
Proteostasis Response to Protein Misfolding in Controlled Hypertension

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Clinical Data Collection and Sample Processing
2.3. Measuring Protein Aggregation with Fluorescent Probes
2.4. Immunoassays
2.5. Fourier-Transform Infra-Red (FTIR) Spectroscopy
2.6. Statistical Analysis
3. Results
3.1. Participant’s Characteristics
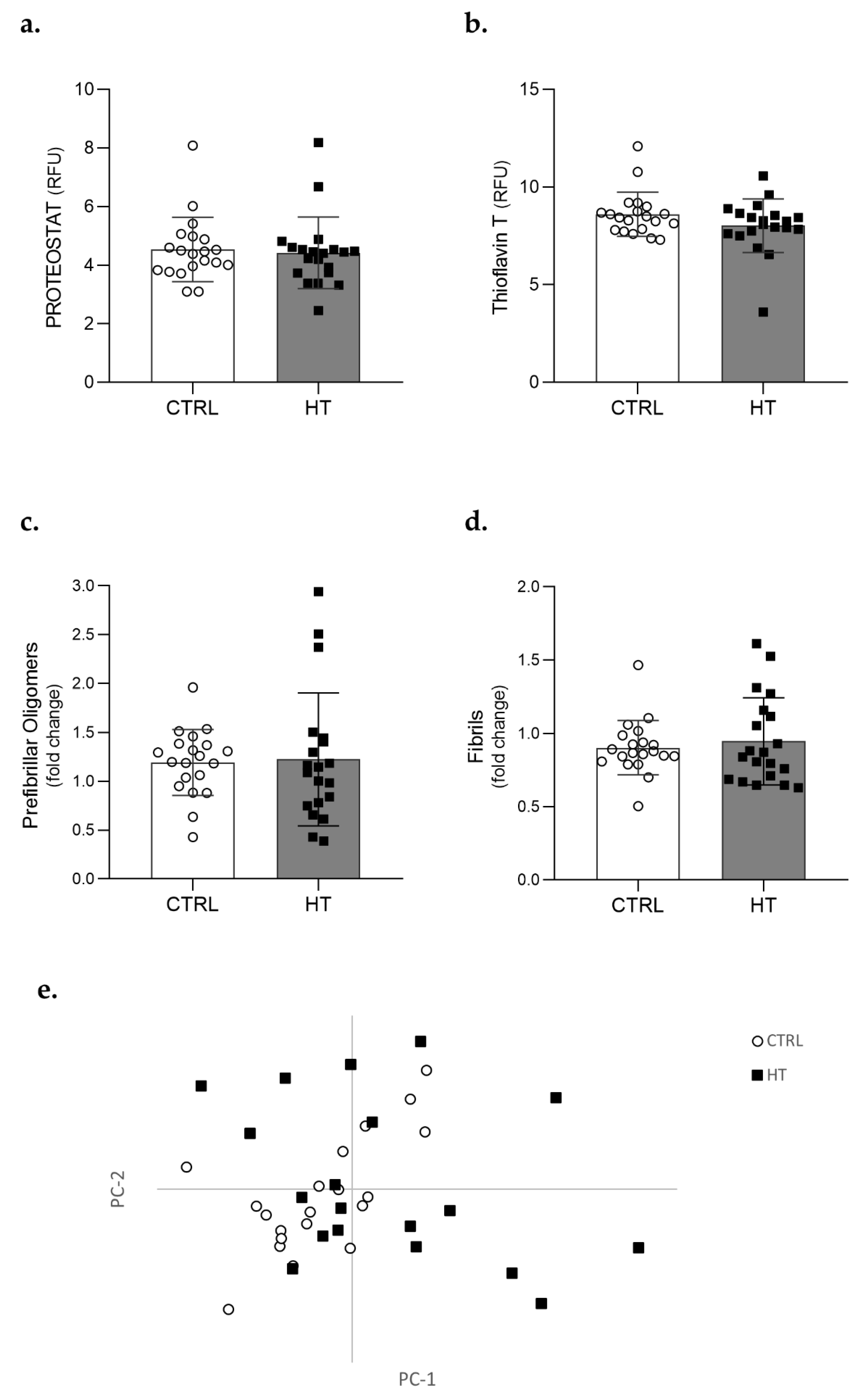
3.2. Protein Aggregation in the Plasma of Individuals with Hypertension
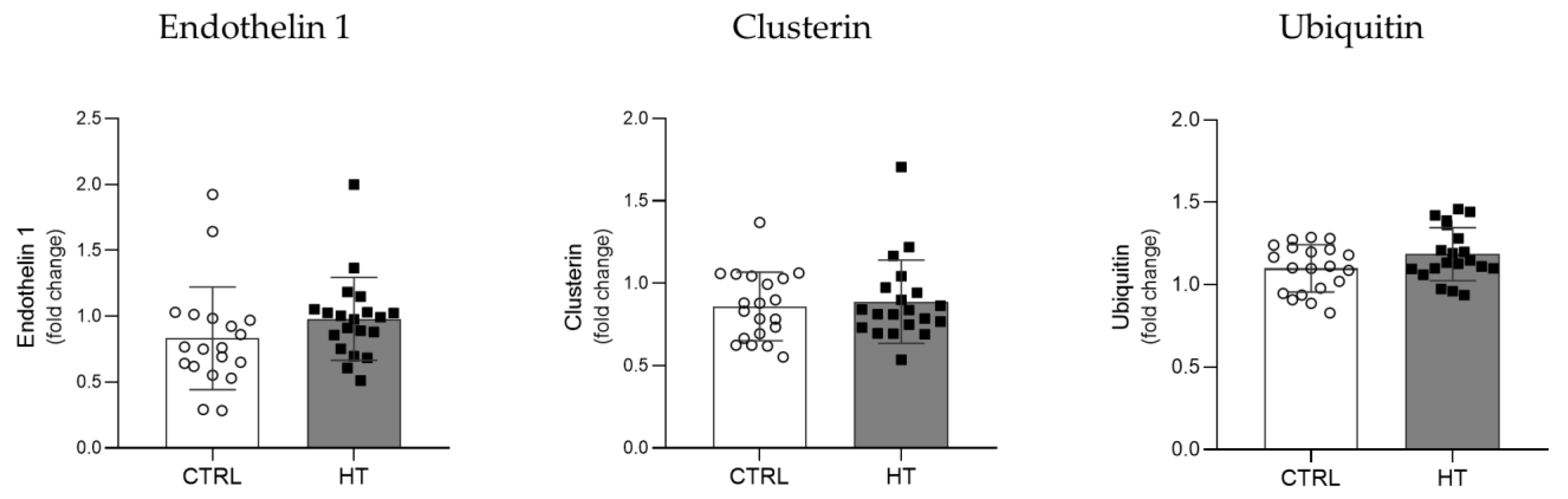
3.3. Proteostasis Biomarkers in the Plasma of HT Individuals
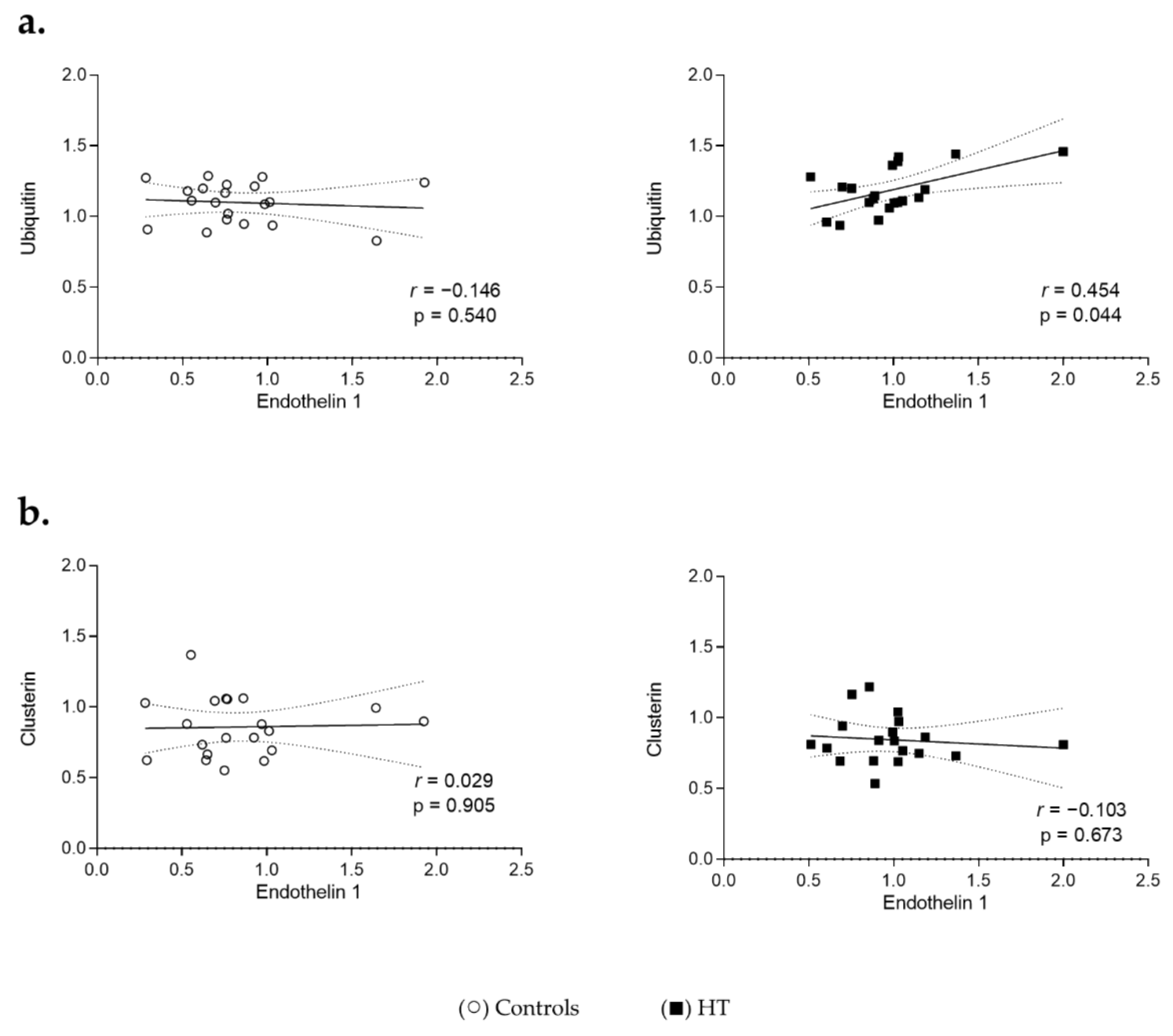
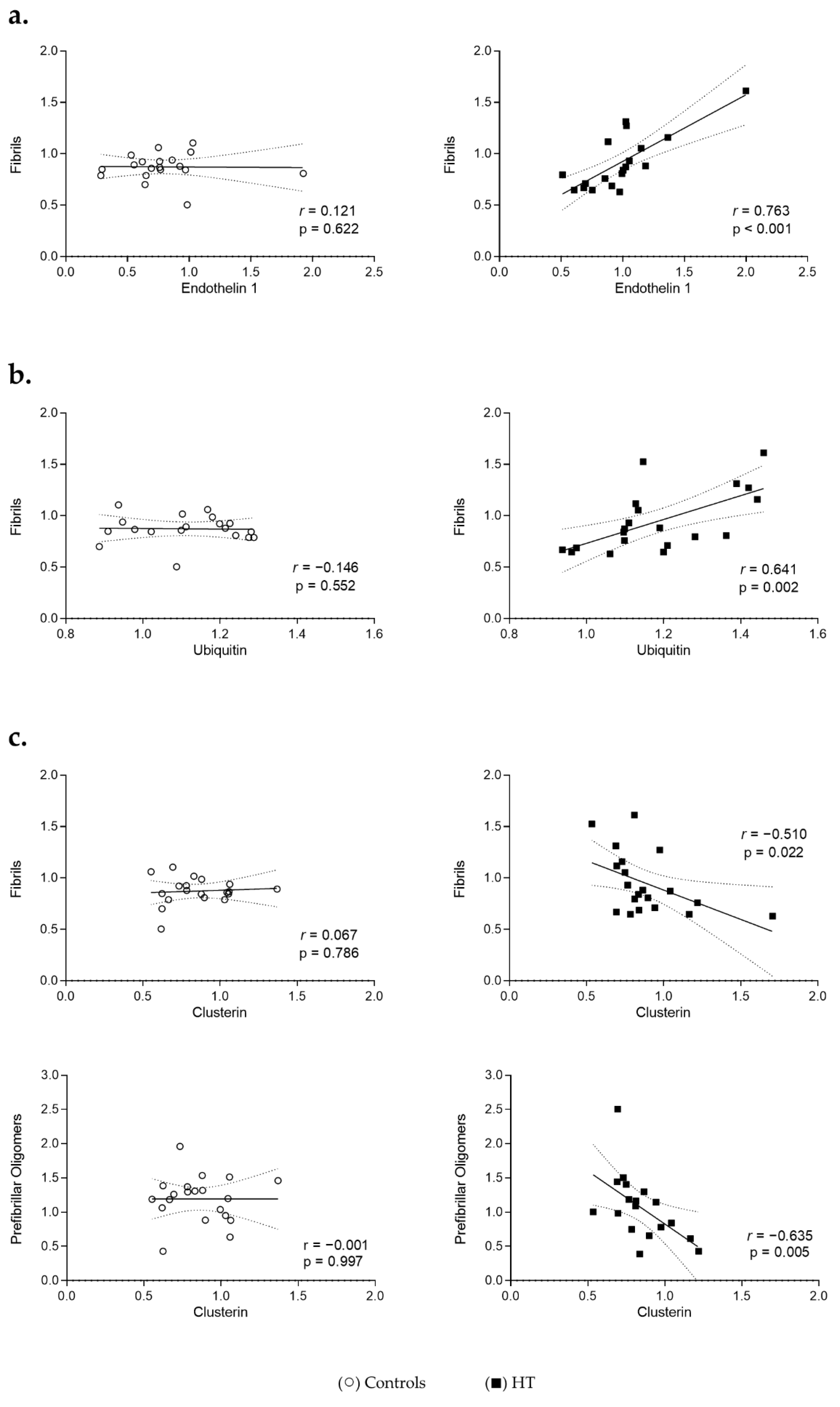
3.4. Proteostasis and Protein Aggregation Associations in the Plasma of HT Individuals
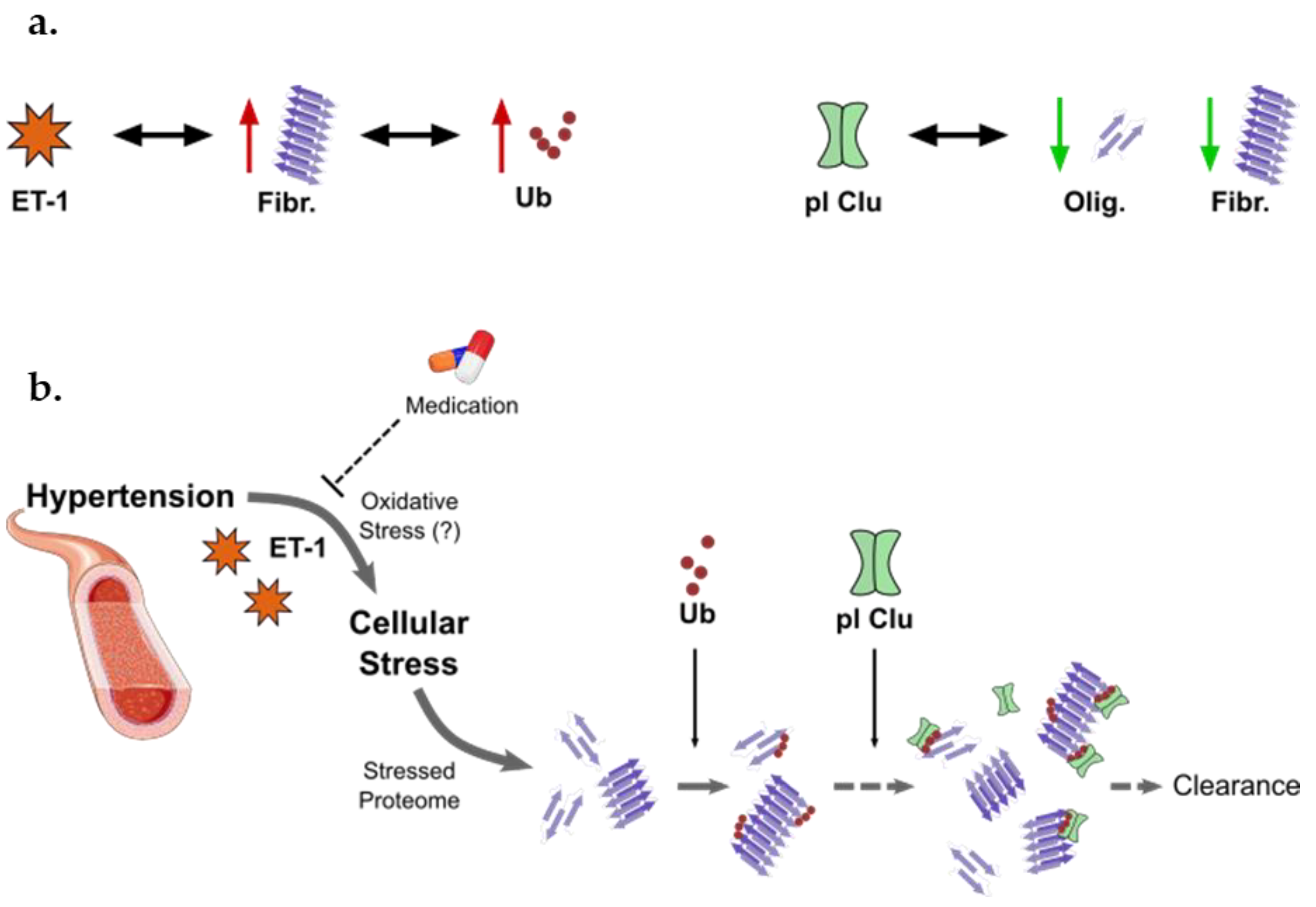
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. World Health Statistics 2020: Monitoring Health for the SDGs, Sustainable Development Goals; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Abbafati, C.; Abbas, K.M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abdelalim, A.; Abdollahi, M.; Abdollahpour, I.; Abegaz, K.H.; Abolhassani, H.; Aboyans, V.; et al. Global Burden of 87 Risk Factors in 204 Countries and Territories, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1223–1249. [Google Scholar] [CrossRef]
- Abbafati, C.; Abbas, K.M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abdelalim, A.; Abdollahi, M.; Abdollahpour, I.; Abegaz, K.H.; Abolhassani, H.; Aboyans, V.; et al. Global Burden of 369 Diseases and Injuries in 204 Countries and Territories, 1990–2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1204–1222. [Google Scholar] [CrossRef]
- Mills, K.T.; Stefanescu, A.; He, J. The Global Epidemiology of Hypertension. Nat. Rev. Nephrol. 2020, 16, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Bentham, J.; Di Cesare, M.; Bixby, H.; Danaei, G.; Cowan, M.J.; Paciorek, C.J.; Singh, G.; Hajifathalian, K.; Bennett, J.E.; et al. Worldwide Trends in Blood Pressure from 1975 to 2015: A Pooled Analysis of 1479 Population-Based Measurement Studies with 19.1 Million Participants. Lancet 2017, 389, 37–55. [Google Scholar] [CrossRef] [Green Version]
- Ostchega, Y.; Fryar, C.D.; Nwankwo, T.; Nguyen, D.T. Hypertension Prevalence Among Adults Aged 18 and Over: United States, 2017–2018. NCHS Data Brief 2020, 364, 1–8. [Google Scholar]
- Harrison, D.G.; Coffman, T.M.; Wilcox, C.S. Pathophysiology of Hypertension: The Mosaic Theory and Beyond. Circ. Res. 2021, 128, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Oparil, S.; Acelajado, M.C.; Bakris, G.L.; Berlowitz, D.R.; Cífková, R.; Dominiczak, A.F.; Grassi, G.; Jordan, J.; Poulter, N.R.; Rodgers, A.; et al. Hypertension. Nat. Rev. Dis. Prim. 2018, 4, 18014. [Google Scholar] [CrossRef] [Green Version]
- Gomes, C.; Ferreira, D.; Carvalho, J.P.F.; Fernandes, J.; Gouveia, M.; Barreto, C.A.V.; Ribeiro, F.; Duque, A.S.; Vieira, S.I. Current Genetic Engineering Strategies for the Production of Antihypertensive ACEI Peptides. Biotechnol. Bioeng. 2020, 117, 2610–2628. [Google Scholar] [CrossRef]
- Tousoulis, D.; Kampoli, A.-M.; Tentolouris, C.; Papageorgiou, N.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr. Vasc. Pharmacol. 2011, 10, 4–18. [Google Scholar] [CrossRef]
- Kostov, K. The Causal Relationship between Endothelin-1 and Hypertension: Focusing on Endothelial Dysfunction, Arterial Stiffness, Vascular Remodeling, and Blood Pressure Regulation. Life 2021, 11, 986. [Google Scholar] [CrossRef]
- Hall, J.E.; Granger, J.P.; Dubinion, J.; George, E.; Hamza, S.; Speed, J.; Hall, M.E. Hypertension: Physiology and Pathophysiology. Compr. Physiol. 2012, 2, 2393–2442. [Google Scholar] [CrossRef] [PubMed]
- Martinez-quinones, P.; Mccarthy, C.G.; Watts, S.W.; Klee, N.S.; Komic, A.; Calmasini, F.B.; Priviero, F.; Warner, A.; Chenghao, Y.; Wenceslau, C.F. Hypertension Induced Morphological and Physiological Changes in Cells of the Arterial Wall. Am. J. Hypertens. 2018, 31, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Cheng, Y.; Zhang, H.; Ba, M.; Chen, P.; Li, H.; Chen, K.; Sha, W.; Zhang, C.; Chen, H. Association between High Blood Pressure and Long Term Cardiovascular Events in Young Adults: Systematic Review and Meta-Analysis. BMJ 2020, 370, m3222. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.; Montezano, A.C.; Touyz, R.M. Vascular Biology of Ageing—Implications in Hypertension. J. Mol. Cell. Cardiol. 2015, 83, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Willis, M.S.; Patterson, C. Proteotoxicity and Cardiac Dysfunction—Alzheimer’s Disease of the Heart? N. Engl. J. Med. 2013, 368, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayyadevara, S.; Mercanti, F.; Wang, X.; Mackintosh, S.G.; Tackett, A.J.; Prayaga, S.V.S.; Romeo, F.; Reis, R.J.S.; Mehta, J.L. Age- and Hypertension-Associated Protein Aggregates in Mouse Heart Have Similar Proteomic Profiles. Hypertension 2016, 67, 1006–1013. [Google Scholar] [CrossRef] [Green Version]
- Kostin, S.; Pool, L.; Elsässer, A.; Hein, S.; Drexler, H.C.A.; Arnon, E.; Hayakawa, Y.; Zimmermann, R.; Bauer, E.; Klövekorn, W.P.; et al. Myocytes Die by Multiple Mechanisms in Failing Human Hearts. Circ. Res. 2003, 92, 715–724. [Google Scholar] [CrossRef] [Green Version]
- Howlett, G.J.; Moore, K.J. Untangling the Role of Amyloid in Atherosclerosis. Curr. Opin. Lipidol. 2006, 17, 541–547. [Google Scholar] [CrossRef]
- González-López, E.; Gallego-Delgado, M.; Guzzo-Merello, G.; De Haro-Del Moral, F.J.; Cobo-Marcos, M.; Robles, C.; Bornstein, B.; Salas, C.; Lara-Pezzi, E.; Alonso-Pulpon, L.; et al. Wild-Type Transthyretin Amyloidosis as a Cause of Heart Failure with Preserved Ejection Fraction. Eur. Heart J. 2015, 36, 2585–2594. [Google Scholar] [CrossRef] [Green Version]
- Gouveia, M.; Xia, K.; Colón, W.; Vieira, S.I.; Ribeiro, F. Protein Aggregation, Cardiovascular Diseases, and Exercise Training: Where Do We Stand? Ageing Res. Rev. 2017, 40, 1–10. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress over the Last Decade. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In Vivo Aspects of Protein Folding and Quality Control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, G.G.; Hipp, M.S.; Ulrich Hartl, F. Functional Modules of the Proteostasis Network. Cold Spring Harb. Perspect. Biol. 2020, 12, a033951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyriakou, P.; Mouselimis, D.; Tsarouchas, A.; Rigopoulos, A.; Bakogiannis, C.; Noutsias, M.; Vassilikos, V. Diagnosis of Cardiac Amyloidosis: A Systematic Review on the Role of Imaging and Biomarkers. BMC Cardiovasc. Disord. 2018, 18, 221. [Google Scholar] [CrossRef]
- From, A.M.; Maleszewski, J.J.; Rihal, C.S. Current Status of Endomyocardial Biopsy. Mayo Clin. Proc. 2011, 86, 1095–1102. [Google Scholar] [CrossRef] [Green Version]
- Williams, B.; Mancia, G.; Spiering, W.; Rosei, E.A.; Azizi, M.; Burnier, M.; Clement, D.L.; Coca, A.; de Simone, G.; Dominiczak, A.; et al. 2018 ESC/ESH Guidelines for the Management of Arterial Hypertension: The Task Force for the management of arterial hypertension of the European Society of Cardiology (ESC) and the European Society of Hypertension (ESH). Eur. Heart J. 2018, 39, 3021–3104. [Google Scholar] [CrossRef]
- Millen, K.R.; Buhimschi, C.S.; Zhao, G.; Rood, K.M.; Tabbah, S.; Buhimschi, I.A. Serum and Urine Thioflavin-T-Enhanced Fluorescence in Severe Preeclampsia. Hypertension 2018, 71, 1185–1192. [Google Scholar] [CrossRef]
- Kim, C.S.; Kim, B.; Choi, H.S.; Bae, E.H.; Ma, S.K.; Han, K.-D.; Kim, S.W. Cumulative Hypertension Burden and Risk of End-Stage Renal Disease. Hypertens. Res. 2021, 44, 1652–1661. [Google Scholar] [CrossRef]
- Trigo, D.; Nadais, A.; da Cruz e Silva, O.A.B. Unravelling Protein Aggregation as an Ageing Related Process or a Neuropathological Response. Ageing Res. Rev. 2019, 51, 67–77. [Google Scholar] [CrossRef]
- Zhang, J.; Cao, L.; Wang, X.; Li, Q.; Zhang, M.; Cheng, C.; Yu, L.; Xue, F.; Sui, W.; Sun, S.; et al. The E3 Ubiquitin Ligase TRIM31 Plays a Critical Role in Hypertensive Nephropathy by Promoting Proteasomal Degradation of MAP3K7 in the TGF-Β1 Signaling Pathway. Cell Death Differ. 2022, 29, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Mccarthy, X.C.G.; Wenceslau, X.C.F.; Calmasini, X.F.B.; Klee, N.S.; Brands, M.W.; Joe, B.; Webb, R.C. Reconstitution of Autophagy Ameliorates Vascular Function and Arterial Stiffening in Spontaneously Hypertensive Rats. Am. J. Physiol. Circ. Physiol. 2019, 317, H1013–H1027. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, O.; Hirohama, D.; Ishizawa, K.; Shibata, S. Role of the Ubiquitin Proteasome System in the Regulation of Blood Pressure: A Review. Int. J. Mol. Sci. 2020, 21, 5358. [Google Scholar] [CrossRef]
- Xia, K.; Trasatti, H.; Wymer, J.P.; Colón, W. Increased Levels of Hyper-Stable Protein Aggregates in Plasma of Older Adults. Age 2016, 38, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magalhães, S.; Trindade, D.; Martins, T.; Martins Rosa, I.; Delgadillo, I.; Goodfellow, B.J.; da Cruz e Silva, O.A.B.; Henriques, A.G.; Nunes, A. Monitoring Plasma Protein Aggregation during Aging Using Conformation-Specific Antibodies and FTIR Spectroscopy. Clin. Chim. Acta 2020, 502, 25–33. [Google Scholar] [CrossRef]
- Hofmann, C.; Katus, H.A.; Doroudgar, S. Protein Misfolding in Cardiac Disease. Circulation 2019, 139, 2085–2088. [Google Scholar] [CrossRef]
- Dobson, C.M. Protein Folding and Misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef]
- Oparil, S.; Zaman, M.A.; Calhoun, D.A. Pathogenesis of Hypertension. Ann. Intern. Med. 2003, 139, 761–776. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabel, C.G. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [Green Version]
- Kayed, R.; Head, E.; Sarsoza, F.; Saing, T.; Cotman, C.W.; Necula, M.; Margol, L.; Wu, J.; Breydo, L.; Thompson, J.L.; et al. Fibril Specific, Conformation Dependent Antibodies Recognize a Generic Epitope Common to Amyloid Fibrils and Fibrillar Oligomers That Is Absent in Prefibrillar Oligomers. Mol. Neurodegener. 2007, 2, 18. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, K.; Narang, R.; Bhatia, J.; Saluja, D. Expression of Heat Shock Protein 70 Gene and Its Correlation with Inflammatory Markers in Essential Hypertension. PLoS ONE 2016, 11, e0151060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Böhm, F.; Pernow, J. The Importance of Endothelin-1 for Vascular Dysfunction in Cardiovascular Disease. Cardiovasc. Res. 2007, 76, 8–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, J.E.; Do Carmo, J.M.; Da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity-Induced Hypertension: Interaction of Neurohumoral and Renal Mechanisms. Circ. Res. 2015, 116, 991–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, H.N.; Rivera-Gonzalez, O.; Gibert, Y.; Speed, J.S. Endothelin-1 in the Pathophysiology of Obesity and Insulin Resistance. Obes. Rev. 2020, 21, e13086. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, S.; Wu, Y.; Song, P.; Zou, M.H. Tyrosine Nitration of PA700 Activates the 26S Proteasome to Induce Endothelial Dysfunction in Mice with Angiotensin II-Induced Hypertension. Hypertension 2009, 54, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Stangl, K.; Stangl, V. The Ubiquitin-Proteasome Pathway and Endothelial (Dys)Function. Cardiovasc. Res. 2010, 85, 281–290. [Google Scholar] [CrossRef]
- Kraft, C.; Peter, M.; Hofmann, K. Selective Autophagy: Ubiquitin-Mediated Recognition and Beyond. Nat. Cell Biol. 2010, 12, 836–841. [Google Scholar] [CrossRef]
- Galves, M.; Rathi, R.; Prag, G.; Ashkenazi, A. Ubiquitin Signaling and Degradation of Aggregate-Prone Proteins. Trends Biochem. Sci. 2019, 44, 872–884. [Google Scholar] [CrossRef]
- Park, S.; Mathis, K.W.; Lee, I.K. The Physiological Roles of Apolipoprotein J/Clusterin in Metabolic and Cardiovascular Diseases. Rev. Endocr. Metab. Disord. 2014, 15, 45–53. [Google Scholar] [CrossRef]
- Zoubeidi, A.; Ettinger, S.; Beraldi, E.; Hadaschik, B.; Zardan, A.; Klomp, L.W.J.; Nelson, C.C.; Rennie, P.S.; Gleave, M.E. Clusterin Facilitates COMMD1 and I-ΚB Degradation to Enhance NF-ΚB Activity in Prostate Cancer Cells. Mol. Cancer Res. 2010, 8, 119–130. [Google Scholar] [CrossRef] [Green Version]
- Nortley, R.; Korte, N.; Izquierdo, P.; Hirunpattarasilp, C.; Mishra, A.; Jaunmuktane, Z.; Kyrargyri, V.; Pfeiffer, T.; Khennouf, L.; Madry, C.; et al. Amyloid β Oligomers Constrict Human Capillaries in Alzheimer’s Disease via Signaling to Pericytes. Science 2019, 365, aac4354. [Google Scholar] [CrossRef] [PubMed]
- Varanda, A.S.; Santos, M.; Soares, A.R.; Vitorino, R.; Oliveira, C.; Santos, M.A.S.; Sofia, A.; Santos, M.; Soares, A.R.; Vitorino, R. Human Cells Adapt to Translational Errors by Modulating Protein Synthesis Rate and Protein Turnover. RNA Biol. 2020, 17, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Scheidt, T.; Łapińska, U.; Kumita, J.R.; Whiten, D.R.; Klenerman, D.; Wilson, M.R.; Cohen, S.I.A.; Linse, S.; Vendruscolo, M.; Dobson, C.M.; et al. Secondary Nucleation and Elongation Occur at Different Sites on Alzheimer’s Amyloid-β Aggregates. Sci. Adv. 2019, 5, eaau3112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turkieh, A.; Porouchani, S.; Beseme, O.; Chwastyniak, M.; Amouyel, P.; Lamblin, N.; Balligand, J.L.; Bauters, C.; Pinet, F. Increased Clusterin Levels after Myocardial Infarction Is Due to a Defect in Protein Degradation Systems Activity. Cell Death Dis. 2019, 10, 608. [Google Scholar] [CrossRef] [Green Version]
- Turkieh, A.; Fertin, M.; Bouvet, M.; Mulder, P.; Drobecq, H.; Lemesle, G.; Lamblin, N.; De Groote, P.; Porouchani, S.; Chwastyniak, M.; et al. Expression and Implication of Clusterin in Left Ventricular Remodeling After Myocardial Infarction. Circ. Heart Fail. 2018, 11, e004838. [Google Scholar] [CrossRef] [Green Version]
- Hassan, N.; Maldonado-Valderrama, J.; Gunning, A.P.; Morris, V.J.; Ruso, J.M. Investigating the Effect of an Arterial Hypertension Drug on the Structural Properties of Plasma Protein. Colloids Surf. B Biointerfaces 2011, 87, 489–497. [Google Scholar] [CrossRef]
- Miyazaki, Y.; Ishikawa, T.; Kamatari, Y.O.; Nakagaki, T.; Takatsuki, H.; Ishibashi, D.; Kuwata, K.; Nishida, N.; Atarashi, R. Identification of Alprenolol Hydrochloride as an Anti-Prion Compound Using Surface Plasmon Resonance Imaging. Mol. Neurobiol. 2019, 56, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Nedaei, H.; Rezaei-Ghaleh, N.; Giller, K.; Becker, S.; Karami, L.; Moosavi-Movahedi, A.A.; Griesinger, C.; Saboury, A.A. The Calcium-Free Form of Atorvastatin Inhibits Amyloid-β(1–42) Aggregation in Vitro. J. Biol. Chem. 2022, 298, 101662. [Google Scholar] [CrossRef]
- Giampietro, C.; Lionetti, M.C.; Costantini, G.; Mutti, F.; Zapperi, S.; La Porta, C.A.M. Cholesterol Impairment Contributes to Neuroserpin Aggregation. Sci. Rep. 2017, 7, 43669. [Google Scholar] [CrossRef] [Green Version]
- Enroth, S.; Maturi, V.; Berggrund, M.; Enroth, S.B.; Moustakas, A.; Johansson, Å.; Gyllensten, U. Systemic and Specific Effects of Antihypertensive and Lipid-Lowering Medication on Plasma Protein Biomarkers for Cardiovascular Diseases. Sci. Rep. 2018, 8, 5531. [Google Scholar] [CrossRef] [Green Version]
- Malik, S.; Siddiqi, M.K.; Majid, N.; Masroor, A.; Moasfar Ali, S.; Khan, R.H. Unravelling the Inhibitory and Cytoprotective Potential of Diuretics towards Amyloid Fibrillation. Int. J. Biol. Macromol. 2020, 150, 1258–1271. [Google Scholar] [CrossRef] [PubMed]
- Cicalese, S.; Okuno, K.; Eguchi, S. Detection of Protein Aggregation and Proteotoxicity Induced by Angiotensin II in Vascular Smooth Muscle Cells. J. Cardiovasc. Pharmacol. 2021, 77, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Gouveia, M.; Schmidt, C.; Teixeira, M.; Magalhães, S.; Nunes, A.; Lopes, M.; Ferreira, R.; Vitorino, R.; Santos, M.; Vieira, S.; et al. Analysis of Plasma Protein Aggregation from Patients with Heart Failure with Preserved Ejection Fraction. J. Hypertens. 2021, 39, e102. [Google Scholar] [CrossRef]
- Teixeira, M.; Gouveia, M.; Duarte, A.; Ferreira, M.; Simões, M.I.; Conceição, M.; Silva, G.; Magalhães, S.; Ferreira, R.; Nunes, A.; et al. Regular Exercise Participation Contributes to Better Proteostasis, Inflammatory and Vasoactive Profiles in Patients with Hypertension. Am. J. Hypertens. 2019, 33, 119–123. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control Group | HT Group | ||
|---|---|---|---|
| Characteristics, Mean ± SD | n = 20 | n = 20 | p Value |
| Male/Female (n) | 8/12 | 5/15 | 0.324 |
| Age (years) | 64.8 ± 6.4 | 65.6 ± 6.1 | 0.750 |
| BMI (kg/m2) | 26.1 ± 3.2 | 30.3 ± 5.8 | 0.009 |
| Waist Circumference (cm) | 92.4 ± 11.6 | 104.2 ± 13.1 | 0.006 |
| Office SBP (mm Hg) | 126.1 ± 11.2 | 126.0 ± 14.1 | 0.979 |
| Office DBP (mm Hg) | 73.7 ± 7.8 | 71.5 ± 8.1 | 0.407 |
| HR (bpm) | 65.6 ± 8.5 | 66.2 ± 10.8 | 0.840 |
| Medical history | No. (%) | No. (%) | p value |
| Diabetes | 1 (5) | 4 (20) | 0.341 |
| Obesity | 3 (12) | 10 (50) | 0.041 |
| Overweight | 11 (55) | 6 (30) | 0.333 |
| Dyslipidaemia | 11 (55) | 16 (80) | 0.176 |
| Medication | No. (%) | No. (%) | p value |
| ACEI | - | 3 (15) | 0.231 |
| ARB | - | 10 (50) | <0.001 |
| Diuretics | - | 13 (65) | <0.001 |
| CCB | - | 4 (20) | 0.106 |
| Β-Blockers | - | 3 (15) | 0.231 |
| Statins | 8 (40) | 10 (50) | 0.751 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teixeira, M.; Trindade, D.; Gouveia, M.; Eller-Borges, R.; Magalhães, S.; Duarte, A.; Ferreira, M.; Simões, M.I.; Conceição, M.; Nunes, A.; et al. Proteostasis Response to Protein Misfolding in Controlled Hypertension. Cells 2022, 11, 1686. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11101686
Teixeira M, Trindade D, Gouveia M, Eller-Borges R, Magalhães S, Duarte A, Ferreira M, Simões MI, Conceição M, Nunes A, et al. Proteostasis Response to Protein Misfolding in Controlled Hypertension. Cells. 2022; 11(10):1686. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11101686
Chicago/Turabian StyleTeixeira, Manuel, Dário Trindade, Marisol Gouveia, Roberta Eller-Borges, Sandra Magalhães, Ana Duarte, Miriam Ferreira, Maria I. Simões, Maria Conceição, Alexandra Nunes, and et al. 2022. "Proteostasis Response to Protein Misfolding in Controlled Hypertension" Cells 11, no. 10: 1686. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11101686