
A Narrative Review on the Role of AMPK on De Novo Lipogenesis in Non-Alcoholic Fatty Liver Disease: Evidence from Human Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Epidemic of NAFLD
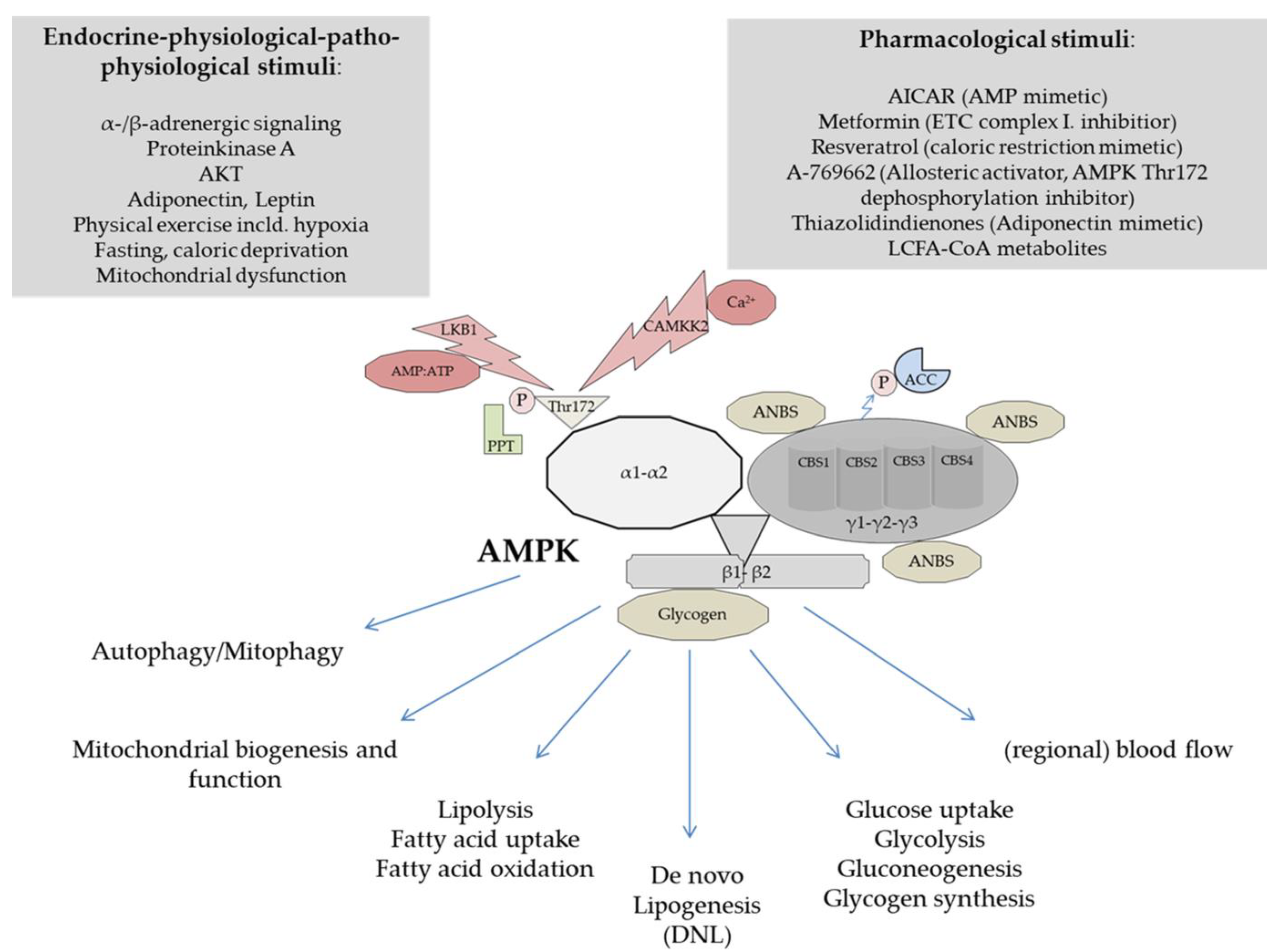
3. Structure and Physiological Regulation of AMPK
4. Interactions of Fatty Acids with Insulin Signaling: A Lipotoxic Event from a Human Perspective
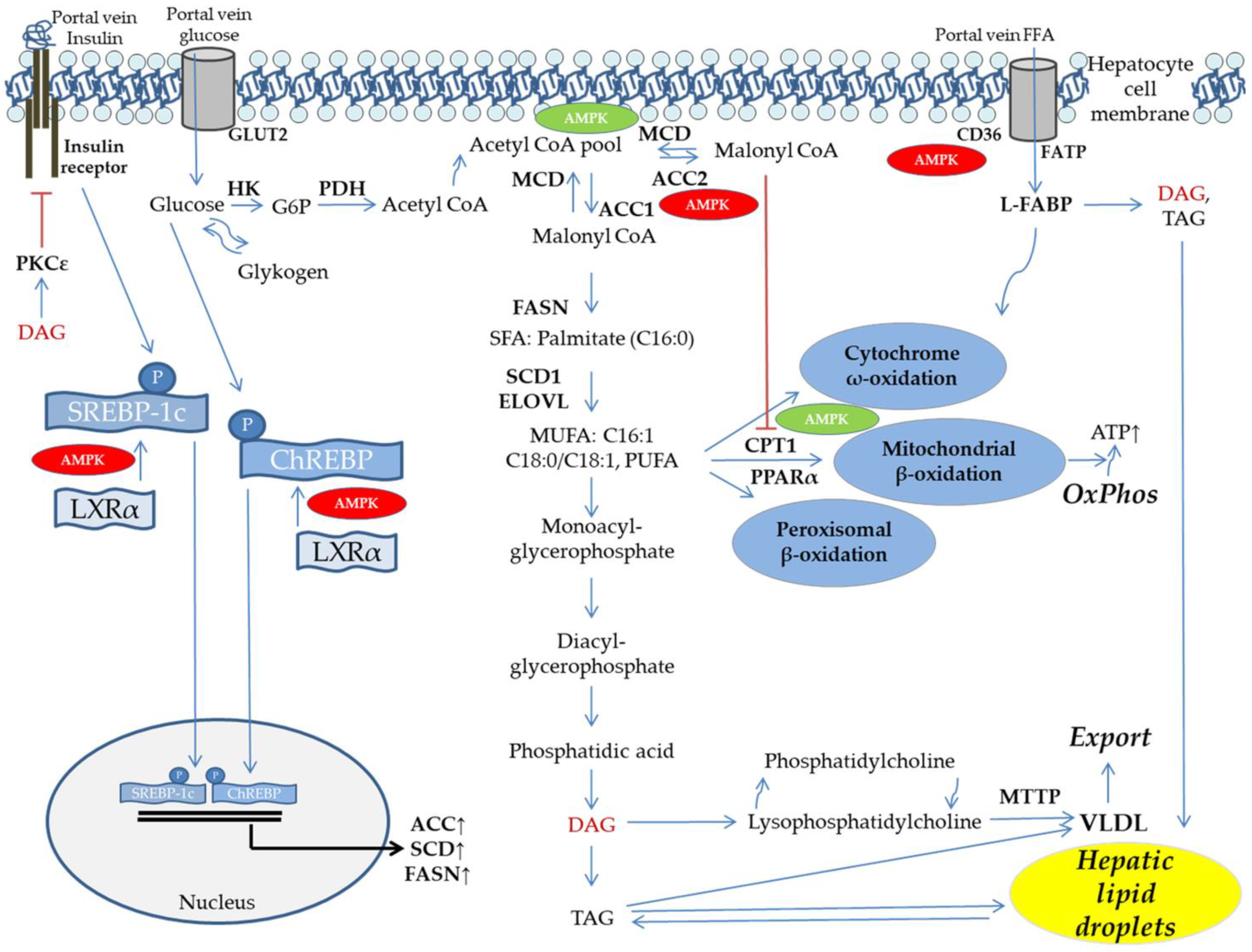
5. DNL and FFA Flux Determine Hepatic Lipid Dysbalance
6. Mitochondrial Function, Fat Oxidation, and Lipid Export
7. The Role of AMPK in Human Fat Depots
8. Skeletal Muscle Is a Predominant Target Tissue of Insulin and Sensitive to AMPK Activation
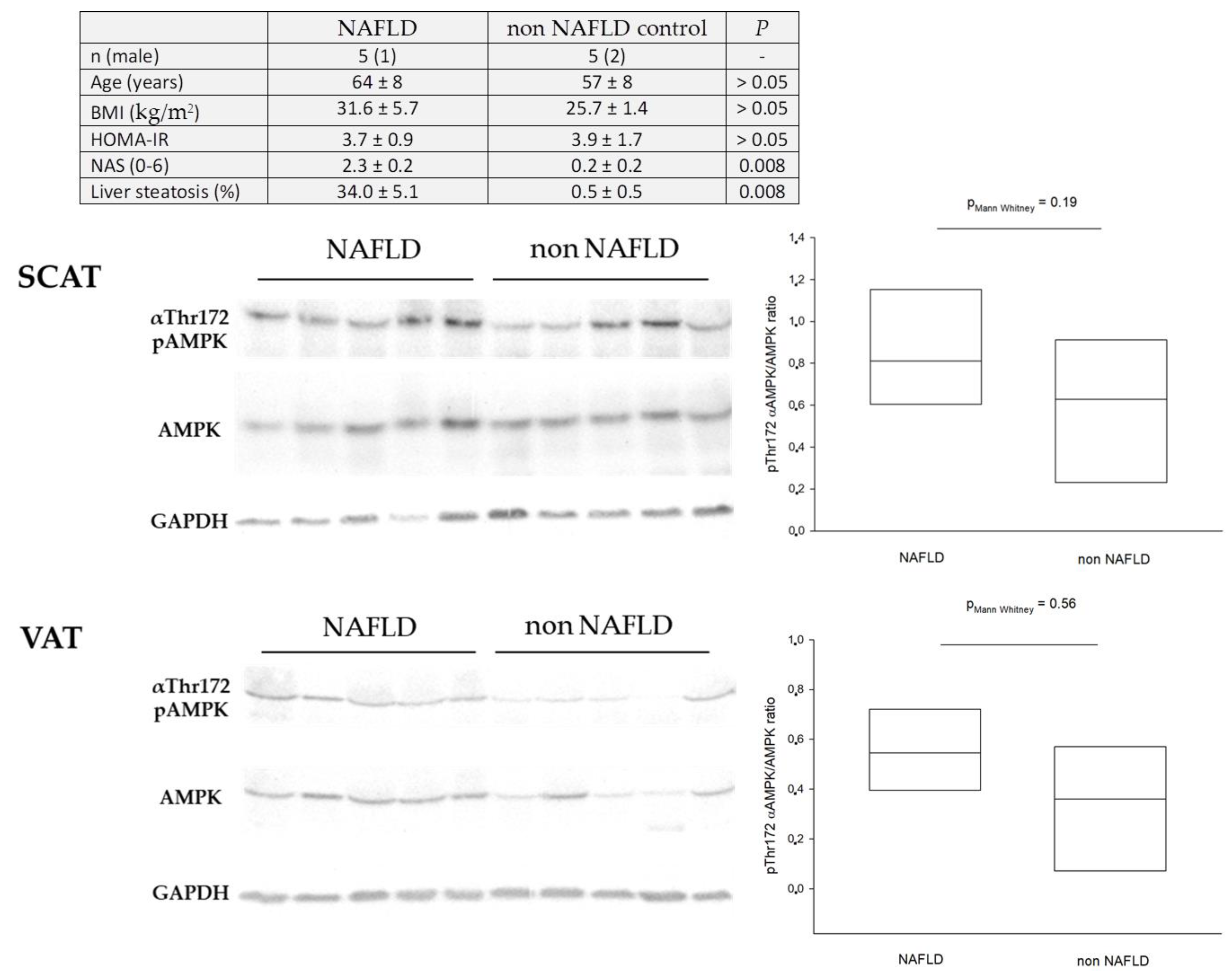
9. Dysregulation of Hepatic AMPK in Humans
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [Green Version]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef] [Green Version]
- Carling, D.; Clarke, P.R.; Zammit, V.A.; Hardie, D.G. Purification and characterization of the AMP-activated protein kinase. Copurification of acetyl-CoA carboxylase kinase and 3-hydroxy-3-methylglutaryl-CoA reductase kinase activities. Eur. J. Biochem. 1989, 186, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G. Regulation of fatty acid and cholesterol metabolism by the AMP-activated protein kinase. Biochim. Biophys. Acta 1992, 1123, 231–238. [Google Scholar] [CrossRef]
- Woods, A.; Munday, M.R.; Scott, J.; Yang, X.; Carlson, M.; Carling, D. Yeast SNF1 is functionally related to mammalian AMP-activated protein kinase and regulates acetyl-CoA carboxylase in vivo. J. Biol. Chem. 1994, 269, 19509–19515. [Google Scholar] [CrossRef]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.-P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single phosphorylation sites in Acc1 and Acc2 regulate lipid homeostasis and the insulin-sensitizing effects of metformin. Nat. Med. 2013, 19, 1649–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, G.F.; Carpentier, A.; Adeli, K.; Giacca, A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr. Rev. 2002, 23, 201–229. [Google Scholar] [CrossRef]
- Gastaldelli, A.; Cusi, K.; Pettiti, M.; Hardies, J.; Miyazaki, Y.; Berria, R.; Buzzigoli, E.; Sironi, A.M.; Cersosimo, E.; Ferrannini, E.; et al. Relationship between hepatic/visceral fat and hepatic insulin resistance in nondiabetic and type 2 diabetic subjects. Gastroenterology 2007, 133, 496–506. [Google Scholar] [CrossRef]
- Zhang, C.; Klett, E.L.; Coleman, R.A. Lipid signals and insulin resistance. Clin. Lipidol. 2013, 8, 659–667. [Google Scholar] [CrossRef] [Green Version]
- Szendroedi, J.; Roden, M. Ectopic lipids and organ function. Curr. Opin. Lipidol. 2009, 20, 50–56. [Google Scholar] [CrossRef]
- Korenblat, K.M.; Fabbrini, E.; Mohammed, B.S.; Klein, S. Liver, muscle, and adipose tissue insulin action is directly related to intrahepatic triglyceride content in obese subjects. Gastroenterology 2008, 134, 1369–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkenfeld, A.L.; Shulman, G.I. Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes. Hepatology 2014, 59, 713–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häring, H.-U. Novel phenotypes of prediabetes? Diabetologia 2016, 59, 1806–1818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Investig. 2016, 126, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Smith, B.K.; Marcinko, K.; Desjardins, E.M.; Lally, J.S.; Ford, R.J.; Steinberg, G.R. Treatment of nonalcoholic fatty liver disease: Role of AMPK. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E730–E740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ipsen, D.H.; Lykkesfeldt, J.; Tveden-Nyborg, P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2018, 75, 3313–3327. [Google Scholar] [CrossRef] [Green Version]
- Bellentani, S. The epidemiology of non-alcoholic fatty liver disease. Liver Int. 2017, 37 (Suppl. 1), 81–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Kopelman, P.G. Obesity as a medical problem. Nature 2000, 404, 635–643. [Google Scholar] [CrossRef]
- Lu, H.; Liu, H.; Hu, F.; Zou, L.; Luo, S.; Sun, L. Independent Association between Nonalcoholic Fatty Liver Disease and Cardiovascular Disease: A Systematic Review and Meta-Analysis. Int. J. Endocrinol. 2013, 2013, 124958. [Google Scholar] [CrossRef]
- Than, N.N.; Newsome, P.N. A concise review of non-alcoholic fatty liver disease. Atherosclerosis 2015, 239, 192–202. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Park, G.-M.; Lee, J.-Y.; Lee, B.U.; Park, J.H.; Kim, B.G.; Jung, S.W.; Du Jeong, I.; Bang, S.-J.; Shin, J.W.; et al. Association between non-alcoholic fatty liver disease and subclinical coronary atherosclerosis: An observational cohort study. J. Hepatol. 2018, 68, 1018–1024. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Kim, K.J.; Yoo, M.E.; Kim, G.; Yoon, H.-J.; Jo, K.; Youn, J.-C.; Yun, M.; Park, J.Y.; Shim, C.Y.; et al. Association of non-alcoholic steatohepatitis with subclinical myocardial dysfunction in non-cirrhotic patients. J. Hepatol. 2018, 68, 764–772. [Google Scholar] [CrossRef] [PubMed]
- Martins, E.; Oliveira, A. NAFLD and cardiovascular disease. Porto Biomed. J. 2018, 3, e2. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Bertolini, L.; Rodella, S.; Lippi, G.; Zoppini, G.; Chonchol, M. Relationship between kidney function and liver histology in subjects with nonalcoholic steatohepatitis. Clin. J. Am. Soc. Nephrol. 2010, 5, 2166–2171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Targher, G.; Byrne, C.D. Non-alcoholic fatty liver disease: An emerging driving force in chronic kidney disease. Nat. Rev. Nephrol. 2017, 13, 297–310. [Google Scholar] [CrossRef] [Green Version]
- Yeung, M.-W.; Wong, G.L.-H.; Choi, K.C.; Luk, A.O.-Y.; Kwok, R.; Shu, S.S.-T.; Chan, A.W.-H.; Lau, E.S.H.; Ma, R.C.W.; Chan, H.L.-Y.; et al. Advanced liver fibrosis but not steatosis is independently associated with albuminuria in Chinese patients with type 2 diabetes. J. Hepatol. 2017. [CrossRef]
- Calzadilla Bertot, L.; Adams, L.A. The Natural Course of Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 774. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.-A.; Lee, H.C.; Choe, J.; Kim, M.-J.; Lee, M.J.; Chang, H.-S.; Bae, I.Y.; Kim, H.-K.; An, J.; Shim, J.H.; et al. Association between non-alcoholic fatty liver disease and cancer incidence rate. J. Hepatol. 2017. [CrossRef]
- Ma, C.; Zhang, Q.; Greten, T.F. Nonalcoholic fatty liver disease promotes hepatocellular carcinoma through direct and indirect effects on hepatocytes. FEBS J. 2018, 285, 752–762. [Google Scholar] [CrossRef] [Green Version]
- Huber, Y.; Labenz, C.; Michel, M.; Wörns, M.-A.; Galle, P.R.; Kostev, K.; Schattenberg, J.M. Tumor Incidence in Patients with Non-Alcoholic Fatty Liver Disease. Dtsch. Arztebl. Int. 2020, 117, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.J.; Cheung, R.; Ahmed, A. Nonalcoholic steatohepatitis is the most rapidly growing indication for liver transplantation in patients with hepatocellular carcinoma in the U.S. Hepatology 2014, 59, 2188–2195. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.; Riaz, D.R.; Shi, G.; Liu, C.; Dai, Y. Outcomes of liver transplantation for nonalcoholic steatohepatitis: A systematic review and meta-analysis. Clin. Gastroenterol. Hepatol. 2014, 12, 394–402.e1. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, M.; Henry, L.; Garg, R.; Kalwaney, S.; Saab, S.; Younossi, Z. Risk of de novo post-transplant type 2 diabetes in patients undergoing liver transplant for non-alcoholic steatohepatitis. BMC Gastroenterol. 2015, 15, 175. [Google Scholar] [CrossRef]
- Golabi, P.; Bush, H.; Stepanova, M.; Locklear, C.T.; Jacobson, I.M.; Mishra, A.; Trimble, G.; Erario, M.; Venkatesan, C.; Younossi, I.; et al. Liver Transplantation (LT) for Cryptogenic Cirrhosis (CC) and Nonalcoholic Steatohepatitis (NASH) Cirrhosis: Data from the Scientific Registry of Transplant Recipients (SRTR): 1994 to 2016. Medicine 2018, 97, e11518. [Google Scholar] [CrossRef] [PubMed]
- Hoppe, S.; von Loeffelholz, C.; Lock, J.F.; Doecke, S.; Sinn, B.V.; Rieger, A.; Malinowski, M.; Pfeiffer, A.F.H.; Neuhaus, P.; Stockmann, M. Nonalcoholic steatohepatits and liver steatosis modify partial hepatectomy recovery. J. Investig. Surg. 2015, 28, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Sommerfeld, O.; von Loeffelholz, C.; Diab, M.; Kiessling, S.; Doenst, T.; Bauer, M.; Sponholz, C. Association between high dose catecholamine support and liver dysfunction following cardiac surgery. J. Card. Surg. 2020, 35, 1228–1236. [Google Scholar] [CrossRef] [PubMed]
- Kwiterovich, P.O.; Sloan, H.R.; Fredrickson, D.S. Glycolipids and other lipid constituents of normal human liver. J. Lipid Res. 1970, 11, 322–330. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Szczepaniak, L.S.; Nurenberg, P.; Leonard, D.; Browning, J.D.; Reingold, J.S.; Grundy, S.; Hobbs, H.H.; Dobbins, R.L. Magnetic resonance spectroscopy to measure hepatic triglyceride content: Prevalence of hepatic steatosis in the general population. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E462–E468. [Google Scholar] [CrossRef] [Green Version]
- Vuppalanchi, R.; Chalasani, N. Nonalcoholic fatty liver disease and nonalcoholic steatohepatitis: Selected practical issues in their evaluation and management. Hepatology 2009, 49, 306–317. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef] [PubMed]
- Teli, M.R.; James, O.F.; Burt, A.D.; Bennett, M.K.; Day, C.P. The natural history of nonalcoholic fatty liver: A follow-up study. Hepatology 1995, 22, 1714–1719. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F.W. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Day, C.P. Natural history of NAFLD: Remarkably benign in the absence of cirrhosis. Gastroenterology 2005, 129, 375–378. [Google Scholar] [CrossRef]
- Pais, R.; Charlotte, F.; Fedchuk, L.; Bedossa, P.; Lebray, P.; Poynard, T.; Ratziu, V. A systematic review of follow-up biopsies reveals disease progression in patients with non-alcoholic fatty liver. J. Hepatol. 2013, 59, 550–556. [Google Scholar] [CrossRef]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs. nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, e1–e9. [Google Scholar] [CrossRef] [Green Version]
- Adams, L.A.; Ratziu, V. Non-alcoholic fatty liver—Perhaps not so benign. J. Hepatol. 2015, 62, 1002–1004. [Google Scholar] [CrossRef] [Green Version]
- McPherson, S.; Hardy, T.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Evidence of NAFLD progression from steatosis to fibrosing-steatohepatitis using paired biopsies: Implications for prognosis and clinical management. J. Hepatol. 2015, 62, 1148–1155. [Google Scholar] [CrossRef]
- Nasr, P.; Ignatova, S.; Kechagias, S.; Ekstedt, M. Natural history of nonalcoholic fatty liver disease: A prospective follow-up study with serial biopsies. Hepatol. Commun. 2018, 2, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Ekstedt, M.; Hagström, H.; Nasr, P.; Fredrikson, M.; Stål, P.; Kechagias, S.; Hultcrantz, R. Fibrosis stage is the strongest predictor for disease-specific mortality in NAFLD after up to 33 years of follow-up. Hepatology 2015, 61, 1547–1554. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.; Stepanova, M.; Sanyal, A.J.; Harrison, S.A.; Ratziu, V.; Abdelmalek, M.F.; Diehl, A.M.; Caldwell, S.; Shiffman, M.L.; Schall, R.A.; et al. The conundrum of cryptogenic cirrhosis: Adverse outcomes without treatment options. J. Hepatol. 2018, 69, 1365–1370. [Google Scholar] [CrossRef] [PubMed]
- Ekstedt, M.; Franzén, L.E.; Mathiesen, U.L.; Thorelius, L.; Holmqvist, M.; Bodemar, G.; Kechagias, S. Long-term follow-up of patients with NAFLD and elevated liver enzymes. Hepatology 2006, 44, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Sanyal, A.J.; George, J. MAFLD: A Consensus-Driven Proposed Nomenclature for Metabolic Associated Fatty Liver Disease. Gastroenterology 2020, 158, 1999–2014.e1. [Google Scholar] [CrossRef]
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Marra, F.; Svegliati-Baroni, G. Lipotoxicity and the gut-liver axis in NASH pathogenesis. J. Hepatol. 2018, 68, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef]
- Akazawa, Y.; Nakao, K. Lipotoxicity pathways intersect in hepatocytes: Endoplasmic reticulum stress, c-Jun N-terminal kinase-1, and death receptors. Hepatol. Res. 2016, 46, 977–984. [Google Scholar] [CrossRef]
- Xu, B.; Jiang, M.; Chu, Y.; Wang, W.; Chen, D.; Li, X.; Zhang, Z.; Zhang, D.; Fan, D.; Nie, Y.; et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2018, 68, 773–782. [Google Scholar] [CrossRef]
- Magee, N.; Zou, A.; Zhang, Y. Pathogenesis of Nonalcoholic Steatohepatitis: Interactions between Liver Parenchymal and Nonparenchymal Cells. BioMed Res. Int. 2016, 2016, 5170402. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Dashty, M. A quick look at biochemistry: Carbohydrate metabolism. Clin. Biochem. 2013, 46, 1339–1352. [Google Scholar] [CrossRef]
- Carling, D.; Aguan, K.; Woods, A.; Verhoeven, A.J.; Beri, R.K.; Brennan, C.H.; Sidebottom, C.; Davison, M.D.; Scott, J. Mammalian AMP-activated protein kinase is homologous to yeast and plant protein kinases involved in the regulation of carbon metabolism. J. Biol. Chem. 1994, 269, 11442–11448. [Google Scholar] [CrossRef]
- Hong, S.-P.; Leiper, F.C.; Woods, A.; Carling, D.; Carlson, M. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc. Natl. Acad. Sci. USA 2003, 100, 8839–8843. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Schaffer, B.E.; Brunet, A. AMPK: An Energy-Sensing Pathway with Multiple Inputs and Outputs. Trends Cell Biol. 2016, 26, 190–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of Cellular Energy Sensing and Restoration of Metabolic Balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viollet, B.; Horman, S.; Leclerc, J.; Lantier, L.; Foretz, M.; Billaud, M.; Giri, S.; Andreelli, F. AMPK inhibition in health and disease. Crit. Rev. Biochem. Mol. Biol. 2010, 45, 276–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultze, S.M.; Hemmings, B.A.; Niessen, M.; Tschopp, O. PI3K/AKT, MAPK and AMPK signalling: Protein kinases in glucose homeostasis. Expert Rev. Mol. Med. 2012, 14, e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, F.A.; MacKintosh, C.; Hardie, D.G. AMP-activated protein kinase: A cellular energy sensor that comes in 12 flavours. FEBS J. 2016, 283, 2987–3001. [Google Scholar] [CrossRef]
- Yan, Y.; Zhou, X.E.; Xu, H.E.; Melcher, K. Structure and Physiological Regulation of AMPK. Int. J. Mol. Sci. 2018, 19, 3534. [Google Scholar] [CrossRef] [Green Version]
- Thornton, C.; Snowden, M.A.; Carling, D. Identification of a novel AMP-activated protein kinase beta subunit isoform that is highly expressed in skeletal muscle. J. Biol. Chem. 1998, 273, 12443–12450. [Google Scholar] [CrossRef] [Green Version]
- Cheung, P.C.; Salt, I.P.; Davies, S.P.; Hardie, D.G.; Carling, D. Characterization of AMP-activated protein kinase gamma-subunit isoforms and their role in AMP binding. Biochem. J. 2000, 346, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Crute, B.E.; Seefeld, K.; Gamble, J.; Kemp, B.E.; Witters, L.A. Functional domains of the alpha1 catalytic subunit of the AMP-activated protein kinase. J. Biol. Chem. 1998, 273, 35347–35354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.-W.; Wong, L.L.-Y.; Tse, E.Y.-T.; Liu, H.-F.; Leong, V.Y.-L.; Lee, J.M.-F.; Hardie, D.G.; Ng, I.O.-L.; Ching, Y.-P. AMPK promotes p53 acetylation via phosphorylation and inactivation of SIRT1 in liver cancer cells. Cancer Res. 2012, 72, 4394–4404. [Google Scholar] [CrossRef] [Green Version]
- Fox, M.M.; Phoenix, K.N.; Kopsiaftis, S.G.; Claffey, K.P. AMP-Activated Protein Kinase α 2 Isoform Suppression in Primary Breast Cancer Alters AMPK Growth Control and Apoptotic Signaling. Genes Cancer 2013, 4, 3–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, S.-M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, S.; Zhai, A.; Zhang, B.; Tian, G. AMPK-Mediated Regulation of Lipid Metabolism by Phosphorylation. Biol. Pharm. Bull. 2018, 41, 985–993. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [Green Version]
- Zadra, G.; Batista, J.L.; Loda, M. Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies. Mol. Cancer Res. 2015, 13, 1059–1072. [Google Scholar] [CrossRef] [Green Version]
- Suter, M.; Riek, U.; Tuerk, R.; Schlattner, U.; Wallimann, T.; Neumann, D. Dissecting the role of 5′-AMP for allosteric stimulation, activation, and deactivation of AMP-activated protein kinase. J. Biol. Chem. 2006, 281, 32207–32216. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G.; Carling, D.; Gamblin, S.J. AMP-activated protein kinase: Also regulated by ADP? Trends Biochem. Sci. 2011, 36, 470–477. [Google Scholar] [CrossRef] [PubMed]
- Gowans, G.J.; Hawley, S.A.; Ross, F.A.; Hardie, D.G. AMP is a true physiological regulator of AMP-activated protein kinase by both allosteric activation and enhancing net phosphorylation. Cell Metab. 2013, 18, 556–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, F.A.; Jensen, T.E.; Hardie, D.G. Differential regulation by AMP and ADP of AMPK complexes containing different γ subunit isoforms. Biochem. J. 2016, 473, 189–199. [Google Scholar] [CrossRef] [Green Version]
- Xiao, B.; Heath, R.; Saiu, P.; Leiper, F.C.; Leone, P.; Jing, C.; Walker, P.A.; Haire, L.; Eccleston, J.F.; Davis, C.T.; et al. Structural basis for AMP binding to mammalian AMP-activated protein kinase. Nature 2007, 449, 496–500. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regulation by ADP. Nature 2011, 472, 230–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, J.W.; Ross, F.A.; Liu, J.K.D.; Hardie, D.G. Regulation of AMP-activated protein kinase by a pseudosubstrate sequence on the gamma subunit. EMBO J. 2007, 26, 806–815. [Google Scholar] [CrossRef]
- Dzamko, N.; van Denderen, B.J.W.; Hevener, A.L.; Jørgensen, S.B.; Honeyman, J.; Galic, S.; Chen, Z.-P.; Watt, M.J.; Campbell, D.J.; Steinberg, G.R.; et al. AMPK beta1 deletion reduces appetite, preventing obesity and hepatic insulin resistance. J. Biol. Chem. 2010, 285, 115–122. [Google Scholar] [CrossRef] [Green Version]
- Gruzman, A.; Babai, G.; Sasson, S. Adenosine Monophosphate-Activated Protein Kinase (AMPK) as a New Target for Antidiabetic Drugs: A Review on Metabolic, Pharmacological and Chemical Considerations. Rev. Diabet. Stud. 2009, 6, 13–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, P.; Rubio, T.; Garcia-Gimeno, M.A. AMPKβ subunits: More than just a scaffold in the formation of AMPK complex. FEBS J. 2013, 280, 3723–3733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, G.R.; O’Neill, H.M.; Dzamko, N.L.; Galic, S.; Naim, T.; Koopman, R.; Jørgensen, S.B.; Honeyman, J.; Hewitt, K.; Chen, Z.-P.; et al. Whole body deletion of AMP-activated protein kinase {beta}2 reduces muscle AMPK activity and exercise capacity. J. Biol. Chem. 2010, 285, 37198–37209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinkosky, S.L.; Scott, J.W.; Desjardins, E.M.; Smith, B.K.; Day, E.A.; Ford, R.J.; Langendorf, C.G.; Ling, N.X.Y.; Nero, T.L.; Loh, K.; et al. Long-chain fatty acyl-CoA esters regulate metabolism via allosteric control of AMPK β1 isoforms. Nat. Metab. 2020, 2, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Woods, A.; Johnstone, S.R.; Dickerson, K.; Leiper, F.C.; Fryer, L.G.D.; Neumann, D.; Schlattner, U.; Wallimann, T.; Carlson, M.; Carling, D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr. Biol. 2003, 13, 2004–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, R.L.; Anderson, K.A.; Franzone, J.M.; Kemp, B.E.; Means, A.R.; Witters, L.A. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J. Biol. Chem. 2005, 280, 29060–29066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, A.; Dickerson, K.; Heath, R.; Hong, S.-P.; Momcilovic, M.; Johnstone, S.R.; Carlson, M.; Carling, D. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005, 2, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Hawley, S.A.; Pan, D.A.; Mustard, K.J.; Ross, L.; Bain, J.; Edelman, A.M.; Frenguelli, B.G.; Hardie, D.G. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005, 2, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Boon, H.; Bosselaar, M.; Praet, S.F.E.; Blaak, E.E.; Saris, W.H.M.; Wagenmakers, A.J.M.; McGee, S.L.; Tack, C.J.; Smits, P.; Hargreaves, M.; et al. Intravenous AICAR administration reduces hepatic glucose output and inhibits whole body lipolysis in type 2 diabetic patients. Diabetologia 2008, 51, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, T.S.; Jessen, N.; Jørgensen, J.O.L.; Møller, N.; Lund, S. Dissecting adipose tissue lipolysis: Molecular regulation and implications for metabolic disease. J. Mol. Endocrinol. 2014, 52, R199–R222. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Tamargo-Gómez, I.; Mariño, G. AMPK: Regulation of Metabolic Dynamics in the Context of Autophagy. Int. J. Mol. Sci. 2018, 19, 3812. [Google Scholar] [CrossRef] [Green Version]
- Salt, I.P.; Hardie, D.G. AMP-Activated Protein Kinase: An Ubiquitous Signaling Pathway with Key Roles in the Cardiovascular System. Circ. Res. 2017, 120, 1825–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, H.; He, L. Current understanding of metformin effect on the control of hyperglycemia in diabetes. J. Endocrinol. 2016, 228, R97–R106. [Google Scholar] [CrossRef] [Green Version]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegarty, B.D.; Turner, N.; Cooney, G.J.; Kraegen, E.W. Insulin resistance and fuel homeostasis: The role of AMP-activated protein kinase. Acta Physiol. 2009, 196, 129–145. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Stark, M.J.; Foster, D.W. Hepatic malonyl-CoA levels of fed, fasted and diabetic rats as measured using a simple radioisotopic assay. J. Biol. Chem. 1978, 253, 8291–8293. [Google Scholar] [CrossRef]
- McGarry, J.D. The mitochondrial carnitine palmitoyltransferase system: Its broadening role in fuel homoeostasis and new insights into its molecular features. Biochem. Soc. Trans. 1995, 23, 321–324. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.K.; Laybutt, D.R.; Dean, D.; Vavvas, D.; Sebokova, E.; Ellis, B.; Klimes, I.; Kraegen, E.W.; Shafrir, E.; Ruderman, N.B. Cytosolic citrate and malonyl-CoA regulation in rat muscle in vivo. Am. J. Physiol. 1999, 276, E1030–E1037. [Google Scholar] [CrossRef]
- Båvenholm, P.N.; Pigon, J.; Saha, A.K.; Ruderman, N.B.; Efendic, S. Fatty acid oxidation and the regulation of malonyl-CoA in human muscle. Diabetes 2000, 49, 1078–1083. [Google Scholar] [CrossRef] [Green Version]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef] [Green Version]
- Ertunc, M.E.; Hotamisligil, G.S. Lipid signaling and lipotoxicity in metaflammation: Indications for metabolic disease pathogenesis and treatment. J. Lipid Res. 2016, 57, 2099–2114. [Google Scholar] [CrossRef] [Green Version]
- Hauck, A.K.; Bernlohr, D.A. Oxidative stress and lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Iqbal, N.; Boden, G. The effects of free fatty acids on gluconeogenesis and glycogenolysis in normal subjects. J. Clin. Investig. 1999, 103, 365–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roden, M. How free fatty acids inhibit glucose utilization in human skeletal muscle. News Physiol. Sci. 2004, 19, 92–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraegen, E.W.; Cooney, G.J.; Turner, N. Muscle insulin resistance: A case of fat overconsumption, not mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2008, 105, 7627–7628. [Google Scholar] [CrossRef] [Green Version]
- Szendroedi, J.; Chmelik, M.; Schmid, A.I.; Nowotny, P.; Brehm, A.; Krssak, M.; Moser, E.; Roden, M. Abnormal hepatic energy homeostasis in type 2 diabetes. Hepatology 2009, 50, 1079–1086. [Google Scholar] [CrossRef]
- Itani, S.I.; Ruderman, N.B.; Schmieder, F.; Boden, G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002, 51, 2005–2011. [Google Scholar] [CrossRef] [Green Version]
- Roden, M.; Bernroider, E. Hepatic glucose metabolism in humans—its role in health and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2003, 17, 365–383. [Google Scholar] [CrossRef]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [Green Version]
- Kumashiro, N.; Erion, D.M.; Zhang, D.; Kahn, M.; Beddow, S.A.; Chu, X.; Still, C.D.; Gerhard, G.S.; Han, X.; Dziura, J.; et al. Cellular mechanism of insulin resistance in nonalcoholic fatty liver disease. Proc. Natl. Acad. Sci. USA 2011, 108, 16381–16385. [Google Scholar] [CrossRef] [Green Version]
- Magkos, F.; Su, X.; Bradley, D.; Fabbrini, E.; Conte, C.; Eagon, J.C.; Varela, J.E.; Brunt, E.M.; Patterson, B.W.; Klein, S. Intrahepatic diacylglycerol content is associated with hepatic insulin resistance in obese subjects. Gastroenterology 2012, 142, 1444–1446.e2. [Google Scholar] [CrossRef] [Green Version]
- Szendroedi, J.; Yoshimura, T.; Phielix, E.; Koliaki, C.; Marcucci, M.; Zhang, D.; Jelenik, T.; Müller, J.; Herder, C.; Nowotny, P.; et al. Role of diacylglycerol activation of PKCθ in lipid-induced muscle insulin resistance in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 9597–9602. [Google Scholar] [CrossRef] [Green Version]
- Ter Horst, K.W.; Gilijamse, P.W.; Versteeg, R.I.; Ackermans, M.T.; Nederveen, A.J.; La Fleur, S.E.; Romijn, J.A.; Nieuwdorp, M.; Zhang, D.; Samuel, V.T.; et al. Hepatic Diacylglycerol-Associated Protein Kinase Cε Translocation Links Hepatic Steatosis to Hepatic Insulin Resistance in Humans. Cell Rep. 2017, 19, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Skyler, J.S.; Bakris, G.L.; Bonifacio, E.; Darsow, T.; Eckel, R.H.; Groop, L.; Groop, P.-H.; Handelsman, Y.; Insel, R.A.; Mathieu, C.; et al. Differentiation of Diabetes by Pathophysiology, Natural History, and Prognosis. Diabetes 2017, 66, 241–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bril, F.; Barb, D.; Portillo-Sanchez, P.; Biernacki, D.; Lomonaco, R.; Suman, A.; Weber, M.H.; Budd, J.T.; Lupi, M.E.; Cusi, K. Metabolic and histological implications of intrahepatic triglyceride content in nonalcoholic fatty liver disease. Hepatology 2017, 65, 1132–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bril, F.; Cusi, K. Liver fat accumulation as a barometer of insulin responsiveness again points to adipose tissue as the culprit. Hepatology 2017, 66, 296–297. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Dufour, S.; Befroy, D.; Lehrke, M.; Hendler, R.E.; Shulman, G.I. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 2005, 54, 603–608. [Google Scholar] [CrossRef] [Green Version]
- Kirk, E.; Reeds, D.N.; Finck, B.N.; Mayurranjan, S.M.; Mayurranjan, M.S.; Patterson, B.W.; Klein, S. Dietary fat and carbohydrates differentially alter insulin sensitivity during caloric restriction. Gastroenterology 2009, 136, 1552–1560. [Google Scholar] [CrossRef] [Green Version]
- Browning, J.D.; Baker, J.A.; Rogers, T.; Davis, J.; Satapati, S.; Burgess, S.C. Short-term weight loss and hepatic triglyceride reduction: Evidence of a metabolic advantage with dietary carbohydrate restriction. Am. J. Clin. Nutr. 2011, 93, 1048–1052. [Google Scholar] [CrossRef]
- Haufe, S.; Engeli, S.; Kast, P.; Böhnke, J.; Utz, W.; Haas, V.; Hermsdorf, M.; Mähler, A.; Wiesner, S.; Birkenfeld, A.L.; et al. Randomized comparison of reduced fat and reduced carbohydrate hypocaloric diets on intrahepatic fat in overweight and obese human subjects. Hepatology 2011, 53, 1504–1514. [Google Scholar] [CrossRef]
- Weickert, M.O.; Loeffelholz, C.V.; Roden, M.; Chandramouli, V.; Brehm, A.; Nowotny, P.; Osterhoff, M.A.; Isken, F.; Spranger, J.; Landau, B.R.; et al. A Thr94Ala mutation in human liver fatty acid-binding protein contributes to reduced hepatic glycogenolysis and blunted elevation of plasma glucose levels in lipid-exposed subjects. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1078–E1084. [Google Scholar] [CrossRef] [Green Version]
- Newberry, E.P.; Xie, Y.; Kennedy, S.; Han, X.; Buhman, K.K.; Luo, J.; Gross, R.W.; Davidson, N.O. Decreased hepatic triglyceride accumulation and altered fatty acid uptake in mice with deletion of the liver fatty acid-binding protein gene. J. Biol. Chem. 2003, 278, 51664–51672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makowski, L.; Hotamisligil, G.S. The role of fatty acid binding proteins in metabolic syndrome and atherosclerosis. Curr. Opin. Lipidol. 2005, 16, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Clore, J.N.; Stillman, J.S.; Li, J.; O’Keefe, S.J.D.; Levy, J.R. Differential effect of saturated and polyunsaturated fatty acids on hepatic glucose metabolism in humans. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E358–E365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Zhang, C.; Luo, X.; Wang, P.; Zhou, W.; Zhong, S.; Xie, Y.; Jiang, Y.; Yang, P.; Tang, R.; et al. CD36 palmitoylation disrupts free fatty acid metabolism and promotes tissue inflammation in non-alcoholic steatohepatitis. J. Hepatol. 2018, 69, 705–717. [Google Scholar] [CrossRef]
- Tamura, S.; Shimomura, I. Contribution of adipose tissue and de novo lipogenesis to nonalcoholic fatty liver disease. J. Clin. Investig. 2005, 115, 1139–1142. [Google Scholar] [CrossRef]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Fabbrini, E.; Tiemann Luecking, C.; Love-Gregory, L.; Okunade, A.L.; Yoshino, M.; Fraterrigo, G.; Patterson, B.W.; Klein, S. Physiological Mechanisms of Weight Gain-Induced Steatosis in People with Obesity. Gastroenterology 2016, 150, 79–81.e2. [Google Scholar] [CrossRef] [Green Version]
- Lawitz, E.J.; Coste, A.; Poordad, F.; Alkhouri, N.; Loo, N.; McColgan, B.J.; Tarrant, J.M.; Nguyen, T.; Han, L.; Chung, C.; et al. Acetyl-CoA Carboxylase Inhibitor GS-0976 for 12 Weeks Reduces Hepatic De Novo Lipogenesis and Steatosis in Patients With Nonalcoholic Steatohepatitis. Clin. Gastroenterol. Hepatol. 2018, 16, 1983–1991.e3. [Google Scholar] [CrossRef]
- Diraison, F.; Dusserre, E.; Vidal, H.; Sothier, M.; Beylot, M. Increased hepatic lipogenesis but decreased expression of lipogenic gene in adipose tissue in human obesity. Am. J. Physiol. Endocrinol. Metab. 2002, 282, E46–E51. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.-M.; Linfoot, P.; Dare, D.; Aghajanian, K. Hepatic de novo lipogenesis in normoinsulinemic and hyperinsulinemic subjects consuming high-fat, low-carbohydrate and low-fat, high-carbohydrate isoenergetic diets. Am. J. Clin. Nutr. 2003, 77, 43–50. [Google Scholar] [CrossRef]
- Hudgins, L.C.; Hellerstein, M.; Seidman, C.; Neese, R.; Diakun, J.; Hirsch, J. Human fatty acid synthesis is stimulated by a eucaloric low fat, high carbohydrate diet. J. Clin. Investig. 1996, 97, 2081–2091. [Google Scholar] [CrossRef] [PubMed]
- Hudgins, L.C.; Seidman, C.E.; Diakun, J.; Hirsch, J. Human fatty acid synthesis is reduced after the substitution of dietary starch for sugar. Am. J. Clin. Nutr. 1998, 67, 631–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques-Lopes, I.; Ansorena, D.; Astiasaran, I.; Forga, L.; Martínez, J.A. Postprandial de novo lipogenesis and metabolic changes induced by a high-carbohydrate, low-fat meal in lean and overweight men. Am. J. Clin. Nutr. 2001, 73, 253–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, M.F.F.; Fielding, B.A.; Frayn, K.N. Metabolic interaction of dietary sugars and plasma lipids with a focus on mechanisms and de novo lipogenesis. Proc. Nutr. Soc. 2007, 66, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Chong, M.F.-F.; Hodson, L.; Bickerton, A.S.; Roberts, R.; Neville, M.; Karpe, F.; Frayn, K.N.; Fielding, B.A. Parallel activation of de novo lipogenesis and stearoyl-CoA desaturase activity after 3 d of high-carbohydrate feeding. Am. J. Clin. Nutr. 2008, 87, 817–823. [Google Scholar] [CrossRef] [Green Version]
- Jensen, T.; Abdelmalek, M.F.; Sullivan, S.; Nadeau, K.J.; Green, M.; Roncal, C.; Nakagawa, T.; Kuwabara, M.; Sato, Y.; Kang, D.-H.; et al. Fructose and sugar: A major mediator of non-alcoholic fatty liver disease. J. Hepatol. 2018, 68, 1063–1075. [Google Scholar] [CrossRef] [Green Version]
- Lammert, O.; Grunnet, N.; Faber, P.; Bjørnsbo, K.S.; Dich, J.; Larsen, L.O.; Neese, R.A.; Hellerstein, M.K.; Quistorff, B. Effects of isoenergetic overfeeding of either carbohydrate or fat in young men. Br. J. Nutr. 2000, 84, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Acheson, K.J.; Schutz, Y.; Bessard, T.; Anantharaman, K.; Flatt, J.P.; Jéquier, E. Glycogen storage capacity and de novo lipogenesis during massive carbohydrate overfeeding in man. Am. J. Clin. Nutr. 1988, 48, 240–247. [Google Scholar] [CrossRef] [Green Version]
- Sanyal, A.J.; Campbell-Sargent, C.; Mirshahi, F.; Rizzo, W.B.; Contos, M.J.; Sterling, R.K.; Luketic, V.A.; Shiffman, M.L.; Clore, J.N. Nonalcoholic steatohepatitis: Association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001, 120, 1183–1192. [Google Scholar] [CrossRef]
- Pérez-Carreras, M.; Del Hoyo, P.; Martín, M.A.; Rubio, J.C.; Martín, A.; Castellano, G.; Colina, F.; Arenas, J.; Solis-Herruzo, J.A. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003, 38, 999–1007. [Google Scholar] [CrossRef]
- Portincasa, P.; Grattagliano, I.; Lauterburg, B.H.; Palmieri, V.O.; Palasciano, G.; Stellaard, F. Liver breath tests non-invasively predict higher stages of non-alcoholic steatohepatitis. Clin. Sci. 2006, 111, 135–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banasch, M.; Ellrichmann, M.; Tannapfel, A.; Schmidt, W.E.; Goetze, O. The non-invasive 13C-methionine breath test detects hepatic mitochondrial dysfunction as a marker of disease activity in non-alcoholic steatohepatitis. Eur. J. Med. Res. 2012, 16, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, A.I.; Szendroedi, J.; Chmelik, M.; Krssák, M.; Moser, E.; Roden, M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care 2011, 34, 448–453. [Google Scholar] [CrossRef] [Green Version]
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T.; et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746. [Google Scholar] [CrossRef] [Green Version]
- Afolabi, P.R.; Scorletti, E.; Smith, D.E.; Almehmadi, A.A.; Calder, P.C.; Byrne, C.D. The characterisation of hepatic mitochondrial function in patients with non-alcoholic fatty liver disease (NAFLD) using the 13C-ketoisocaproate breath test. J. Breath Res. 2018, 12, 46002. [Google Scholar] [CrossRef] [Green Version]
- Petersen, K.F.; Befroy, D.E.; Dufour, S.; Rothman, D.L.; Shulman, G.I. Assessment of Hepatic Mitochondrial Oxidation and Pyruvate Cycling in NAFLD by (13)C Magnetic Resonance Spectroscopy. Cell Metab. 2016, 24, 167–171. [Google Scholar] [CrossRef] [Green Version]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef] [Green Version]
- Iozzo, P.; Bucci, M.; Roivainen, A.; Någren, K.; Järvisalo, M.J.; Kiss, J.; Guiducci, L.; Fielding, B.; Naum, A.G.; Borra, R.; et al. Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology 2010, 139, 846–856.e6. [Google Scholar] [CrossRef]
- Dasarathy, S.; Kasumov, T.; Edmison, J.M.; Gruca, L.L.; Bennett, C.; Duenas, C.; Marczewski, S.; McCullough, A.J.; Hanson, R.W.; Kalhan, S.C. Glycine and urea kinetics in nonalcoholic steatohepatitis in human: Effect of intralipid infusion. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G567–G575. [Google Scholar] [CrossRef] [Green Version]
- Geng, Y.; Faber, K.N.; de Meijer, V.E.; Blokzijl, H.; Moshage, H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol. Int. 2021, 15, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Bedossa, P.; Tordjman, J.; Aron-Wisnewsky, J.; Poitou, C.; Oppert, J.-M.; Torcivia, A.; Bouillot, J.-L.; Paradis, V.; Ratziu, V.; Clément, K. Systematic review of bariatric surgery liver biopsies clarifies the natural history of liver disease in patients with severe obesity. Gut 2017, 66, 1688–1696. [Google Scholar] [CrossRef]
- Martínez-Agustin, O.; Hernández-Morante, J.J.; Martínez-Plata, E.; Sánchez de Medina, F.; Garaulet, M. Differences in AMPK expression between subcutaneous and visceral adipose tissue in morbid obesity. Regul. Pept. 2010, 163, 31–36. [Google Scholar] [CrossRef]
- Gauthier, M.-S.; O’Brien, E.L.; Bigornia, S.; Mott, M.; Cacicedo, J.M.; Xu, X.J.; Gokce, N.; Apovian, C.; Ruderman, N. Decreased AMP-activated protein kinase activity is associated with increased inflammation in visceral adipose tissue and with whole-body insulin resistance in morbidly obese humans. Biochem. Biophys. Res. Commun. 2011, 404, 382–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.J.; Gauthier, M.-S.; Hess, D.T.; Apovian, C.M.; Cacicedo, J.M.; Gokce, N.; Farb, M.; Valentine, R.J.; Ruderman, N.B. Insulin sensitive and resistant obesity in humans: AMPK activity, oxidative stress, and depot-specific changes in gene expression in adipose tissue. J. Lipid Res. 2012, 53, 792–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grisouard, J.; Timper, K.; Radimerski, T.M.; Frey, D.M.; Peterli, R.; Kola, B.; Korbonits, M.; Herrmann, P.; Krähenbühl, S.; Zulewski, H.; et al. Mechanisms of metformin action on glucose transport and metabolism in human adipocytes. Biochem. Pharmacol. 2010, 80, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 112, 1796–1808. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, W.; Kasuga, M. Cell signaling. Fat stress and liver resistance. Science 2008, 322, 1483–1484. [Google Scholar] [CrossRef] [PubMed]
- Elbein, S.C.; Kern, P.A.; Rasouli, N.; Yao-Borengasser, A.; Sharma, N.K.; Das, S.K. Global gene expression profiles of subcutaneous adipose and muscle from glucose-tolerant, insulin-sensitive, and insulin-resistant individuals matched for BMI. Diabetes 2011, 60, 1019–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Kusminski, C.M.; Scherer, P.E. Adipose tissue remodeling and obesity. J. Clin. Investig. 2011, 121, 2094–2101. [Google Scholar] [CrossRef] [Green Version]
- Yin, W.; Mu, J.; Birnbaum, M.J. Role of AMP-activated protein kinase in cyclic AMP-dependent lipolysis in 3T3-L1 adipocytes. J. Biol. Chem. 2003, 278, 43074–43080. [Google Scholar] [CrossRef] [Green Version]
- Daval, M.; Diot-Dupuy, F.; Bazin, R.; Hainault, I.; Viollet, B.; Vaulont, S.; Hajduch, E.; Ferré, P.; Foufelle, F. Anti-lipolytic action of AMP-activated protein kinase in rodent adipocytes. J. Biol. Chem. 2005, 280, 25250–25257. [Google Scholar] [CrossRef] [Green Version]
- Daval, M.; Foufelle, F.; Ferré, P. Functions of AMP-activated protein kinase in adipose tissue. J. Physiol. 2006, 574, 55–62. [Google Scholar] [CrossRef]
- Koh, H.-J.; Hirshman, M.F.; He, H.; Li, Y.; Manabe, Y.; Balschi, J.A.; Goodyear, L.J. Adrenaline is a critical mediator of acute exercise-induced AMP-activated protein kinase activation in adipocytes. Biochem. J. 2007, 403, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, M.; Abbott, M.J.; Tang, T.; Hudak, C.S.S.; Kim, Y.; Bruss, M.; Hellerstein, M.K.; Lee, H.-Y.; Samuel, V.T.; Shulman, G.I.; et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011, 13, 739–748. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-J.; Tang, T.; Abbott, M.; Viscarra, J.A.; Wang, Y.; Sul, H.S. AMPK Phosphorylates Desnutrin/ATGL and Hormone-Sensitive Lipase To Regulate Lipolysis and Fatty Acid Oxidation within Adipose Tissue. Mol. Cell. Biol. 2016, 36, 1961–1976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zechner, R.; Madeo, F.; Kratky, D. Cytosolic lipolysis and lipophagy: Two sides of the same coin. Nat. Rev. Mol. Cell Biol. 2017, 18, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.M.; Azuma, K.; Kelley, C.; Pencek, R.; Radikova, Z.; Laymon, C.; Price, J.; Goodpaster, B.H.; Kelley, D.E. PET imaging reveals distinctive roles for different regional adipose tissue depots in systemic glucose metabolism in nonobese humans. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1134–E1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mottillo, E.P.; Desjardins, E.M.; Crane, J.D.; Smith, B.K.; Green, A.E.; Ducommun, S.; Henriksen, T.I.; Rebalka, I.A.; Razi, A.; Sakamoto, K.; et al. Lack of Adipocyte AMPK Exacerbates Insulin Resistance and Hepatic Steatosis through Brown and Beige Adipose Tissue Function. Cell Metab. 2016, 24, 118–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferré, P.; Foufelle, F. SREBP-1c transcription factor and lipid homeostasis: Clinical perspective. Horm. Res. 2007, 68, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Castle, J.C.; Hara, Y.; Raymond, C.K.; Garrett-Engele, P.; Ohwaki, K.; Kan, Z.; Kusunoki, J.; Johnson, J.M. ACC2 is expressed at high levels in human white adipose and has an isoform with a novel N-terminus corrected. PLoS ONE 2009, 4, e4369. [Google Scholar] [CrossRef]
- Goodpaster, B.H.; Sparks, L.M. Metabolic Flexibility in Health and Disease. Cell Metab. 2017, 25, 1027–1036. [Google Scholar] [CrossRef] [Green Version]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef] [Green Version]
- Højlund, K.; Mustard, K.J.; Staehr, P.; Hardie, D.G.; Beck-Nielsen, H.; Richter, E.A.; Wojtaszewski, J.F.P. AMPK activity and isoform protein expression are similar in muscle of obese subjects with and without type 2 diabetes. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E239–E244. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Smith, A.C.; van Denderen, B.J.W.; Chen, Z.; Murthy, S.; Campbell, D.J.; Heigenhauser, G.J.F.; Dyck, D.J.; Kemp, B.E. AMP-activated protein kinase is not down-regulated in human skeletal muscle of obese females. J. Clin. Endocrinol. Metab. 2004, 89, 4575–4580. [Google Scholar] [CrossRef] [Green Version]
- Højlund, K.; Staehr, P.; Hansen, B.F.; Green, K.A.; Hardie, D.G.; Richter, E.A.; Beck-Nielsen, H.; Wojtaszewski, J.F.P. Increased phosphorylation of skeletal muscle glycogen synthase at NH2-terminal sites during physiological hyperinsulinemia in type 2 diabetes. Diabetes 2003, 52, 1393–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koistinen, H.A.; Galuska, D.; Chibalin, A.V.; Yang, J.; Zierath, J.R.; Holman, G.D.; Wallberg-Henriksson, H. 5-amino-imidazole carboxamide riboside increases glucose transport and cell-surface GLUT4 content in skeletal muscle from subjects with type 2 diabetes. Diabetes 2003, 52, 1066–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuthbertson, D.J.; Babraj, J.A.; Mustard, K.J.W.; Towler, M.C.; Green, K.A.; Wackerhage, H.; Leese, G.P.; Baar, K.; Thomason-Hughes, M.; Sutherland, C.; et al. 5-aminoimidazole-4-carboxamide 1-beta-D-ribofuranoside acutely stimulates skeletal muscle 2-deoxyglucose uptake in healthy men. Diabetes 2007, 56, 2078–2084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babraj, J.A.; Mustard, K.; Sutherland, C.; Towler, M.C.; Chen, S.; Smith, K.; Green, K.; Leese, G.; Hardie, D.G.; Rennie, M.J.; et al. Blunting of AICAR-induced human skeletal muscle glucose uptake in type 2 diabetes is dependent on age rather than diabetic status. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1042–E1048. [Google Scholar] [CrossRef] [Green Version]
- Musi, N.; Fujii, N.; Hirshman, M.F.; Ekberg, I.; Fröberg, S.; Ljungqvist, O.; Thorell, A.; Goodyear, L.J. AMP-activated protein kinase (AMPK) is activated in muscle of subjects with type 2 diabetes during exercise. Diabetes 2001, 50, 921–927. [Google Scholar] [CrossRef] [Green Version]
- Roepstorff, C.; Vistisen, B.; Donsmark, M.; Nielsen, J.N.; Galbo, H.; Green, K.A.; Hardie, D.G.; Wojtaszewski, J.F.P.; Richter, E.A.; Kiens, B. Regulation of hormone-sensitive lipase activity and Ser563 and Ser565 phosphorylation in human skeletal muscle during exercise. J. Physiol. 2004, 560, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Watt, M.J.; Steinberg, G.R.; Chan, S.; Garnham, A.; Kemp, B.E.; Febbraio, M.A. Beta-adrenergic stimulation of skeletal muscle HSL can be overridden by AMPK signaling. FASEB J. 2004, 18, 1445–1446. [Google Scholar] [CrossRef] [PubMed]
- Steenberg, D.E.; Jørgensen, N.B.; Birk, J.B.; Sjøberg, K.A.; Kiens, B.; Richter, E.A.; Wojtaszewski, J.F.P. Exercise training reduces the insulin-sensitizing effect of a single bout of exercise in human skeletal muscle. J. Physiol. 2019, 597, 89–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibala, M.J.; McGee, S.L.; Garnham, A.P.; Howlett, K.F.; Snow, R.J.; Hargreaves, M. Brief intense interval exercise activates AMPK and p38 MAPK signaling and increases the expression of PGC-1alpha in human skeletal muscle. J. Appl. Physiol. 2009, 106, 929–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, N.J.; Parker, B.L.; Chaudhuri, R.; Fisher-Wellman, K.H.; Kleinert, M.; Humphrey, S.J.; Yang, P.; Holliday, M.; Trefely, S.; Fazakerley, D.J.; et al. Global Phosphoproteomic Analysis of Human Skeletal Muscle Reveals a Network of Exercise-Regulated Kinases and AMPK Substrates. Cell Metab. 2015, 22, 922–935. [Google Scholar] [CrossRef] [Green Version]
- Golabi, P.; Locklear, C.T.; Austin, P.; Afdhal, S.; Byrns, M.; Gerber, L.; Younossi, Z.M. Effectiveness of exercise in hepatic fat mobilization in non-alcoholic fatty liver disease: Systematic review. World J. Gastroenterol. 2016, 22, 6318–6327. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, M.; Suraamornkul, S.; Piper, P.; Hardies, L.J.; Glass, L.; Cersosimo, E.; Pratipanawatr, T.; Miyazaki, Y.; Defronzo, R.A. Decreased plasma adiponectin concentrations are closely related to hepatic fat content and hepatic insulin resistance in pioglitazone-treated type 2 diabetic patients. J. Clin. Endocrinol. Metab. 2004, 89, 200–206. [Google Scholar] [CrossRef] [Green Version]
- Brown, M.S.; Goldstein, J.L. Selective versus total insulin resistance: A pathogenic paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef] [Green Version]
- Kohjima, M.; Enjoji, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2007, 20, 351–358. [Google Scholar] [CrossRef] [Green Version]
- Kohjima, M.; Higuchi, N.; Kato, M.; Kotoh, K.; Yoshimoto, T.; Fujino, T.; Yada, M.; Yada, R.; Harada, N.; Enjoji, M.; et al. SREBP-1c, regulated by the insulin and AMPK signaling pathways, plays a role in nonalcoholic fatty liver disease. Int. J. Mol. Med. 2008, 21, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Von Loeffelholz, C.; Döcke, S.; Lock, J.F.; Lieske, S.; Horn, P.; Kriebel, J.; Wahl, S.; Singmann, P.; de Las Heras Gala, T.; Grallert, H.; et al. Increased lipogenesis in spite of upregulated hepatic 5′AMP-activated protein kinase in human non-alcoholic fatty liver. Hepatol. Res. 2017, 47, 890–901. [Google Scholar] [CrossRef]
- Piro, S.; Spadaro, L.; Russello, M.; Spampinato, D.; Oliveri, C.E.; Vasquez, E.; Benigno, R.; Brancato, F.; Purrello, F.; Rabuazzo, A.M. Molecular determinants of insulin resistance, cell apoptosis and lipid accumulation in non-alcoholic steatohepatitis. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 545–552. [Google Scholar] [CrossRef]
- Angelini, G.; Castagneto Gissey, L.; Del Corpo, G.; Giordano, C.; Cerbelli, B.; Severino, A.; Manco, M.; Basso, N.; Birkenfeld, A.L.; Bornstein, S.R.; et al. New insight into the mechanisms of ectopic fat deposition improvement after bariatric surgery. Sci. Rep. 2019, 9, 17315. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.; Saltiel, A.R. From overnutrition to liver injury: AMP-activated protein kinase in nonalcoholic fatty liver diseases. J. Biol. Chem. 2020, 295, 12279–12289. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.-W.; Addy, C.; Kusunoki, J.; Anderson, N.N.; Deja, S.; Fu, X.; Burgess, S.C.; Li, C.; Ruddy, M.; Chakravarthy, M.; et al. Acetyl CoA Carboxylase Inhibition Reduces Hepatic Steatosis but Elevates Plasma Triglycerides in Mice and Humans: A Bedside to Bench Investigation. Cell Metab. 2017, 26, 394–406.e6. [Google Scholar] [CrossRef] [PubMed]
- Stiede, K.; Miao, W.; Blanchette, H.S.; Beysen, C.; Harriman, G.; Harwood, H.J.; Kelley, H.; Kapeller, R.; Schmalbach, T.; Westlin, W.F. Acetyl-coenzyme A carboxylase inhibition reduces de novo lipogenesis in overweight male subjects: A randomized, double-blind, crossover study. Hepatology 2017, 66, 324–334. [Google Scholar] [CrossRef] [Green Version]
- Goedeke, L.; Bates, J.; Vatner, D.F.; Perry, R.J.; Wang, T.; Ramirez, R.; Li, L.; Ellis, M.W.; Zhang, D.; Wong, K.E.; et al. Acetyl-CoA Carboxylase Inhibition Reverses NAFLD and Hepatic Insulin Resistance but Promotes Hypertriglyceridemia in Rodents. Hepatology 2018, 68, 2197–2211. [Google Scholar] [CrossRef] [Green Version]
- Loomba, R.; Kayali, Z.; Noureddin, M.; Ruane, P.; Lawitz, E.J.; Bennett, M.; Wang, L.; Harting, E.; Tarrant, J.M.; McColgan, B.J.; et al. GS-0976 Reduces Hepatic Steatosis and Fibrosis Markers in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1463–1473.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, N.; Cohen, D.E. Trimming the Fat: Acetyl-CoA Carboxylase Inhibition for the Management of NAFLD. Hepatology 2018, 68, 2062–2065. [Google Scholar] [CrossRef] [Green Version]
- Esquejo, R.M.; Salatto, C.T.; Delmore, J.; Albuquerque, B.; Reyes, A.; Shi, Y.; Moccia, R.; Cokorinos, E.; Peloquin, M.; Monetti, M.; et al. Activation of Liver AMPK with PF-06409577 Corrects NAFLD and Lowers Cholesterol in Rodent and Primate Preclinical Models. EBioMedicine 2018, 31, 122–132. [Google Scholar] [CrossRef] [Green Version]
- Von Loeffelholz, C.; Lieske, S.; Neuschäfer-Rube, F.; Willmes, D.M.; Raschzok, N.; Sauer, I.M.; König, J.; Fromm, M.F.; Horn, P.; Chatzigeorgiou, A.; et al. The human longevity gene homolog INDY and interleukin-6 interact in hepatic lipid metabolism. Hepatology 2017, 66, 616–630. [Google Scholar] [CrossRef] [Green Version]
- Birkenfeld, A.L.; Lee, H.-Y.; Guebre-Egziabher, F.; Alves, T.C.; Jurczak, M.J.; Jornayvaz, F.R.; Zhang, D.; Hsiao, J.J.; Martin-Montalvo, A.; Fischer-Rosinsky, A.; et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 2011, 14, 184–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, P.; Sun, X.; Chaggan, C.; Liao, Z.; Wong, K.; He, F.; Singh, S.; Loomba, R.; Karin, M.; Witztum, J.L.; et al. An AMPK-caspase-6 axis controls liver damage in nonalcoholic steatohepatitis. Science 2020, 367, 652–660. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
von Loeffelholz, C.; Coldewey, S.M.; Birkenfeld, A.L. A Narrative Review on the Role of AMPK on De Novo Lipogenesis in Non-Alcoholic Fatty Liver Disease: Evidence from Human Studies. Cells 2021, 10, 1822. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10071822
von Loeffelholz C, Coldewey SM, Birkenfeld AL. A Narrative Review on the Role of AMPK on De Novo Lipogenesis in Non-Alcoholic Fatty Liver Disease: Evidence from Human Studies. Cells. 2021; 10(7):1822. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10071822
Chicago/Turabian Stylevon Loeffelholz, Christian, Sina M. Coldewey, and Andreas L. Birkenfeld. 2021. "A Narrative Review on the Role of AMPK on De Novo Lipogenesis in Non-Alcoholic Fatty Liver Disease: Evidence from Human Studies" Cells 10, no. 7: 1822. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10071822