
Amyotrophic Lateral Sclerosis (ALS): Stressed by Dysfunctional Mitochondria-Endoplasmic Reticulum Contacts (MERCs)


Abstract
:1. Introduction
2. Important ALS Genes and Cytosolic Stress
3. Aggregation-Derived Cytosolic Stress Triggers ER Dysfunction
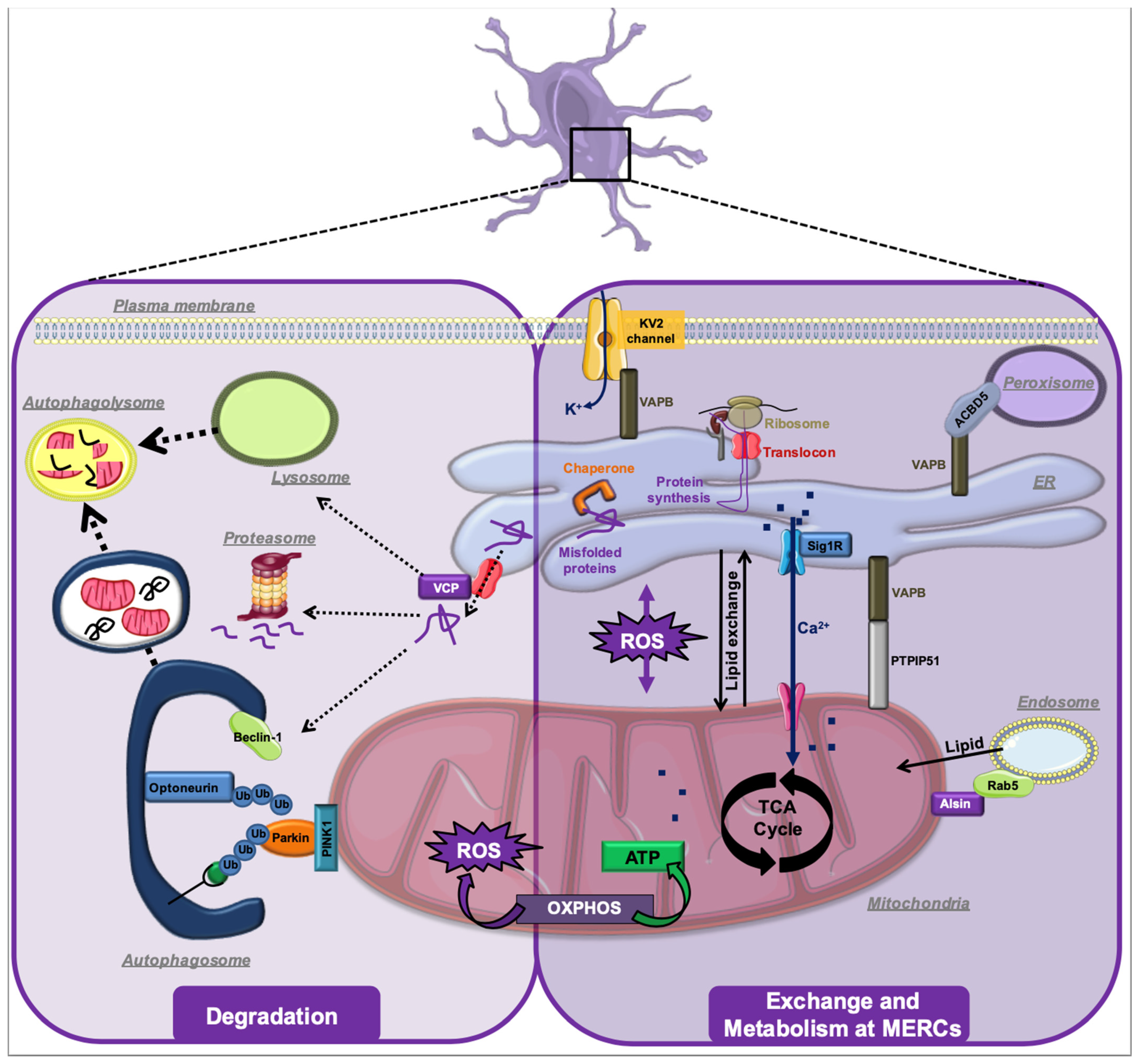
4. ALS-Associated Stress Signaling Converges on Membrane Contact Sites
5. MERCs in Other Types of Neurodegeneration
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fu, H.; Hardy, J.; Duff, K.E. Selective vulnerability in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1350–1358. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegeneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef] [PubMed]
- Gerovska, D.; Irizar, H.; Otaegi, D.; Ferrer, I.; De Munain, A.L.; Araúzo-Bravo, M.J. Genealogy of the neurodegenerative diseases based on a meta-analysis of age-stratified incidence data. Sci. Rep. 2020, 10, 18923. [Google Scholar] [CrossRef] [PubMed]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef] [Green Version]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef]
- Hetz, C.; Saxena, S. ER stress and the unfolded protein response in neurodegeneration. Nat. Rev. Neurol. 2017, 13, 477–491. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 710, 132933. [Google Scholar] [CrossRef]
- Van Dam, L.; Dansen, T.B. Cross-talk between redox signalling and protein aggregation. Biochem. Soc. Trans. 2020, 48, 379–397. [Google Scholar] [CrossRef] [Green Version]
- Kirstein, J.; Morito, D.; Kakihana, T.; Sugihara, M.; Minnen, A.; Hipp, M.S.; Nussbaum-Krammer, C.; Kasturi, P.; Hartl, F.U.; Nagata, K.; et al. Proteotoxic stress and ageing triggers the loss of redox homeostasis across cellular compartments. EMBO J. 2015, 34, 2334–2349. [Google Scholar] [CrossRef] [Green Version]
- Saxena, S.; Cabuy, E.; Caroni, P. A role for motoneuron subtype–selective ER stress in disease manifestations of FALS mice. Nat. Neurosci. 2009, 12, 627–636. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic Reticulum Stress and Oxidative Stress: A Vicious Cycle or a Double-Edged Sword? Antioxid. Redox Signal. 2007, 9, 2277–2294. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Harding, H.; Zhang, Y.; Bertolotti, A.; Zeng, H.; Ron, D. Perk Is Essential for Translational Regulation and Cell Survival during the Unfolded Protein Response. Mol. Cell 2000, 5, 897–904. [Google Scholar] [CrossRef]
- Scorrano, L.; De Matteis, M.A.; Emr, S.; Giordano, F.; Hajnóczky, G.; Kornmann, B.; Lackner, L.L.; Levine, T.P.; Pellegrini, L.; Reinisch, K.; et al. Coming together to define membrane contact sites. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Prinz, W.A.; Toulmay, A.; Balla, T. The functional universe of membrane contact sites. Nat. Rev. Mol. Cell Biol. 2019, 21, 7–24. [Google Scholar] [CrossRef]
- Chung, J.; Torta, F.; Masai, K.; Lucast, L.; Czapla, H.; Tanner, L.B.; Narayanaswamy, P.; Wenk, M.R.; Nakatsu, F.; De Camilli, P. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science 2015, 349, 428–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raturi, A.; Gutiérrez, T.; Ortiz-Sandoval, C.; Ruangkittisakul, A.; Herrera-Cruz, M.S.; Rockley, J.P.; Gesson, K.; Ourdev, D.; Lou, P.-H.; Lucchinetti, E.; et al. TMX1 determines cancer cell metabolism as a thiol-based modulator of ER–mitochondria Ca2+ flux. J. Cell Biol. 2016, 214, 433–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria–ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef]
- Anastasia, I.; Ilacqua, N.; Raimondi, A.; Lemieux, P.; Ghandehari-Alavijeh, R.; Faure, G.; Mekhedov, S.L.; Williams, K.J.; Caicci, F.; Valle, G.; et al. Mitochondria-rough-ER contacts in the liver regulate systemic lipid homeostasis. Cell Rep. 2021, 34, 108873. [Google Scholar] [CrossRef]
- Gomez-Suaga, P.; Pailusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C.J. The ER-Mitochondria Tethering Complex VAPB-PTPIP51 Regulates Autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoica, R.; De Vos, K.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J.; et al. ER–mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 3996. [Google Scholar] [CrossRef] [Green Version]
- Simmen, T.; Aslan, J.E.; Thomas, L.; Wan, L.; Xiang, Y. PACS-2 controls endoplasmic reticulum-mitochondria communication and Bid-mediated apoptosis. EMBO J. 2005, 24, 717–729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elbaz-Alon, Y.; Shinder, V.; Stiller, S.B.; Shimoni, E.; Geiger, T.; Schuldiner, M. Lam6 Regulates the Extent of Contacts between Organelles. Cell Rep. 2015, 12, 7–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, E.J. Newly made phosphatidylserine and phosphatidylethanolamine are preferentially translocated between rat liver mitochondria and endoplasmic reticulum. J. Biol. Chem. 1991, 266, 89–97. [Google Scholar] [CrossRef]
- Gellerich, F.N.; Gizatullina, Z.; Trumbeckaite, S.; Nguyen, H.P.; Pallas, T.; Arandarcikaite, O.; Vielhaber, S.; Seppet, E.; Striggow, F. The regulation of OXPHOS by extramitochondrial calcium. Biochim. Biophys. Acta 2010, 1797, 1018–1027. [Google Scholar] [CrossRef] [Green Version]
- Orrell, R.W. Motor neuron disease: Systematic reviews of treatment for ALS and SMA. Br. Med. Bull. 2009, 93, 145–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetz, C.G. Amyotrophic lateral sclerosis: Early contributions of Jean-Martin Charcot. Muscle Nerve 2000, 23, 336–343. [Google Scholar] [CrossRef]
- GBD 2016 Motor Neuron Disease Collaborators. Global, regional, and national burden of motor neuron diseases 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 1083–1097. [Google Scholar] [CrossRef] [Green Version]
- Logroscino, G.; Traynor, B.J.; Hardiman, O.; Chio, A.; Mitchell, D.; Swingler, R.J.; Millul, A.; Benn, E.; Beghi, E.; Eurals, F. Incidence of amyotrophic lateral sclerosis in Europe. J. Neurol. Neurosurg. Psychiatry 2009, 81, 385–390. [Google Scholar] [CrossRef]
- Calvo, A.C.; Manzano, R.; Mendonça, D.M.F.; Muñoz, M.J.; Zaragoza, P.; Osta, R. Amyotrophic Lateral Sclerosis: A Focus on Disease Progression. BioMed Res. Int. 2014, 2014, 925101. [Google Scholar] [CrossRef]
- Masrori, P.; Van Damme, P. Amyotrophic lateral sclerosis: A clinical review. Eur. J. Neurol. 2020, 27, 1918–1929. [Google Scholar] [CrossRef]
- Perry, J.J.P.; Pratt, A.; Getzoff, E.D. Amyotrophic lateral sclerosis: Update and new developments. Degener. Neurol. Neuromuscul. Dis. 2012, 2, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.C.; Hentati, A.; Donaldson, D.H.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.C.; Shaw, P.J. Oxidative stress in ALS: Key role in motor neuron injury and therapeutic target. Free Radic. Biol. Med. 2010, 48, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Okado-Matsumoto, A.; Fridovich, I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu, Zn-SOD in mitochondria. J. Biol. Chem. 2001, 276, 38388–38393. [Google Scholar] [CrossRef] [Green Version]
- Cleveland, D.; Laing, N.; Hurse, P.V., Jr.; Brown, R.H. Toxic mutants in Charcot’s sclerosis. Nat. Cell Biol. 1995, 378, 342–343. [Google Scholar] [CrossRef] [PubMed]
- Bruijn, L.I.; Becher, M.W.; Lee, M.K.; Anderson, K.L.; Jenkins, N.A.; Copeland, N.G.; Sisodia, S.S.; Rothstein, J.D.; Borchelt, D.R.; Price, D.L.; et al. ALS-Linked SOD1 Mutant G85R Mediates Damage to Astrocytes and Promotes Rapidly Progressive Disease with SOD1-Containing Inclusions. Neuron 1997, 18, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Bruijn, L.I.; Houseweart, M.K.; Kato, S.; Anderson, K.L.; Anderson, S.D.; Ohama, E.; Reaume, A.G.; Scott, R.W.; Cleveland, D. Aggregation and Motor Neuron Toxicity of an ALS-Linked SOD1 Mutant Independent from Wild-Type SOD1. Science 1998, 281, 1851–1854. [Google Scholar] [CrossRef] [Green Version]
- Bozzo, F.; Mirra, A.; Carrì, M. Oxidative stress and mitochondrial damage in the pathogenesis of ALS: New perspectives. Neurosci. Lett. 2017, 636, 3–8. [Google Scholar] [CrossRef]
- Carri, M.T.; Valle, C.; Bozzo, F.; Cozzolina, M. Oxidative stress and mitochondrial damage: Importance in non-SOD1 ALS. Front. Cell. Neurosci. 2015, 9, 41. [Google Scholar] [CrossRef] [Green Version]
- Keith, J.L.; Swinkin, E.; Gao, A.; Alminawi, S.; Zhang, M.; McGoldrick, P.; McKeever, P.; Robertson, J.; Rogaeva, E.; Zinman, L. Neuropathologic description of CHCHD10 mutated amyotrophic lateral sclerosis. Neurol. Genet. 2019, 6, e394. [Google Scholar] [CrossRef] [Green Version]
- Boylan, K. Familial Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 807–830. [Google Scholar] [CrossRef] [Green Version]
- Mejzini, R.; Flynn, L.; Pitout, I.L.; Fletcher, S.; Wilton, S.; Akkari, P.A. ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now? Front. Neurosci. 2019, 13, 1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muddapu, V.R.; Dharshini, S.A.P.; Chakravarthy, V.S.; Gromiha, M.M. Neurodegenerative Diseases—Is Metabolic Deficiency the Root Cause? Front. Neurosci. 2020, 14, 213. [Google Scholar] [CrossRef] [PubMed]
- Saez-Atienzar, S.; Bandres-Ciga, S.; Langston, R.G.; Kim, J.J.; Choi, S.W.; Reynolds, R.H.; Abramzon, Y.; Dewan, R.; Ahmed, S.; Landers, J.E.; et al. Genetic analysis of amyotrophic lateral sclerosis identifies contributing pathways and cell types. Sci. Adv. 2021, 7, eabd9036. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O.; Kiernan, M.C.; Chiò, A.; Rix-Brooks, B.; Berg, L.H.V.D. Amyotrophic lateral sclerosis: Moving towards a new classification system. Lancet Neurol. 2016, 15, 1182–1194. [Google Scholar] [CrossRef]
- Kim, G.; Gautier, O.; Tassoni-Tsuchida, E.; Ma, X.R.; Gitler, A.D. ALS Genetics: Gains, Losses, and Implications for Future Therapies. Neuron 2020, 108, 822–842. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, R.; Sone, J.; Atsuta, N.; Tohnai, G.; Watanabe, H.; Yokoi, D.; Ito, M. Next-generation sequencing of 28 ALS-related genes in a Japanese ALS cohort. Neurobiol. Aging 2016, 39, 219.e1. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC Hexanucleotide Repeat in Noncoding Region of C9ORF72 Causes Chromosome 9p-Linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Sánchez, J.S.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A Hexanucleotide Repeat Expansion in C9ORF72 Is the Cause of Chromosome 9p21-Linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Gendron, T.F.; Petrucelli, L. Disease Mechanisms of C9ORF72 Repeat Expansions. Cold Spring Harb. Perspect. Med. 2017, 8, a024224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fratta, P.; Mizielinska, S.; Nicoll, A.J.; Zloh, M.; Fisher, E.; Parkinson, G.; Isaacs, A.M. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci. Rep. 2012, 2, 1016. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Serano, A.; Gerbino, V.; Giorgi, A.; Di Francesco, L.; Nencini, M.; Bozzo, F. Nuclear accumulation of mRNAs underlies G4C2-repeat-induced translational repression in a cellular model of C9orf72 ALS. J. Cell Sci. 2015, 128, 1787–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freibaum, B.D.; Lu, Y.; Lopez-Gonzalez, R.; Kim, N.C.; Almeida, S.; Lee, K.-H.; Badders, N.; Valentine, M.; Miller, B.L.; Wong, P.C.; et al. GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nat. Cell Biol. 2015, 525, 129–133. [Google Scholar] [CrossRef]
- Jovičić, A.; Mertens, J.; Boeynaems, S.; Bogaert, E.; Chai, N.; Yamada, S.B.; Paul, J.W.; Sun, S.; Herdy, J.R.; Bieri, G.; et al. Modifiers of C9orf72 dipeptide repeat toxicity connect nucleocytoplasmic transport defects to FTD/ALS. Nat. Neurosci. 2015, 18, 1226–1229. [Google Scholar] [CrossRef] [Green Version]
- Ash, P.E.; Bieniek, K.; Gendron, T.F.; Caulfield, T.; Lin, W.-L.; DeJesus-Hernandez, M.; van Blitterswijk, M.; Jansen-West, K.; Paul, J.W.; Rademakers, R.; et al. Unconventional Translation of C9ORF72 GGGGCC Expansion Generates Insoluble Polypeptides Specific to c9FTD/ALS. Neuron 2013, 77, 639–646. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Weng, S.-M.; Arzberger, T.; May, S.; Rentzsch, K.; Kremmer, E.; Schmid, B.; Kretzschmar, H.A.; Cruts, M.; Van Broeckhoven, C.; et al. The C9orf72 GGGGCC Repeat Is Translated into Aggregating Dipeptide-Repeat Proteins in FTLD/ALS. Science 2013, 339, 1335–1338. [Google Scholar] [CrossRef]
- Hayes, L.R.; Duan, L.; Bowen, K.; Kalab, P.; Rothstein, J.D. C9orf72 arginine-rich dipeptide repeat proteins disrupt karyopherin-mediated nuclear import. eLife 2020, 9, 9. [Google Scholar] [CrossRef]
- Van Blitterswijk, M.; Gendron, T.F.; Baker, M.C.; Finch, M.A. Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol. 2015, 130, 863–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boivin, M.; Pfister, V.; Gaucherot, A.; Ruffenach, F.; Negroni, L.; Sellier, C.; Charlet-Berguerand, N. Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. EMBO J. 2020, 39, e100574. [Google Scholar] [CrossRef]
- Laflamme, C.; Kumar, R.; Schwartz, J.; Chen, C.X.; You, Z.; Gileadi, O. Implementation of an antibody characterization procedure and application to the major ALS/FTD disease gene C9ORF72. Elife 2019, 8, e48363. [Google Scholar] [CrossRef]
- Sullivan, P.M.; Zhou, X.; Robins, A.M. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 2016, 4, 51. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Liu, H.; Itoh, K.; Oh, S.; Zhao, L.; Murata, D.; Sesaki, H.; Hartung, T.; Na, C.H.; Wang, J. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 2021, 33, 531–546.e9. [Google Scholar] [CrossRef]
- Corbier, C.; Sellier, C. C9ORF72 is a GDP/GTP exchange factor for Rab8 and Rab39 and regulates autophagy. Small GTPases 2017, 8, 181–186. [Google Scholar] [CrossRef] [Green Version]
- Ho, W.Y.; Tai, Y.K.; Chang, J.-C.; Liang, J.; Tyan, S.-H.; Chen, S.; Guan, J.-L.; Zhou, H.; Shen, H.-M.; Koo, E.; et al. The ALS-FTD-linked gene product, C9orf72, regulates neuronal morphogenesis via autophagy. Autophagy 2019, 15, 827–842. [Google Scholar] [CrossRef] [PubMed]
- McAlpine, W.; Sun, L.; Wang, K.-W.; Liu, A.; Jain, R.; Miguel, M.S.; Wang, J.; Zhang, Z.; Hayse, B.; McAlpine, S.G.; et al. Excessive endosomal TLR signaling causes inflammatory disease in mice with defective SMCR8-WDR41-C9ORF72 complex function. Proc. Natl. Acad. Sci. USA 2018, 115, E11523–E11531. [Google Scholar] [CrossRef] [Green Version]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Russ, C.; Davis, A. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in Frontotemporal Lobar Degeneration and Amyotrophic Lateral Sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Arai, S.; Song, X.; Reichart, D.; Du, K.; Pascual, G.; Tempst, P.; Rosenfeld, M.G.; Glass, C.K.; Kurokawa, R. Induced ncRNAs allosterically modify RNA-binding proteins in cis to inhibit transcription. Nat. Cell Biol. 2008, 454, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Baloh, R.H. How do the RNA-binding proteins TDP-43 and FUS relate to amyotrophic lateral sclerosis and frontotemporal degeneration, and to each other? Curr. Opin. Neurol. 2012, 25, 701–707. [Google Scholar] [CrossRef]
- Giordana, M.T.; Piccinini, M.; Grifoni, S.; DE Marco, G.; Vercellino, M.; Magistrello, M.; Pellerino, A.; Buccinnà, B.; Lupino, E.; Rinaudo, M.T. TDP-43 Redistribution is an Early Event in Sporadic Amyotrophic Lateral Sclerosis. Brain Pathol. 2010, 20, 351–360. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.S.; Snead, D.; Shorter, J. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef] [Green Version]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Cleveland, D.W. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Charbaut, E.; Chauvin, S.; Enslen, H.; Zamaroczy, S.; Sobel, A. Two separate motifs cooperate to target stathmin-related proteins to the Golgi complex. J. Cell Sci. 2005, 118, 2313–2323. [Google Scholar] [CrossRef] [Green Version]
- Klim, J.R.; Williams, L.A.; Limone, F.; Juan, I.G.S.; Davis-Dusenbery, B.N.; Mordes, D.A.; Burberry, A.; Steinbaugh, M.; Gamage, K.K.; Kirchner, R.; et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 2019, 22, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Melamed, Z.; López-Erauskin, J.; Baughn, M.; Zhang, O.; Drenner, K.; Sun, Y.; Freyermuth, F.; McMahon, M.A.; Beccari, M.S.; Artates, J.W.; et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat. Neurosci. 2019, 22, 180–190. [Google Scholar] [CrossRef]
- Ito, D.; Seki, M.; Tsunoda, Y.; Uchiyama, H.; Suzuki, N. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann. Neurol. 2010, 69, 152–162. [Google Scholar] [CrossRef]
- Shiihashi, G.; Ito, D.; Yagi, T.; Nihei, Y.; Ebine, T.; Suzuki, N. Mislocated FUS is sufficient for gain-of-toxic-function amyotrophic lateral sclerosis phenotypes in mice. Brain 2016, 139, 2380–2394. [Google Scholar] [CrossRef]
- Lagier-Tourenne, C.; Hutt, K.R.; Vu, A.Q.; Huelga, S.C.; Klutario, M.C. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142. [Google Scholar] [CrossRef]
- Mureşan, V.; Muresan, Z.L. Shared Molecular Mechanisms in Alzheimer’s Disease and Amyotrophic Lateral Sclerosis: Neurofilament-Dependent Transport of sAPP, FUS, TDP-43 and SOD1, with Endoplasmic Reticulum-Like Tubules. Neurodegener. Dis. 2015, 16, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Niu, L.; Ma, T.; Yang, F.; Yan, B.; Tang, X.; Yin, H.; Wu, Q.; Huang, Y.; Yao, Z.-P.; Wang, J.; et al. Atlastin-mediated membrane tethering is critical for cargo mobility and exit from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2019, 116, 14029–14038. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.; Qi, L. Quality Control in the Endoplasmic Reticulum: Crosstalk between ERAD and UPR pathways. Trends Biochem. Sci. 2018, 43, 593–605. [Google Scholar] [CrossRef]
- Molinari, M.; Calanca, V.; Galli, C.; Lucca, P.; Paganetti, P. Role of EDEM in the Release of Misfolded Glycoproteins from the Calnexin Cycle. Science 2003, 299, 1397–1400. [Google Scholar] [CrossRef]
- Oda, Y.; Hosokawa, N.; Wada, I.; Nagata, K. EDEM As an Acceptor of Terminally Misfolded Glycoproteins Released from Calnexin. Science 2003, 299, 1394–1397. [Google Scholar] [CrossRef]
- Song, B.-L.; Sever, N.; DeBose-Boyd, R.A. Gp78, a Membrane-Anchored Ubiquitin Ligase, Associates with Insig-1 and Couples Sterol-Regulated Ubiquitination to Degradation of HMG CoA Reductase. Mol. Cell 2005, 19, 829–840. [Google Scholar] [CrossRef]
- Fang, S.; Ferrone, M.; Yang, C.; Jensen, J.P.; Tiwari, S.; Weissman, A.M. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 2001, 98, 14422–14427. [Google Scholar] [CrossRef] [Green Version]
- St-Pierre, P.; Dang, T.; Joshi, B.; Nabi, I.R. Peripheral Endoplasmic Reticulum Localization of Gp78 Ubiquitin Ligase Activity. J. Cell Sci. 2012, 125, 1727–1737. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Meyer, H.; Rapoport, T.A. The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nat. Cell Biol. 2001, 414, 652–656. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harding, H.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.; Hughes, P.D.; Michalak, E.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [Green Version]
- Reimertz, C.; Kogel, D.; Rami, A.; Prehn, J.H.M. Gene expression during ER stress-induced apoptosis in neurons: Induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J. Cell. Biol. 2003, 162, 587–597. [Google Scholar] [CrossRef]
- Birk, J.; Meyer, M.; Aller, I.; Hansen, H.G.; Odermatt, A.; Dick, T.P.; Meyer, A.J.; Appenzeller-Herzog, C. Endoplasmic reticulum: Reduced and oxidized glutathione revisited. J. Cell Sci. 2013, 126, 1604–1617. [Google Scholar] [CrossRef] [Green Version]
- Lizak, B.; Birk, J.; Zana, M. Ca2+ mobilization-dependent reduction of the endoplasmic reticulum lumen is due to influx of cytosolic glutathione. BMC Biol. 2020, 18, 19. [Google Scholar] [CrossRef]
- Andreu, C.I.; Woehlbier, U.; Torres-Gutierrez, M.A.; Hetz, C. Protein disulfide isomerases in neurodegeneration: From disease mechanisms to biomedical applications. FEBS Lett. 2012, 586, 2826–2834. [Google Scholar] [CrossRef] [Green Version]
- Uehara, T.; Nakamura, T.; Yao, D.; Shi, Z.-Q.; Gu, Z.; Ma, Y.; Masliah, E.; Nomura, Y.; Lipton, S.A. S-Nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nat. Cell Biol. 2006, 441, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhang, X.; Li, C.; Guan, T.; Shang, H.; Cui, L.; Li, X.-M.; Kong, J. S-nitrosylated protein disulfide isomerase contributes to mutant SOD1 aggregates in amyotrophic lateral sclerosis. J. Neurochem. 2013, 124, 45–58. [Google Scholar] [CrossRef] [PubMed]
- Woehlbier, U.; Colombo, A.; Saaranen, M.J.; Pérez, V.; Ojeda, J.; Bustos, F.J.; Andreu, I.C.; Torres, M.; Valenzuela, V.; Medinas, D.B.; et al. ALS -linked protein disulfide isomerase variants cause motor dysfunction. EMBO J. 2016, 35, 845–865. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Thielen, P.; Matus, S.; Nassif, M.; Court, F.; Kiffin, R.; Martinez, G.; Cuervo, A.M.; Brown, R.H.; Glimcher, L.H. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009, 23, 2294–2306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Popko, B.; Roos, R.P. An enhanced integrated stress response ameliorates mutant SOD1-induced ALS. Hum. Mol. Genet. 2014, 23, 2629–2638. [Google Scholar] [CrossRef] [PubMed]
- Tsaytler, P.; Harding, H.; Ron, D.; Bertolotti, A. Selective Inhibition of a Regulatory Subunit of Protein Phosphatase 1 Restores Proteostasis. Science 2011, 332, 91–94. [Google Scholar] [CrossRef] [PubMed]
- Bella, E.D.; Bersano, E.; Antonini, G.; Borghero, G.; Capasso, M.; Caponnetto, C.; Chiò, A.; Corbo, M.; Filosto, M.; Giannini, F.; et al. The unfolded protein response in amyotrophic later sclerosis: Results of a phase 2 trial. Brain 2021. [Google Scholar] [CrossRef]
- Dzhashiashvili, Y.; Monckton, C.P.; Shah, H.S.; Kunjamma, R.B.; Popko, B. The UPR-PERK pathway is not a promising therapeutic target for mutant SOD1-induced ALS. Neurobiol. Dis. 2019, 127, 527–544. [Google Scholar] [CrossRef]
- Gutiérrez, T.; Simmen, T. Endoplasmic reticulum chaperones tweak the mitochondrial calcium rheostat to control metabolism and cell death. Cell Calcium 2018, 70, 64–75. [Google Scholar] [CrossRef]
- Bravo, R.; Vicencio, J.M.; Parra, V.; Troncoso, R.; Munoz, J.P.; Bui, M.; Quiroga, C.; Rodriguez, A.E.; Verdejo, H.E.; Ferreira, J.; et al. Increased ER–mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J. Cell Sci. 2011, 124, 2511. [Google Scholar] [CrossRef] [Green Version]
- Area-Gomez, E.; Schon, E.A. On the Pathogenesis of Alzheimer’s Disease: The MAM Hypothesis. FASEB J. 2017, 31, 864–867. [Google Scholar] [CrossRef] [Green Version]
- Schon, E.A.; Area-Gomez, E. Is Alzheimer’s Disease a Disorder of Mitochondria-Associated Membranes? J. Alzheimer’s Dis. 2010, 20, S281–S292. [Google Scholar] [CrossRef] [Green Version]
- Zampese, E.; Fasolato, K.; Pozzan, T.; Pizzo, P. Presenilin-2 modulation of ER-mitochondria interactions: FAD mutations, mechanisms and pathological consequences. Commun. Integr. Biol. 2011, 4, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef] [Green Version]
- Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Montesinos, J.; Velasco, K.R.; Agrawal, R.R.; Xu, Y.; Chan, R.B.; Di Paolo, G.; Mehler, M.F.; et al. Increased localization of APP -C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 2017, 36, 3356–3371. [Google Scholar] [CrossRef]
- Montesinos, J.; Pera, M.; Larrea, D.; Guardia-Laguarta, C.; Agrawal, R.R.; Velasco, K.R.; Area-Gomez, E. The Alzheimer’s disease-associated C99 fragment of APP regulates cellular cholesterol trafficking. EMBO J. 2020, 39, e103791. [Google Scholar] [CrossRef] [PubMed]
- Mourelatos, Z.; Adler, H.; Hirano, A.; Donnenfeld, H.; Gonatas, J.O.; Gonatas, N.K.; Mourelatos, Z.; Adler, H.; Hirano, A.; Donnenfeld, H.; et al. Fragmentation of the Golgi apparatus of motor neurons in amyotrophic lateral sclerosis revealed by organelle-specific antibodies. Proc. Natl. Acad. Sci. USA 1990, 87, 4393–4395. [Google Scholar] [CrossRef] [Green Version]
- Oyanagi, K.; Yamazaki, M.; Takahashi, H.; Watabe, K.; Wada, M.; Komori, T.; Morita, T.; Mizutani, T. Spinal anterior horn cells in sporadic amyotrophic lateral sclerosis show ribosomal detachment from, and cisternal distention of the rough endoplasmic reticulum. Neuropathol. Appl. Neurobiol. 2008, 34, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Simmen, T. Mechanistic Connections between Endoplasmic Reticulum (ER) Redox Control and Mitochondrial Metabolism. Cells 2019, 8, 1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Makar, M.; Wang, M.X.; Ai, H. Monitoring thioredoxin redox with a genetically encoded red fluorescent biosensor. Nat. Chem. Biol. 2017, 13, 1045–1052. [Google Scholar] [CrossRef] [Green Version]
- Filadi, R.; Theurey, P.; Pizzo, P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium 2017, 62, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Simmen, T.; Herrera-Cruz, M.S. Plastic mitochondria-endoplasmic reticulum (ER) contacts use chaperones and tethers to mould their structure and signaling. Curr. Opin. Cell Biol. 2018, 53, 61–69. [Google Scholar] [CrossRef]
- Booth, D.M.; Enyedi, B.; Geiszt, M. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol. Cell 2016, 63, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganguly, G.; Chakrabarti, S.; Chatterjee, U.; Saso, L. Proteinopathy, oxidative stress and mitochondrial dysfunction: Cross talk in Alzheimer’s disease and Parkinson’s disease. Drug Des. Dev. Ther. 2017, 11, 797–810. [Google Scholar] [CrossRef] [Green Version]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lévy, E.; El Banna, N.; Baïlle, D.; Heneman-Masurel, A.; Truchet, S.; Rezaei, H.; Huang, M.-E.; Béringue, V.; Martin, D.; Vernis, L. Causative Links between Protein Aggregation and Oxidative Stress: A Review. Int. J. Mol. Sci. 2019, 20, 3896. [Google Scholar] [CrossRef] [Green Version]
- Lim, Y.A.; Rhein, V.; Meier, F. Abeta and human amylin share a common toxicity pathway via mitochondrial dysfunction. Proteomics 2010, 10, 1621–1633. [Google Scholar] [CrossRef]
- Singh, S.; Bhowmick, D.C.; Pany, S.; Joe, M.; Zaghlula, N.; Jeremic, A.M. Apoptosis signal regulating kinase-1 and NADPH oxidase mediate human amylin evoked redox stress and apoptosis in pancreatic beta-cells. Biochim. Biophys. Acta 2018, 1860, 1721–1733. [Google Scholar] [CrossRef]
- Mogk, A.; Bukau, B.; Kampinga, H.H. Cellular Handling of Protein Aggregates by Disaggregation Machines. Mol. Cell 2018, 69, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, B.T. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef]
- Zhou, C.; Unruh, J.R. Organelle-based aggregation and retention of damaged proteins in asymmetrically dividing cells. Cell 2014, 159, 530–542. [Google Scholar] [CrossRef] [Green Version]
- Fasana, E.; Fossati, M.; Ruggiano, A.; Brambillasca, S.; Hoogenraad, C.C.; Navone, F.; Francolini, M.; Borgese, N. A VAPB mutant linked to amyotrophic lateral sclerosis generates a novel form of organized smooth endoplasmic reticulum. FASEB J. 2010, 24, 1419–1430. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.L.; Silva, H.C.A. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am. J. Hum. Genet. 2004, 75, 822–831. [Google Scholar] [CrossRef] [Green Version]
- Soussan, L.; Burakov, D.; Daniels, M.P.; Toister-Achituv, M.; Porat, A.; Yarden, Y.; Elazar, Z. Erg30, a Vap-33–Related Protein, Functions in Protein Transport Mediated by Copi Vesicles. J. Cell Biol. 1999, 146, 301–312. [Google Scholar] [CrossRef]
- Johnson, B.; Leek, A.N.; Solé, L.; Maverick, E.E.; Levine, T.P.; Tamkun, M.M. Kv2 potassium channels form endoplasmic reticulum/plasma membrane junctions via interaction with VAPA and VAPB. Proc. Natl. Acad. Sci. USA 2018, 115, E7331–E7340. [Google Scholar] [CrossRef] [Green Version]
- Kirmiz, M.; Vierra, N.C.; Palacio, S.; Trimmer, J.S. Identification of VAPA and VAPB as Kv2 Channel-Interacting Proteins Defining Endoplasmic Reticulum–Plasma Membrane Junctions in Mammalian Brain Neurons. J. Neurosci. 2018, 38, 7562–7584. [Google Scholar] [CrossRef] [Green Version]
- De Vos, K.J.; Morotz, G.M.; Stoica, R.; Tudor, E.L. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef] [Green Version]
- Loewen, C.J.; Roy, A.; Levine, T.P. A conserved ER targeting motif in three families of lipid binding proteins and in Opi1p binds VAP. EMBO J. 2003, 22, 2025–2035. [Google Scholar] [CrossRef] [Green Version]
- Di Mattia, T.; Wilhelm, L.P.; Ikhlef, S.; Wendling, C.; Spehner, D.; Nominé, Y.; Giordano, F.; Mathelin, C.; Drin, G.; Tomasetto, C.; et al. Identification of MOSPD2, a novel scaffold for endoplasmic reticulum membrane contact sites. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Di Mattia, T.; Martinet, A.; Ikhlef, S.; McEwen, A.G.; Nominé, Y.; Wendling, C.; Poussin-Courmontagne, P.; Voilquin, L.; Eberling, P.; Ruffenach, F.; et al. FFAT motif phosphorylation controls formation and lipid transfer function of inter-organelle contacts. EMBO J. 2020, 39, e104369. [Google Scholar] [CrossRef]
- Murphy, S.E.; Levine, T.P. VAP, a Versatile Access Point for the Endoplasmic Reticulum: Review and analysis of FFAT-like motifs in the VAPome. Biochim. Biophys. Acta 2016, 1861, 952–961. [Google Scholar] [CrossRef]
- Lau, D.H.W.; Paillusson, S.; Hartopp, N.; Rupawala, H.; Mórotz, G.M.; Gomez-Suaga, P.; Greig, J.; Troakes, C.; Noble, W.; Miller, C.C.J. Disruption of endoplasmic reticulum-mitochondria tethering proteins in post-mortem Alzheimer’s disease brain. Neurobiol. Dis. 2020, 143, 105020. [Google Scholar] [CrossRef] [PubMed]
- Costello, J.; Castro, I.G.; Hacker, C.; Schrader, T.A.; Metz, J.; Zeuschner, D.; Azadi, A.S.; Godinho, L.F.; Costina, V.; Findeisen, P.; et al. ACBD5 and VAPB mediate membrane associations between peroxisomes and the ER. J. Cell Biol. 2017, 216, 331–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, R.; Cheng, D.; Coyaud, É.; Freeman, S.; Di Pietro, E.; Wang, Y.; Vissa, A.; Yip, C.M.; Fairn, G.D.; Braverman, N.; et al. VAPs and ACBD5 tether peroxisomes to the ER for peroxisome maintenance and lipid homeostasis. J. Cell Biol. 2017, 216, 367–377. [Google Scholar] [CrossRef]
- Abdel-Khalik, J.; Yutuc, E.; Crick, P.J.; Gustafsson, J.-Å.; Warner, M.; Roman, G.; Talbot, K.; Gray, E.; Griffiths, W.J.; Turner, M.R.; et al. Defective cholesterol metabolism in amyotrophic lateral sclerosis. J. Lipid Res. 2017, 58, 267–278. [Google Scholar] [CrossRef] [Green Version]
- Lev, S.; Benhalevy, D.; Peretti, D.; Dahan, N. The VAP protein family: From cellular functions to motor neuron disease. Trends Cell Biol. 2008, 18, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Papiani, G.; Ruggiano, A.; Fossati, M.; Raimondi, A.; Bertoni, G.; Francolini, M.; Benfante, R.; Navone, F.; Borgese, N. Restructured Endoplasmic Reticulum, Generated by Mutant, Amyotrophic Lateral Sclerosis-Linked VAPB, is Cleared by the Proteasome. J. Cell Sci. 2012, 125, 3601–3611. [Google Scholar] [CrossRef] [Green Version]
- Stoica, R.; Pailusson, S.; Gomez-Suaga, P.; Mitchell, S.C. ALS/FTD-associated FUS activates GSK-3beta to disrupt the VAPB-PTPIP51 interaction and ER-mitochondria associations. EMBO Rep. 2016, 17, 1326–1342. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, N.; Lu, B. Mechanisms and roles of mitophagy in neurodegenerative diseases. CNS Neurosci. Ther. 2019, 25, 859–875. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nat. Cell Biol. 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Toth, R.P.; Atkin, J.D. Dysfunction of Optineurin in Amyotrophic Lateral Sclerosis and Glaucoma. Front. Immunol. 2018, 9, 1017. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L.F. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. USA 2014, 111, E4439–E4448. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Henati, A.; Deng, H.-X.; Sasaki, T.; Hirano, M. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 2001, 29, 160–165. [Google Scholar] [CrossRef]
- Kanekura, K.; Hashimoto, Y.; Niikura, T.; Aiso, S.; Matsuoka, M.; Nishimoto, I. Alsin, the Product of ALS2 Gene, Suppresses SOD1 Mutant Neurotoxicity through RhoGEF Domain by Interacting with SOD1 Mutants. J. Biol. Chem. 2004, 279, 19247–19256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, F.; Ferguson, C.; Hyman, A.A.; Parton, R.G.; Zerial, M. Rab5 and Alsin regulate stress-activated cytoprotective signaling on mitochondria. Elife 2018, 7, e32282. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, S.; Nahm, M.; Li, T.-N.; Lin, H.-C.; Kim, Y.D.; Lee, J.; Yao, C.-K.; Lee, S. ALS2 regulates endosomal trafficking, postsynaptic development, and neuronal survival. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wójcik, C.; Rowicka, M.; Kudlicki, A.; Nowis, D.; McConnell, E.; Kujawa, M.; DeMartino, G.N. Valosin-containing Protein (p97) Is a Regulator of Endoplasmic Reticulum Stress and of the Degradation of N-End Rule and Ubiquitin-Fusion Degradation Pathway Substrates in Mammalian Cells. Mol. Biol. Cell 2006, 17, 4606–4618. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Peng, G.; Wang, Y.; Fang, S.; Karbowski, M. The AAA-ATPase p97 is essential for outer mitochondrial membrane protein turnover. Mol. Biol. Cell 2011, 22, 291–300. [Google Scholar] [CrossRef]
- Papadopoulos, C.; Kirchner, P.; Bug, M.; Grum, D.; Koerver, L.; Schulze, N.; Poehler, R.; Dressler, A.; Fengler, S.; Arhzaouy, K.; et al. VCP/p97 cooperates with YOD 1, UBXD 1 and PLAA to drive clearance of ruptured lysosomes by autophagy. EMBO J. 2017, 36, 135–150. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Gao, G.; Shankar, J.; Joshi, B.; Foster, L.J.; Nabi, I.R. p38 MAP kinase–dependent phosphorylation of the Gp78 E3 ubiquitin ligase controls ER–mitochondria association and mitochondria motility. Mol. Biol. Cell 2015, 26, 3828–3840. [Google Scholar] [CrossRef]
- Wang, P.T.; Garcin, P.O.; Fu, M.; Masoudi, M.; Pante, N. Distinct mechanisms controlling rough and smooth endoplasmic reticulum contacts with mitochondria. J. Cell. Sci. 2015, 128, 2759–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sano, R.; Patterson, A.; Gomero, E. GM1-ganglioside accumulation at the mitochondria-associated ER membranes links ER stress to Ca2+-dependent mitochondrial apoptosis. Mol. Cell. 2009, 36, 500–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartolome, F.; Wu, H.-C.; Burchell, V.S.; Preza, E.; Wray, S.; Mahoney, C.; Fox, N.; Calvo, A.; Canosa, A.; Moglia, C.; et al. Pathogenic VCP Mutations Induce Mitochondrial Uncoupling and Reduced ATP Levels. Neuron 2013, 78, 57–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamasaki, M.; Furuta, N.; Matsuda, A.; Nezu, A.; Yamamoto, A.; Fujita, N.; Oomori, H.; Noda, T.; Haraguchi, T.; Hiraoka, Y.; et al. Autophagosomes form at ER–mitochondria contact sites. Nat. Cell Biol. 2013, 495, 389–393. [Google Scholar] [CrossRef]
- Ju, J.-S.; Fuentealba, R.; Miller, S.E.; Jackson, E.; Piwnica-Worms, D.; Baloh, R.H.; Weihl, C.C. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 2009, 187, 875–888. [Google Scholar] [CrossRef]
- Liu, G.; Byrd, A.; Warner, A.N.; Pei, F. Cdc48/VCP and Endocytosis Regulate TDP-43 and FUS Toxicity and Turnover. Mol. Cell. Biol. 2020, 40, 4. [Google Scholar] [CrossRef]
- Hill, S.M.; Wrobel, L.; Ashkenazi, A.; Fernandez-Estevez, M.; Tan, K.; Bürli, R.W.; Rubinsztein, D.C. VCP/p97 regulates Beclin-1-dependent autophagy initiation. Nat. Chem. Biol. 2021, 17, 1–8. [Google Scholar] [CrossRef]
- Kim, N.C.; Tresse, E.; Kolaitis, R.-M.; Molliex, A.; Thomas, R.E.; Alami, N.H.; Wang, B.; Joshi, A.; Smith, R.B.; Ritson, G.P.; et al. VCP Is Essential for Mitochondrial Quality Control by PINK1/Parkin and this Function Is Impaired by VCP Mutations. Neuron 2013, 78, 403. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Qiu, H. Valosin-Containing Protein, a Calcium-Associated ATPase Protein, in Endoplasmic Reticulum and Mitochondrial Function and Its Implications for Diseases. Int. J. Mol. Sci. 2020, 21, 3842. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Horn, S.; Belkacemi, A.; Kojer, K.; Petrungaro, C.; Habich, M.; Ali, M.; Küttner, V.; Bien, M.; Kauff, F.; et al. Protein import and oxidative folding in the mitochondrial intermembrane space of intact mammalian cells. Mol. Biol. Cell 2013, 24, 2160–2170. [Google Scholar] [CrossRef]
- Straub, I.R.; Weraarpachai, W.; Shoubridge, A.E. Multi-OMICS study of a CHCHD10 variant causing ALS demonstrates metabolic rewiring and activation of endoplasmic reticulum and mitochondrial unfolded protein responses. Hum. Mol. Genet. 2021, 30, 687–705. [Google Scholar] [CrossRef]
- Vollrath, J.T.; Sechi, A.; Dreser, A.; Katona, I.; Wiemuth, D.; Vervoorts, J.; Dohmen, M.J.W.; Chandrasekar, A.; Prause, J.U.; Brauers, E.; et al. Loss of function of the ALS protein SigR1 leads to ER pathology associated with defective autophagy and lipid raft disturbances. Cell Death Dis. 2014, 5, e1290. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Illieva, H.; Tamada, H.; Nomura, H.; Komine, O. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef]
- Dreser, A.; Vollrath, J.T.; Sechi, A.; Johann, S.; Roos, A.; Yamoah, A.; Katona, I.; Bohlega, S.; Wiemuth, D.; Tian, Y.; et al. The ALS-linked E102Q mutation in Sigma receptor-1 leads to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins. Cell Death Differ. 2017, 24, 1655–1671. [Google Scholar] [CrossRef] [PubMed]
- Carreras-Sureda, A.; Jaña, F.; Urra, H.; Durand, S.; Mortenson, D.E.; Sagredo, A.; Bustos, G.; Hazari, Y.; Ramos-Fernández, E.; Sassano, M.L.; et al. Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 2019, 21, 755–767. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Rubio, N.; Garg, A.; Bultynck, G.; Rizzuto, R.; Decuypere, J.-P.; Piette, J.; Linehan, C.; Gupta, S.; Samali, A.; et al. PERK is required at the ER-mitochondrial contact sites to convey apoptosis after ROS-based ER stress. Cell Death Differ. 2012, 19, 1880–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekris, L.M.; Yu, K.-E.; Bird, T.D.; Tsuang, D. Genetics of Alzheimer disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15, 1–22. [Google Scholar] [CrossRef]
- Swerdlow, R.H. Mitochondria and Mitochondrial Cascades in Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 62, 1403–1416. [Google Scholar] [CrossRef] [Green Version]
- Bubber, P.; Haroutunian, V.; Fisch, G.; Blass, J.P.; Gibson, G.E. Mitochondrial abnormalities in Alzheimer brain: Mechanistic implications. Ann. Neurol. 2005, 57, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.C.; Ladd, A.C.; Bennett, J.P.; Rice, A.C.; Ladd, A.C.; Bennett, J.P. Postmortem Alzheimer’s Disease Hippocampi Show Oxidative Phosphorylation Gene Expression Opposite that of Isolated Pyramidal Neurons. J. Alzheimer’s Dis. 2015, 45, 1051–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hangen, E.; Féraud, O.; Lachkar, S.; Mou, H.; Doti, N.; Fimia, G.M.; Lam, N.-V.; Zhu, C.; Godin, I.; Muller, K.; et al. Interaction between AIF and CHCHD4 Regulates Respiratory Chain Biogenesis. Mol. Cell 2015, 58, 1001–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef]
- Gendron, T.F.; Petrucelli, L. The role of tau in neurodegeneration. Mol. Neurodegener. 2009, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, D.M.; Selkoe, D.J. A beta oligomers—A decade of discovery. J. Neurochem. 2007, 101, 1172–1184. [Google Scholar] [CrossRef]
- Gerakis, Y.; Hetz, C. Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer’s disease. FEBS J. 2018, 285, 995–1011. [Google Scholar] [CrossRef] [Green Version]
- Schreiner, B.; Hedskog, L. Amyloid-beta peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J. Alzheimers Dis. 2015, 43, 369–374. [Google Scholar] [CrossRef]
- Zatti, G.; Burgo, A.; Giacomello, M.; Barbiero, L.; Ghidoni, R.; Sinigaglia, G.; Florean, C.; Bagnoli, S.; Binetti, G.; Sorbi, S.; et al. Presenilin mutations linked to familial Alzheimer’s disease reduce endoplasmic reticulum and Golgi apparatus calcium levels. Cell Calcium 2006, 39, 539–550. [Google Scholar] [CrossRef]
- Zampese, E.; Fasolato, C.; Kipanyula, M.J.; Bortolozzi, M.; Pozzan, T.; Pizzo, P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc. Natl. Acad. Sci. USA 2011, 108, 2777–2782. [Google Scholar] [CrossRef] [Green Version]
- Area-Gomez, E.; de Groof, J.C.; Boldogh, I. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [Green Version]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and gamma-secretase: Structure, function, and role in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar] [CrossRef]
- Citron, M.; Westaway, D.; Xia, W.; Carlson, G.; Diehl, T.; Lee, M. Mutant presenilins of Alzheimer’s disease increase production of 42-residue amyloid beta-protein in both transfected cells and transgenic mice. Nat. Med. 1997, 3, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Cheung, K.-H.; Mei, L.; Mak, D.-O.D.; Hayashi, I.; Iwatsubo, T.; Kang, D.E.; Foskett, J.K. Gain-of-Function Enhancement of IP3 Receptor Modal Gating by Familial Alzheimer’s Disease-Linked Presenilin Mutants in Human Cells and Mouse Neurons. Sci. Signal. 2010, 3, ra22. [Google Scholar] [CrossRef] [Green Version]
- Yi, M.; Weaver, D.; Hajnoczky, G. Control of mitochondrial motility and distribution by the calcium signal: A homeostatic circuit. J. Cell. Biol. 2004, 167, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Rigotto, G.; Valente, G.; Basso, E. Defective Mitochondrial Pyruvate Flux Affects Cell Bioenergetics in Alzheimer’s Disease-Related Models. Cell Rep. 2020, 30, 2332–2348.e10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abisambra, J.F.; Jinwal, U.K.; Blair, L.J.; O’Leary, J.C., 3rd; Li, Q.; Brady, S.; Wang, L.; Guidi, C.E.; Zhang, B.; Nordhues, B.A.; et al. Tau Accumulation Activates the Unfolded Protein Response by Impairing Endoplasmic Reticulum-Associated Degradation. J. Neurosci. 2013, 33, 9498–9507. [Google Scholar] [CrossRef] [Green Version]
- Endo, M.; Mori, M.; Akira, S.; Gotoh, T. C/EBP homologous protein (CHOP) is crucial for the induction of caspase-11 and the pathogenesis of lipopolysaccharide-induced inflammation. J. Immunol. 2006, 176, 6245–6253. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Joe, E.; Kim, H.J. Endoplasmic reticulum stress-induced IRE1alpha activation mediates cross-talk of GSK-3beta and XBP-1 to regulate inflammatory cytokine production. J. Immunol. 2015, 194, 4498–4506. [Google Scholar] [CrossRef] [Green Version]
- Stone, S.; Lin, W. The unfolded protein response in multiple sclerosis. Front. Neurosci. 2015, 9, 264. [Google Scholar] [CrossRef] [Green Version]
- Haile, Y.; Deng, X.; Ortiz-Sandoval, C.; Tahbaz, N.; Janowicz, A.; Lu, J.-Q.; Kerr, B.J.; Gutowski, N.J.; Holley, J.E.; Eggleton, P.; et al. Rab32 connects ER stress to mitochondrial defects in multiple sclerosis. J. Neuroinflamm. 2017, 14, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, L.J. Mitochondrial Pathobiology in Parkinson’s Disease and Amyotrophic Lateral Sclerosis. J. Alzheimers Dis. 2010, 20, S335–S356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandres-Ciga, S.; Diez-Fairen, M.; Kim, J.J.; Singleton, A.B. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol. Dis. 2020, 137, 104782. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, R.H.; International Parkinson’s Disease Genomics Consortium (IPDGC); Botía, J.; Nalls, M.A.; Hardy, J.; Taliun, S.A.G.; Ryten, M.; System Genomics of Parkinson’s Disease (SGPD). Moving beyond neurons: The role of cell type-specific gene regulation in Parkinson’s disease heritability. Park. Dis. 2019, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Bryois, J.; Skene, N.; Hansen, T.F.; Kogelman, L.J.A.; Watson, H.J.; Liu, Z.; Brueggeman, L.; Breen, G.; Bulik, C.M.; Arenas, E.; et al. Genetic identification of cell types underlying brain complex traits yields insights into the etiology of Parkinson’s disease. Nat. Genet. 2020, 52, 482–493. [Google Scholar] [CrossRef]
- Dernie, F. Mitophagy in Parkinson’s disease: From pathogenesis to treatment target. Neurochem. Int. 2020, 138, 104756. [Google Scholar] [CrossRef]
- Ganjam, G.K.; Bolte, K.; Matschke, L.A.; Neitemeier, S.; Dolga, A.; Höllerhage, M.; Höglinger, G.; Adamczyk, A.; Decher, N.; Oertel, W.H.; et al. Mitochondrial damage by α-synuclein causes cell death in human dopaminergic neurons. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Li, W.-W.; Yang, R.; Guo, J.-C.; Ren, H.-M.; Zha, X.-L.; Cheng, J.-S.; Cai, D.-F. Localization of α-synuclein to mitochondria within midbrain of mice. NeuroReport 2007, 18, 1543–1546. [Google Scholar] [CrossRef]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rüb, C.; Liu, Y.; Magrané, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. Synuclein Is Localized to Mitochondria-Associated ER Membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P.-Y. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef] [Green Version]
- Im, J.-Y.; Lee, K.-W.; Woo, J.-M.; Junn, E.; Mouradian, M.M. DJ-1 induces thioredoxin 1 expression through the Nrf2 pathway. Hum. Mol. Genet. 2012, 21, 3013–3024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.-S.; Davis, R.L.; Sue, C.M. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr. Neurol. Neurosci. Rep. 2018, 18, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimprich, A.; Benet-Pagès, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A Mutation in VPS35, Encoding a Subunit of the Retromer Complex, Causes Late-Onset Parkinson Disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.T.; Huang, X.; Nguyen, D.; Shammas, M.K. Loss of CHCHD2 and CHCHD10 activates OMA1 peptidase to disrupt mitochondrial cristae phenocopying patient mutations. Hum. Mol. Genet. 2020, 29, 1547–1567. [Google Scholar] [CrossRef]
- Weihofen, A.; Thomas, K.J.; Ostaszewski, B.L.; Cookson, M.R.; Selkoe, D.J. Pink1 Forms a Multiprotein Complex with Miro and Milton, Linking Pink1 Function to Mitochondrial Trafficking. Biochemistry 2009, 48, 2045–2052. [Google Scholar] [CrossRef] [Green Version]
- Liu, W.; Duan, X.; Xu, L.; Shang, W.; Zhao, J.; Wang, L.; Li, J.-C.; Chen, C.-H.; Liu, J.-P.; Tong, C. Chchd2 regulates mitochondrial morphology by modulating the levels of Opa1. Cell Death Differ. 2020, 27, 2014–2029. [Google Scholar] [CrossRef]
- Liu, Y.; Clegg, H.V.; Leslie, P.; Di, J.; Tollini, A.L.; He, Y.; Kim, T.-H.; Jin, A.; Graves, L.M.; Zheng, J.; et al. CHCHD2 inhibits apoptosis by interacting with Bcl-x L to regulate Bax activation. Cell Death Differ. 2014, 22, 1035–1046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Wu, B.P.; Nguyen, D.; Liu, Y.-T.; Marani, M.; Hench, J.; Bénit, P.; Kozjak-Pavlovic, V.; Rustin, P.; Frank, S.; et al. CHCHD2 accumulates in distressed mitochondria and facilitates oligomerization of CHCHD10. Hum. Mol. Genet. 2018, 27, 3881–3900. [Google Scholar] [CrossRef] [PubMed]
- Straub, I.R.; Janer, A.; Weraarpachai, W.; Zinman, L.; Robertson, J.; Rogaeva, E.; Shoubridge, A.E. Loss of CHCHD10-CHCHD2 complexes required for respiration underlies the pathogenicity of a CHCHD10 mutation in ALS. Hum. Mol. Genet. 2017, 27, 178–189. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328. [Google Scholar] [CrossRef]
- Ortiz-Sandoval, C.G.; Hughes, S.C.; Dacks, J.B.; Simmen, T. Interaction with the effector dynamin-related protein 1 (Drp1) is an ancient function of Rab32 subfamily proteins. Cell Logist. 2014, 4, e986399. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wilson, G.R.; Stephenson, S.E.M.; Bozaoglu, K.; Farrer, M.J.; Lockhart, P.J. The emerging role of Rab GTPases in the pathogenesis of Parkinson’s disease. Mov. Disord. 2018, 33, 196–207. [Google Scholar] [CrossRef] [PubMed]
- Waschbüsch, D.; Michels, H.; Strassheim, S.; Ossendorf, E.; Kessler, D.; Gloeckner, C.J.; Barnekow, A. LRRK2 Transport Is Regulated by Its Novel Interacting Partner Rab32. PLoS ONE 2014, 9, e111632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betarbet, R.; Sherer, T.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef]
- Johnson, M.E.; Bobrovskaya, L. An update on the rotenone models of Parkinson’s disease: Their ability to reproduce the features of clinical disease and model gene-environment interactions. Neurotoxicology 2015, 46, 101–116. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Locus | Gene | Protein | Protein Function | Role in ALS |
|---|---|---|---|---|
| 21q22.1 | SOD1 | Superoxide dismutase-1 (SOD1) | UPR activation ERAD inhibition Antioxidant defense Autophagy enhancement | Mutants cause toxic SOD1 clusters aggregation within astrocytes or motor neurons. |
| 2q33.2 | ALS2 | Alsin | A guanine-nucleotide exchange factor (GEF) to activate the GTPase Rab5. | Mutants influence the functions of endosomes in the subsynaptic reticulum. |
| 9p13-p12 | VCP | Valosin-containing protein (VCP) or Transitional endoplasmic reticulum ATPase (TER ATPase) | Ubiquitin/protein degradation Secretory protein trafficking | Mutants disrupt the control of OXPHOS and reduce autophagic clearance of TDP-43 and FUS aggregates. |
| 10p13 | OPTN | Optineurin | Selective autophagic adaptor Protein aggregates clearance | Mutants (e.g., E478G, R96L) are associated with both fALS and sALS. |
| 20q13.3 | VAPB | Vesicle-associated membrane protein (VAMP)-associated protein B (VAPB) | Vesicle trafficking ATF6 sensor interaction and XBP1 inhibition Lipid metabolism Microtubule organization | A dominantly inherited mutant, P56S-VAPB, causes fALS. |
| 9p13.3 | SIGMAR1 | Sigma-1 receptor (SIGMAR1) | Calcium signaling Lipid metabolism | A missense mutation in SIGMAR1 (e.g., G304C) causes fALS; Lack of SIGMAR1 induces motoneuron hyperexcitability and exacerabates ALS pathology. |
| 16p11.2 | FUS | Fused in sarcoma (FUS) | Transcriptional activation Protein and RNA binding | Mutations in FUS cause fALS and lead to the cytosolic deposition of FUS in the brain and spinal cord of ALS-FUS patients. |
| 1p36.2 | TARDBP | TAR-DNA binding protein (TDP43) | Transcriptional repression DNA and RNA binding | TARDBP gene rearrangement has been implicated in the pathogenesis of ALS; Mutations in the TARDBP gene (e.g., M337V and Q331K) are related to ALS. |
| 22q11.23 | CHCHD10 | Coiled-coil-helix-coiled-coil domain protein 10 (CHCHD10) | Mitochondrial Cristae morphology maintenance Oxidative phosphorylation | Mutants are associated with ALS as well as in other mitochondrial diseases. |
| 17q25 | P4HB | Protein disulfide-isomerase A1 (PDIA1) | Disulfide bonds formation, breakage and rearrangement Inhibition of misfolded proteins aggregation | Together with other ER stress markers, PDIA1 is greatly elevated in ALS spinal cord. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Bassot, A.; Giuliani, F.; Simmen, T. Amyotrophic Lateral Sclerosis (ALS): Stressed by Dysfunctional Mitochondria-Endoplasmic Reticulum Contacts (MERCs). Cells 2021, 10, 1789. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10071789
Chen J, Bassot A, Giuliani F, Simmen T. Amyotrophic Lateral Sclerosis (ALS): Stressed by Dysfunctional Mitochondria-Endoplasmic Reticulum Contacts (MERCs). Cells. 2021; 10(7):1789. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10071789
Chicago/Turabian StyleChen, Junsheng, Arthur Bassot, Fabrizio Giuliani, and Thomas Simmen. 2021. "Amyotrophic Lateral Sclerosis (ALS): Stressed by Dysfunctional Mitochondria-Endoplasmic Reticulum Contacts (MERCs)" Cells 10, no. 7: 1789. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10071789