, U.; Lidh, E.; Straseviciene, J.; Backlund, F.; Kjellkvist, E.; Berggren, E.; Lundberg, I.; Bergqvist, I.; et al. PAN-AMPK activator O304 improves glucose homeostasis and microvascular perfusion in mice and type 2 diabetes patients. JCI Insight 2018, 3, e99114. [Google Scholar] [CrossRef] [PubMed]
- Myers, R.W.; Guan, H.-P.; Ehrhart, J.; Petrov, A.; Prahalada, S.; Tozzo, E.; Yang, X.; Kurtz, M.M.; Trujillo, M.; Gonzalez Trotter, D.; et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science 2017, 357, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Li, J.; Wang, M.; Qu, K.; Zhu, H. A novel AMPK activator improves hepatic lipid metabolism and leukocyte trafficking in experimental hepatic steatosis. J. Pharmacol. Sci. 2019, 140, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Jorquera, G.; Echiburú, B.; Crisosto, N.; Sotomayor-Zárate, R.; Maliqueo, M.; Cruz, G. Metformin during Pregnancy: Effects on Offspring Development and Metabolic Function. Front. Pharmacol. 2020, 11, 653. [Google Scholar] [CrossRef] [PubMed]
- Given, J.E.; Loane, M.; Garne, E.; Addor, M.-C.; Bakker, M.; Bertaut-Nativel, B.; Gatt, M.; Klungsoyr, K.; Lelong, N.; Morgan, M.; et al. Metformin exposure in first trimester of pregnancy and risk of all or specific congenital anomalies: Exploratory case-control study. BMJ 2018, 361, k2477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyer, S.; Balani, J.; Shehata, H. Metformin in Pregnancy: Mechanisms and Clinical Applications. Int. J. Mol. Sci. 2018, 19, 1954. [Google Scholar] [CrossRef] [Green Version]
- Lonard, D.M.; O’Malley, B.W. Nuclear receptor coregulators: Modulators of pathology and therapeutic targets. Nat. Rev. Endocrinol. 2012, 8, 598–604. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasper, P.; Breuer, S.; Hoffmann, T.; Vohlen, C.; Janoschek, R.; Schmitz, L.; Appel, S.; Fink, G.; Hünseler, C.; Quaas, A.; et al. Maternal Exercise Mediates Hepatic Metabolic Programming via Activation of AMPK-PGC1α Axis in the Offspring of Obese Mothers. Cells 2021, 10, 1247. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051247
Kasper P, Breuer S, Hoffmann T, Vohlen C, Janoschek R, Schmitz L, Appel S, Fink G, Hünseler C, Quaas A, et al. Maternal Exercise Mediates Hepatic Metabolic Programming via Activation of AMPK-PGC1α Axis in the Offspring of Obese Mothers. Cells. 2021; 10(5):1247. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051247
Chicago/Turabian StyleKasper, Philipp, Saida Breuer, Thorben Hoffmann, Christina Vohlen, Ruth Janoschek, Lisa Schmitz, Sarah Appel, Gregor Fink, Christoph Hünseler, Alexander Quaas, and et al. 2021. "Maternal Exercise Mediates Hepatic Metabolic Programming via Activation of AMPK-PGC1α Axis in the Offspring of Obese Mothers" Cells 10, no. 5: 1247. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051247