
Alterations of Mitochondrial Network by Cigarette Smoking and E-Cigarette Vaping

 , and
, and 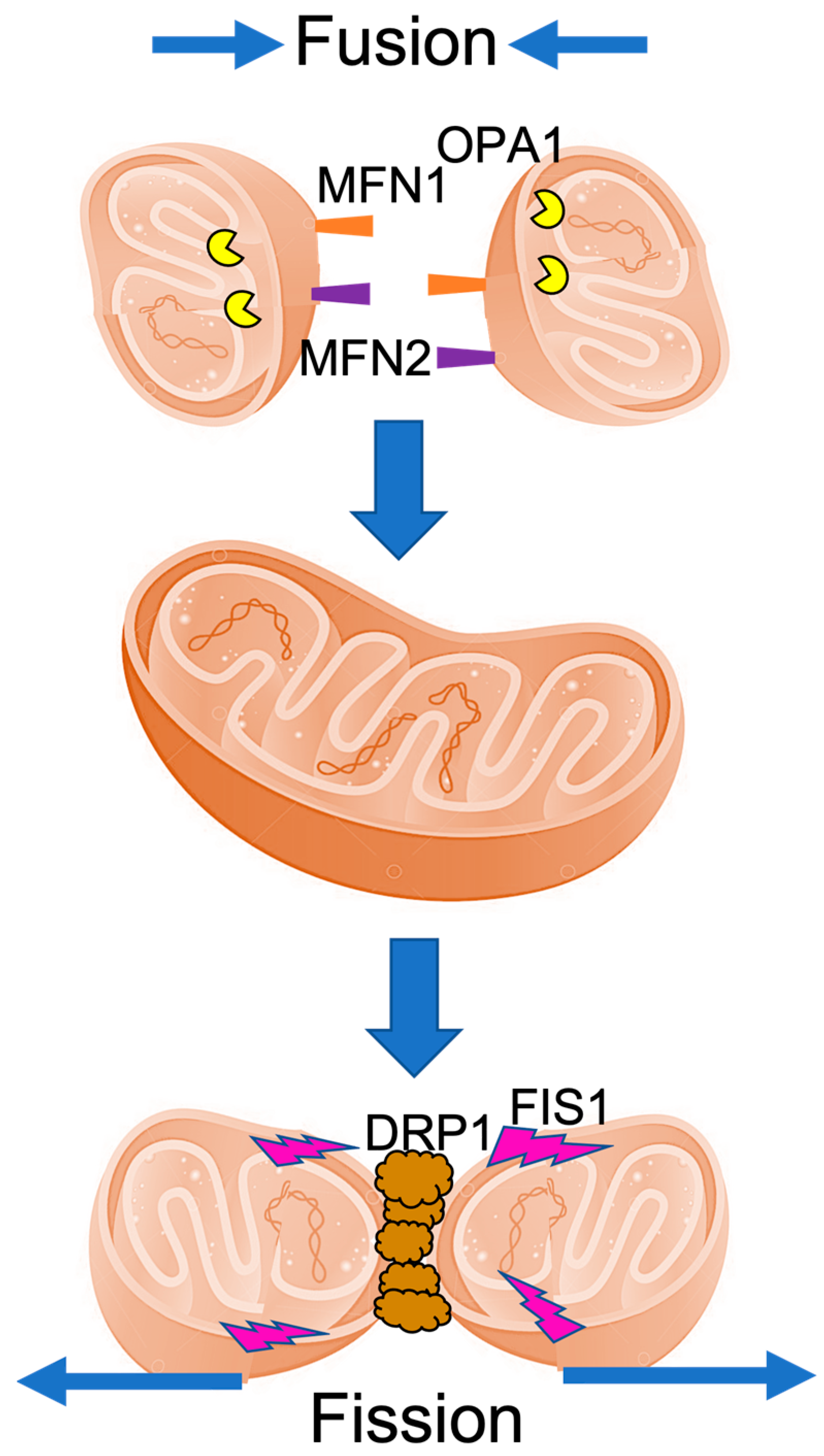{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Mitochondrial Fusion and Fission Machinery
3. Cigarette Smoke and E-Cigarette Vape Extraction
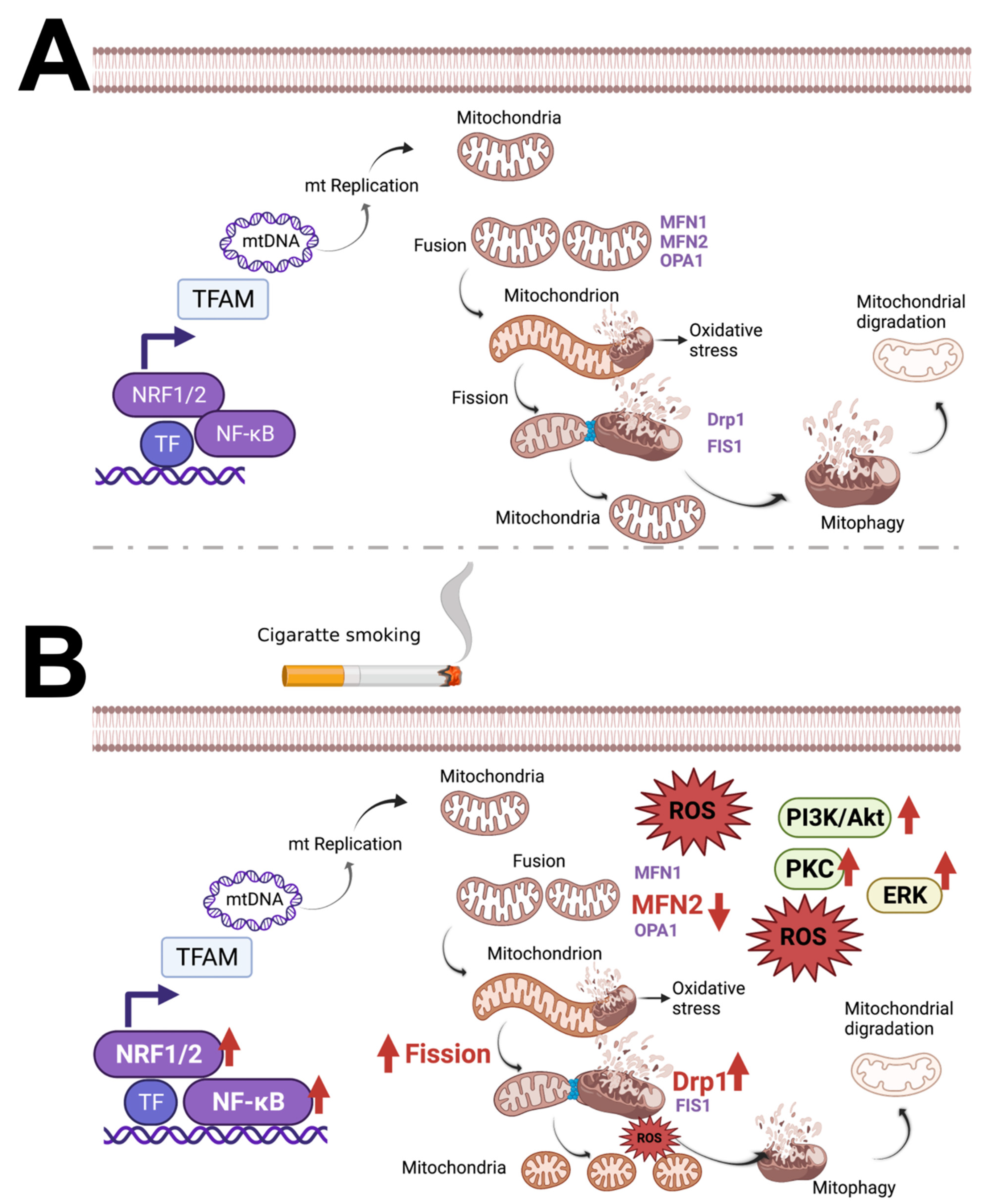
3.1. Cigarette Smoke (CS) and Cigarette Smoke Extract (CSE) Trigger Mitochondrial ROS
3.2. The Role of Nicotine in Mitochondrial Dysfunction
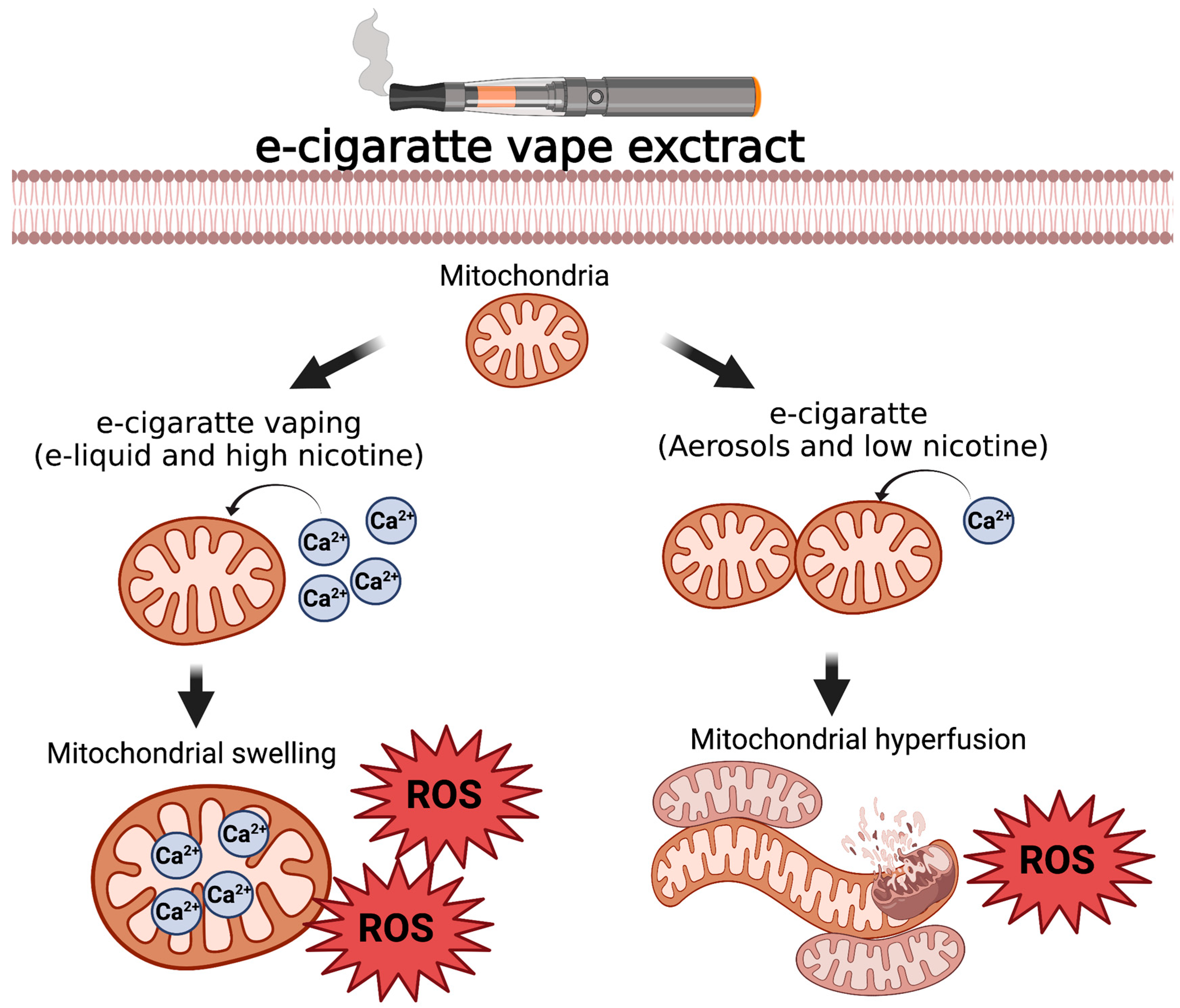
3.3. Constituents of Fluids Used in ENDS Are Cytotoxic and Impair Mitochondrial Function
4. Maternal Health
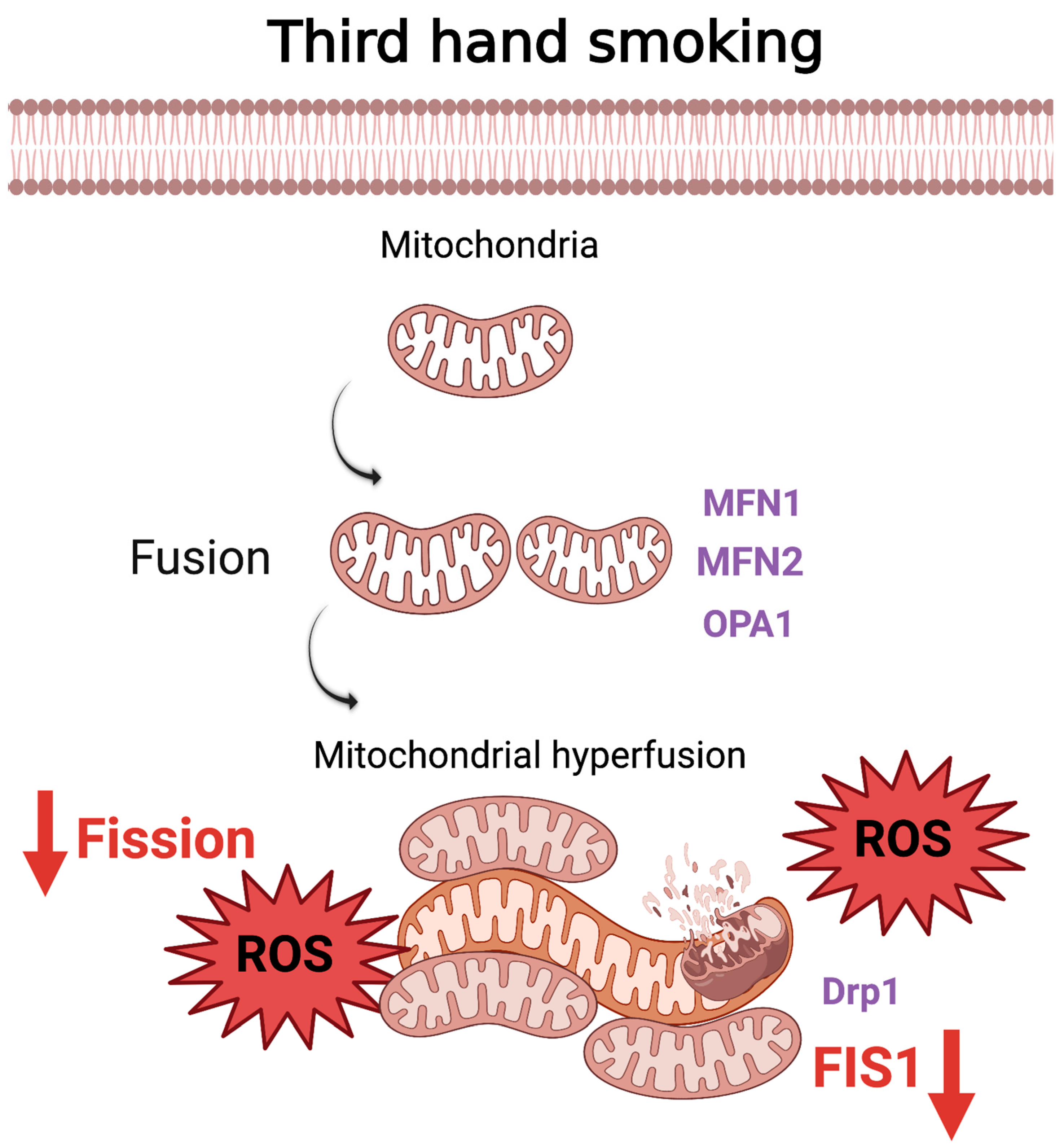
5. Thirdhand Smoke
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- WHO. Tobacco. 2020. Available online: https://www.who.int/news-room/fact-sheets/detail/tobacco (accessed on 17 April 2022).
- WHO. Electronic Nicotine and Non-Nicotine Delivery Systems: A Brief. 2020. Available online: https://www.euro.who.int/en/health-topics/disease-prevention/tobacco/publications/2020/electronic-nicotine-and-non-nicotine-delivery-systems-a-brief-2020 (accessed on 17 April 2022).
- Grana, R.; Benowitz, N.; Glantz, S.A. E-cigarettes: A scientific review. Circulation 2014, 129, 1972–1986. [Google Scholar] [CrossRef] [PubMed]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Tobacco Smoke and Involuntary Smoking; IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 2004; Volume 83.
- Jassem, E.; Szymanowska, A.; Siemińska, A.; Jassem, J. Smoking and lung cancer. Pneumonol. Alergol. Pol. 2009, 77, 469–473. [Google Scholar] [PubMed]
- Loeb, L.A.; Ernster, V.L.; Warner, K.E.; Abbotts, J.; Laszlo, J. Smoking and lung cancer: An overview. Cancer Res. 1984, 44, 5940–5958. [Google Scholar] [PubMed]
- Macacu, A.; Autier, P.; Boniol, M.; Boyle, P. Active and passive smoking and risk of breast cancer: A meta-analysis. Breast Cancer Res. Treat. 2015, 154, 213–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrose, J.A.; Barua, R.S. The pathophysiology of cigarette smoking and cardiovascular disease: An update. J. Am. Coll. Cardiol. 2004, 43, 1731–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leone, A.; Giannini, D.; Bellotto, C.; Balbarini, A. Passive smoking and coronary heart disease. Curr. Vasc. Pharm. 2004, 2, 175–182. [Google Scholar] [CrossRef]
- Rigotti, N.A.; Pasternak, R.C. Cigarette smoking and coronary heart disease: Risks and management. Cardiol. Clin. 1996, 14, 51–68. [Google Scholar] [CrossRef]
- Pan, B.; Jin, X.; Jun, L.; Qiu, S.; Zheng, Q.; Pan, M. The relationship between smoking and stroke: A meta-analysis. Medicine 2019, 98, e14872. [Google Scholar] [CrossRef]
- Shah, R.S.; Cole, J.W. Smoking and stroke: The more you smoke the more you stroke. Expert Rev. Cardiovasc. Ther. 2010, 8, 917–932. [Google Scholar] [CrossRef]
- Li, G.; Saad, S.; Oliver, B.G.; Chen, H. Heat or Burn? Impacts of Intrauterine Tobacco Smoke and E-Cigarette Vapor Exposure on the Offspring’s Health Outcome. Toxics 2018, 6, 43. [Google Scholar] [CrossRef] [Green Version]
- Arcavi, L.; Benowitz, N.L. Cigarette Smoking and Infection. Arch. Intern. Med. 2004, 164, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.L.; Oliver, B.G.; Chen, H. What lessons have we learnt about the impact of maternal cigarette smoking from animal models? Clin. Exp. Pharmacol. Physiol. 2020, 47, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Talhout, R.; Schulz, T.; Florek, E.; van Benthem, J.; Wester, P.; Opperhuizen, A. Hazardous compounds in tobacco smoke. Int. J. Environ. Res. Public Health 2011, 8, 613–628. [Google Scholar] [CrossRef] [PubMed]
- Konstantinou, E.; Fotopoulou, F.; Drosos, A.; Dimakopoulou, N.; Zagoriti, Z.; Niarchos, A.; Makrynioti, D.; Kouretas, D.; Farsalinos, K.; Lagoumintzis, G.; et al. Tobacco-specific nitrosamines: A literature review. Food Chem. Toxicol. 2018, 118, 198–203. [Google Scholar] [CrossRef]
- Hang, B.; Sarker, A.H.; Havel, C.; Saha, S.; Hazra, T.K.; Schick, S.; Jacob, P., 3rd; Rehan, V.K.; Chenna, A.; Sharan, D.; et al. Thirdhand smoke causes DNA damage in human cells. Mutagenesis 2013, 28, 381–391. [Google Scholar] [CrossRef]
- Kim, S.; Oancea, S.C. Electronic cigarettes may not be a “safer alternative” of conventional cigarettes during pregnancy: Evidence from the nationally representative PRAMS data. BMC Pregnancy Childbirth 2020, 20, 557. [Google Scholar] [CrossRef]
- Collins, L.; Glasser, A.M.; Abudayyeh, H.; Pearson, J.L.; Villanti, A.C. E-Cigarette Marketing and Communication: How E-Cigarette Companies Market E-Cigarettes and the Public Engages with E-cigarette Information. Nicotine Tob. Res. 2019, 21, 14–24. [Google Scholar] [CrossRef]
- Margham, J.; McAdam, K.; Forster, M.; Liu, C.; Wright, C.; Mariner, D.; Proctor, C. Chemical Composition of Aerosol from an E-Cigarette: A Quantitative Comparison with Cigarette Smoke. Chem. Res. Toxicol. 2016, 29, 1662–1678. [Google Scholar] [CrossRef]
- Spindel, E.R.; McEvoy, C.T. The Role of Nicotine in the Effects of Maternal Smoking during Pregnancy on Lung Development and Childhood Respiratory Disease. Implications for Dangers of E-Cigarettes. Am. J. Respir. Crit. Care Med. 2016, 193, 486–494. [Google Scholar] [CrossRef] [Green Version]
- Franzen, K.F.; Willig, J.; Cayo Talavera, S.; Meusel, M.; Sayk, F.; Reppel, M.; Dalhoff, K.; Mortensen, K.; Droemann, D. E-cigarettes and cigarettes worsen peripheral and central hemodynamics as well as arterial stiffness: A randomized, double-blinded pilot study. Vasc. Med. 2018, 23, 419–425. [Google Scholar] [CrossRef]
- Fetterman, J.L.; Hamburg, N.M. A cautionary note on electronic cigarettes and vascular health. Vasc. Med. 2018, 23, 426–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thirión-Romero, I.; Pérez-Padilla, R.; Zabert, G.; Barrientos-Gutiérrez, I. Respiratory impact of electronic cigarettes and “low-risk” tobacco. Rev. Investig. Clin. 2019, 71, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Ralho, A.; Coelho, A.; Ribeiro, M.; Paula, A.; Amaro, I.; Sousa, J.; Marto, C.; Ferreira, M.; Carrilho, E. Effects of Electronic Cigarettes on Oral Cavity: A Systematic Review. J. Evid. Based Dent. Pract. 2019, 19, 101318. [Google Scholar] [CrossRef] [PubMed]
- Collins, Y.; Chouchani, E.T.; James, A.M.; Menger, K.E.; Cochemé, H.M.; Murphy, M.P. Mitochondrial redox signalling at a glance. J. Cell Sci. 2012, 125, 801–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, J.N.; Leung, M.C.; Rooney, J.P.; Sendoel, A.; Hengartner, M.O.; Kisby, G.E.; Bess, A.S. Mitochondria as a target of environmental toxicants. Toxicol. Sci. 2013, 134, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Jia, L.; Liu, Z.; Sun, L.; Miller, S.S.; Ames, B.N.; Cotman, C.W.; Liu, J. Acrolein, a toxicant in cigarette smoke, causes oxidative damage and mitochondrial dysfunction in RPE cells: Protection by (R)-alpha-lipoic acid. Investig. Ophthalmol. Vis. Sci. 2007, 48, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Stridh, H.; Orrenius, S.; Hampton, M.B. Caspase Involvement in the Induction of Apoptosis by the Environmental Toxicants Tributyltin and Triphenyltin. Toxicol. Appl. Pharmacol. 1999, 156, 141–146. [Google Scholar] [CrossRef]
- Tsubouchi, K.; Araya, J.; Kuwano, K. PINK1-PARK2-mediated mitophagy in COPD and IPF pathogeneses. Inflamm. Regen. 2018, 38, 18. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Wei, P.Z.; Szeto, C.C. Mitochondrial dysfunction in diabetic kidney disease. Clin. Chim. Acta 2019, 496, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics—The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef] [PubMed]
- West, A.P. Mitochondrial dysfunction as a trigger of innate immune responses and inflammation. Toxicology 2017, 391, 54–63. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Craigen, W.J.; Scaglia, F. Mitochondrial DNA maintenance defects. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Duanmu, X.; Zeng, L.; Liu, B.; Song, Z. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells 2019, 8, 379. [Google Scholar] [CrossRef] [Green Version]
- Zanna, C.; Ghelli, A.; Porcelli, A.M.; Karbowski, M.; Youle, R.J.; Schimpf, S.; Wissinger, B.; Pinti, M.; Cossarizza, A.; Vidoni, S.; et al. OPA1 mutations associated with dominant optic atrophy impair oxidative phosphorylation and mitochondrial fusion. Brain 2007, 131, 352–367. [Google Scholar] [CrossRef] [Green Version]
- Sweeney, M.G.; Bundey, S.; Brockington, M.; Poulton, K.R.; Winer, J.B.; Harding, A.E. Mitochondrial myopathy associated with sudden death in young adults and a novel mutation in the mitochondrial DNA leucine transfer RNA(UUR) gene. QJM 1993, 86, 709–713. [Google Scholar]
- Gerbitz, K.D.; Gempel, K.; Brdiczka, D. Mitochondria and diabetes. Genetic, biochemical, and clinical implications of the cellular energy circuit. Diabetes 1996, 45, 113–126. [Google Scholar] [CrossRef]
- Venditti, P.; Di Stefano, L.; Di Meo, S. Mitochondrial metabolism of reactive oxygen species. Mitochondrion 2013, 13, 71–82. [Google Scholar] [CrossRef]
- Mishra, P.; Chan, D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 634–646. [Google Scholar] [CrossRef] [Green Version]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Annesley, S.J.; Fisher, P.R. Mitochondria in Health and Disease. Cells 2019, 8, 680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archer, S.L. Mitochondrial fission and fusion in human diseases. N. Engl. J. Med. 2014, 370, 1074. [Google Scholar] [CrossRef] [PubMed]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar] [CrossRef] [Green Version]
- Detmer, S.A.; Chan, D.C. Functions and dysfunctions of mitochondrial dynamics. Nat. Rev. Mol. Cell Biol. 2007, 8, 870–879. [Google Scholar] [CrossRef]
- Yoon, Y.; Galloway, C.A.; Jhun, B.S.; Yu, T. Mitochondrial dynamics in diabetes. Antioxid. Redox Signal. 2011, 14, 439–457. [Google Scholar] [CrossRef]
- Olichon, A.; Guillou, E.; Delettre, C.; Landes, T.; Arnauné-Pelloquin, L.; Emorine, L.J.; Mils, V.; Daloyau, M.; Hamel, C.; Amati-Bonneau, P.; et al. Mitochondrial dynamics and disease, OPA1. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2006, 1763, 500–509. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Chan, D.C. Mitochondrial dynamics--fusion, fission, movement, and mitophagy—In neurodegenerative diseases. Hum. Mol. Genet. 2009, 18, R169–R176. [Google Scholar] [CrossRef]
- Wakabayashi, J.; Zhang, Z.; Wakabayashi, N.; Tamura, Y.; Fukaya, M.; Kensler, T.W.; Iijima, M.; Sesaki, H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009, 186, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Detmer, S.A.; Ewald, A.J.; Griffin, E.E.; Fraser, S.E.; Chan, D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003, 160, 189–200. [Google Scholar] [CrossRef]
- Shen, Q.; Yamano, K.; Head, B.P.; Kawajiri, S.; Cheung, J.T.; Wang, C.; Cho, J.H.; Hattori, N.; Youle, R.J.; van der Bliek, A.M. Mutations in Fis1 disrupt orderly disposal of defective mitochondria. Mol. Biol. Cell 2014, 25, 145–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Vermulst, M.; Wang, Y.E.; Chomyn, A.; Prolla, T.A.; McCaffery, J.M.; Chan, D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010, 141, 280–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, C.S.; Osellame, L.D.; Laine, D.; Koutsopoulos, O.S.; Frazier, A.E.; Ryan, M.T. MiD49 and MiD51, new components of the mitochondrial fission machinery. EMBO Rep. 2011, 12, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Liu, L.; Wu, S.; Xing, D. Drp1, Mff, Fis1, and MiD51 are coordinated to mediate mitochondrial fission during UV irradiation-induced apoptosis. FASEB J. 2016, 30, 466–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eura, Y.; Ishihara, N.; Oka, T.; Mihara, K. Identification of a novel protein that regulates mitochondrial fusion by modulating mitofusin (Mfn) protein function. J. Cell Sci. 2006, 119, 4913–4925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravamudan, B.; Kiel, A.; Freeman, M.; Delmotte, P.; Thompson, M.; Vassallo, R.; Sieck, G.C.; Pabelick, C.M.; Prakash, Y.S. Cigarette smoke-induced mitochondrial fragmentation and dysfunction in human airway smooth muscle. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 306, L840–L854. [Google Scholar] [CrossRef] [Green Version]
- Song, Z.; Chen, H.; Fiket, M.; Alexander, C.; Chan, D.C. OPA1 processing controls mitochondrial fusion and is regulated by mRNA splicing, membrane potential, and Yme1L. J. Cell Biol. 2007, 178, 749–755. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, Q.; Li, Y.; Li, C.; Zhu, Y.; Xia, F.; Xu, S.; Li, W. PTEN-Induced Putative Kinase 1 (PINK1)/Parkin-Mediated Mitophagy Protects PC12 Cells Against Cisplatin-Induced Neurotoxicity. Med. Sci. Monit. 2019, 25, 8797–8806. [Google Scholar] [CrossRef]
- Wang, N.; Zhu, P.; Huang, R.; Wang, C.; Sun, L.; Lan, B.; He, Y.; Zhao, H.; Gao, Y. PINK1: The guard of mitochondria. Life Sci. 2020, 259, 118247. [Google Scholar] [CrossRef]
- Quinn, P.M.J.; Moreira, P.I.; Ambrósio, A.F.; Alves, C.H. PINK1/PARKIN signalling in neurodegeneration and neuroinflammation. Acta Neuropathol. Commun. 2020, 8, 189. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarou, M. Keeping the immune system in check: A role for mitophagy. Immunol. Cell Biol. 2015, 93, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Jeong, Y.Y. Mitophagy in Alzheimer’s Disease and Other Age-Related Neurodegenerative Diseases. Cells 2020, 9, 150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and Mitophagy in Cardiovascular Disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Killackey, S.A.; Philpott, D.J.; Girardin, S.E. Mitophagy pathways in health and disease. J. Cell Biol. 2020, 219, e202004029. [Google Scholar] [CrossRef]
- Teague, S.V.; Pinkerton, K.E.; Goldsmith, M.; Gebremichael, A.; Chang, S.; Jenkins, R.A.; Moneyhun, J.H. Sidestream Cigarette Smoke Generation and Exposure System for Environmental Tobacco Smoke Studies. Inhal. Toxicol. 1994, 6, 79–93. [Google Scholar] [CrossRef]
- Abouassali, O.; Chang, M.; Chidipi, B.; Martinez, J.L.; Reiser, M.; Kanithi, M.; Soni, R.; McDonald, T.V.; Herweg, B.; Saiz, J.; et al. In vitro and in vivo cardiac toxicity of flavored electronic nicotine delivery systems. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H133–H143. [Google Scholar] [CrossRef]
- Hamanaka, R.B.; Chandel, N.S. Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 2010, 35, 505–513. [Google Scholar] [CrossRef] [Green Version]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef] [Green Version]
- Pryde, K.R.; Hirst, J. Superoxide is produced by the reduced flavin in mitochondrial complex I: A single, unified mechanism that applies during both forward and reverse electron transfer. J. Biol. Chem. 2011, 286, 18056–18065. [Google Scholar] [CrossRef] [Green Version]
- Kudin, A.P.; Bimpong-Buta, N.Y.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Y.; Liu, Y.; Jiang, Y.; Mei, S.; Liu, Y.; Feng, J.; Guo, L.; Du, J.; Graves, D.T.; Liu, Y. Cigarette Smoke Exposure Inhibits Osteoclast Apoptosis via the mtROS Pathway. J. Dent. Res. 2021, 100, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Sundar, I.K.; Maremanda, K.P.; Rahman, I. Mitochondrial dysfunction is associated with Miro1 reduction in lung epithelial cells by cigarette smoke. Toxicol. Lett. 2019, 317, 92–101. [Google Scholar] [CrossRef]
- Manevski, M.; Muthumalage, T.; Devadoss, D.; Sundar, I.K.; Wang, Q.; Singh, K.P.; Unwalla, H.J.; Chand, H.S.; Rahman, I. Cellular stress responses and dysfunctional Mitochondrial-cellular senescence, and therapeutics in chronic respiratory diseases. Redox Biol. 2020, 33, 101443. [Google Scholar] [CrossRef]
- Wang, Z.; White, A.; Wang, X.; Ko, J.; Choudhary, G.; Lange, T.; Rounds, S.; Lu, Q. Mitochondrial Fission Mediated Cigarette Smoke-induced Pulmonary Endothelial Injury. Am. J. Respir. Cell Mol. Biol. 2020, 63, 637–651. [Google Scholar] [CrossRef]
- Pozuelos, G.L.; Kagda, M.S.; Schick, S.; Girke, T.; Volz, D.C.; Talbot, P. Experimental Acute Exposure to Thirdhand Smoke and Changes in the Human Nasal Epithelial Transcriptome: A Randomized Clinical Trial. JAMA Netw. Open 2019, 2, e196362. [Google Scholar] [CrossRef] [Green Version]
- Tan, D.; Goerlitz, D.S.; Dumitrescu, R.G.; Han, D.; Seillier-Moiseiwitsch, F.; Spernak, S.M.; Orden, R.A.; Chen, J.; Goldman, R.; Shields, P.G. Associations between cigarette smoking and mitochondrial DNA abnormalities in buccal cells. Carcinogenesis 2008, 29, 1170–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baglole, C.J.; Bushinsky, S.M.; Garcia, T.M.; Kode, A.; Rahman, I.; Sime, P.J.; Phipps, R.P. Differential induction of apoptosis by cigarette smoke extract in primary human lung fibroblast strains: Implications for emphysema. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L19–L29. [Google Scholar] [CrossRef] [Green Version]
- van der Toorn, M.; Rezayat, D.; Kauffman, H.F.; Bakker, S.J.; Gans, R.O.; Koëter, G.H.; Choi, A.M.; van Oosterhout, A.J.; Slebos, D.J. Lipid-soluble components in cigarette smoke induce mitochondrial production of reactive oxygen species in lung epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2009, 297, L109–L114. [Google Scholar] [CrossRef] [Green Version]
- Ballweg, K.; Mutze, K.; Königshoff, M.; Eickelberg, O.; Meiners, S. Cigarette smoke extract affects mitochondrial function in alveolar epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L895–L907. [Google Scholar] [CrossRef]
- Hara, H.; Araya, J.; Ito, S.; Kobayashi, K.; Takasaka, N.; Yoshii, Y.; Wakui, H.; Kojima, J.; Shimizu, K.; Numata, T.; et al. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2013, 305, L737–L746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, E.J.; Park, Y.J.; Lee, S.J.; Lee, K.; Yoon, C. Whole cigarette smoke condensates induce ferroptosis in human bronchial epithelial cells. Toxicol. Lett. 2019, 303, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, R.F.; Zarrintan, S.; Brandenburg, S.M.; Kol, A.; de Bruin, H.G.; Jafari, S.; Dijk, F.; Kalicharan, D.; Kelders, M.; Gosker, H.R.; et al. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res. 2013, 14, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, M.A.; Reddy, G.; Quinn, M.J.; Millikan Bell, A. Toxicological assessment of electronic cigarette vaping: An emerging threat to force health, readiness and resilience in the U.S. Army. Drug Chem. Toxicol. 2021, 1–37. [Google Scholar] [CrossRef] [PubMed]
- Godoy, J.A.; Valdivieso, A.G.; Inestrosa, N.C. Nicotine Modulates Mitochondrial Dynamics in Hippocampal Neurons. Mol. Neurobiol. 2018, 55, 8965–8977. [Google Scholar] [CrossRef]
- Guo, L.; Li, L.; Wang, W.; Pan, Z.; Zhou, Q.; Wu, Z. Mitochondrial reactive oxygen species mediates nicotine-induced hypoxia-inducible factor-1α expression in human non-small cell lung cancer cells. Biochim. Biophys. Acta 2012, 1822, 852–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chernyavsky, A.I.; Shchepotin, I.B.; Galitovkiy, V.; Grando, S.A. Mechanisms of tumor-promoting activities of nicotine in lung cancer: Synergistic effects of cell membrane and mitochondrial nicotinic acetylcholine receptors. BMC Cancer 2015, 15, 152. [Google Scholar] [CrossRef] [Green Version]
- Bruin, J.E.; Petre, M.A.; Lehman, M.A.; Raha, S.; Gerstein, H.C.; Morrison, K.M.; Holloway, A.C. Maternal nicotine exposure increases oxidative stress in the offspring. Free Radic. Biol. Med. 2008, 44, 1919–1925. [Google Scholar] [CrossRef]
- Hirata, N.; Yamada, S.; Asanagi, M.; Sekino, Y.; Kanda, Y. Nicotine induces mitochondrial fission through mitofusin degradation in human multipotent embryonic carcinoma cells. Biochem. Biophys. Res. Commun. 2016, 470, 300–305. [Google Scholar] [CrossRef]
- Picca, A.; Mankowski, R.T.; Burman, J.L.; Donisi, L.; Kim, J.S.; Marzetti, E.; Leeuwenburgh, C. Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 2018, 15, 543–554. [Google Scholar] [CrossRef]
- Zhu, S.H.; Sun, J.Y.; Bonnevie, E.; Cummins, S.E.; Gamst, A.; Yin, L.; Lee, M. Four hundred and sixty brands of e-cigarettes and counting: Implications for product regulation. Tob. Control 2014, 23 (Suppl. 3), iii3–iii9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, N.J.; Lawton, R.I.; Hershberger, P.A.; Goniewicz, M.L. Flavourings significantly affect inhalation toxicity of aerosol generated from electronic nicotine delivery systems (ENDS). Tob. Control 2016, 25, ii81–ii87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, S.Y.; Akers, A.T.; Henderson, B.J. Flavors Enhance Nicotine Vapor Self-administration in Male Mice. Nicotine Tob. Res. 2021, 23, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Avelar, A.J.; Akers, A.T.; Baumgard, Z.J.; Cooper, S.Y.; Casinelli, G.P.; Henderson, B.J. Why flavored vape products may be attractive: Green apple tobacco flavor elicits reward-related behavior, upregulates nAChRs on VTA dopamine neurons, and alters midbrain dopamine and GABA neuron function. Neuropharmacology 2019, 158, 107729. [Google Scholar] [CrossRef] [PubMed]
- Basma, H.; Tatineni, S.; Dhar, K.; Qiu, F.; Rennard, S.; Lowes, B.D. Electronic cigarette extract induced toxic effect in iPS-derived cardiomyocytes. BMC Cardiovasc. Disord. 2020, 20, 357. [Google Scholar] [CrossRef]
- Jabba, S.V.; Diaz, A.N.; Erythropel, H.C.; Zimmerman, J.B.; Jordt, S.-E. Chemical Adducts of Reactive Flavor Aldehydes Formed in E-Cigarette Liquids Are Cytotoxic and Inhibit Mitochondrial Function in Respiratory Epithelial Cells. Nicotine Tob. Res. 2020, 22, S25–S34. [Google Scholar] [CrossRef]
- Williams, M.; Ventura, J.; Loza, A.; Wang, Y.; Talbot, P. Chemical Elements in Electronic Cigarette Solvents and Aerosols Inhibit Mitochondrial Reductases and Induce Oxidative Stress. Nicotine Tob. Res. 2020, 22, S14–S24. [Google Scholar] [CrossRef]
- Zhao, D.; Aravindakshan, A.; Hilpert, M.; Olmedo, P.; Rule, A.M.; Navas-Acien, A.; Aherrera, A. Metal/Metalloid Levels in Electronic Cigarette Liquids, Aerosols, and Human Biosamples: A Systematic Review. Environ. Health Perspect. 2020, 128, 36001. [Google Scholar] [CrossRef] [Green Version]
- Northrup, T.F.; Klawans, M.R.; Villarreal, Y.R.; Abramovici, A.; Suter, M.A.; Mastrobattista, J.M.; Moreno, C.A.; Aagaard, K.M.; Stotts, A.L. Family Physicians’ Perceived Prevalence, Safety, and Screening for Cigarettes, Marijuana, and Electronic-Nicotine Delivery Systems (ENDS) Use during Pregnancy. J. Am. Board Fam. Med. 2017, 30, 743–757. [Google Scholar] [CrossRef] [Green Version]
- Calder, R.; Gant, E.; Bauld, L.; McNeill, A.; Robson, D.; Brose, L.S. Vaping in Pregnancy: A Systematic Review. Nicotine Tob. Res. 2021, 23, 1451–1458. [Google Scholar] [CrossRef]
- Li, G.; Chan, Y.L.; Wang, B.; Saad, S.; George, J.; Oliver, B.G.; Chen, H. E-cigarettes damage the liver and alter nutrient metabolism in pregnant mice and their offspring. Ann. N. Y. Acad. Sci. 2020, 1475, 64–77. [Google Scholar] [CrossRef] [PubMed]
- Lei, W.; Lerner, C.; Sundar, I.K.; Rahman, I. Myofibroblast differentiation and its functional properties are inhibited by nicotine and e-cigarette via mitochondrial OXPHOS complex III. Sci. Rep. 2017, 7, 43213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sailer, S.; Sebastiani, G.; Andreu-Férnández, V.; García-Algar, O. Impact of Nicotine Replacement and Electronic Nicotine Delivery Systems on Fetal Brain Development. Int. J. Environ. Res. Public Health 2019, 16, 5113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahedi, A.; Phandthong, R.; Chaili, A.; Leung, S.; Omaiye, E.; Talbot, P. Mitochondrial Stress Response in Neural Stem Cells Exposed to Electronic Cigarettes. iScience 2019, 16, 250–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahl, V.; Lin, S.; Xu, N.; Davis, B.; Wang, Y.H.; Talbot, P. Comparison of electronic cigarette refill fluid cytotoxicity using embryonic and adult models. Reprod. Toxicol. 2012, 34, 529–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behar, R.Z.; Davis, B.; Wang, Y.; Bahl, V.; Lin, S.; Talbot, P. Identification of toxicants in cinnamon-flavored electronic cigarette refill fluids. Toxicol. In Vitro 2014, 28, 198–208. [Google Scholar] [CrossRef] [Green Version]
- Bahl, V.; Johnson, K.; Phandthong, R.; Zahedi, A.; Schick, S.F.; Talbot, P. From the Cover: Thirdhand Cigarette Smoke Causes Stress-Induced Mitochondrial Hyperfusion and Alters the Transcriptional Profile of Stem Cells. Toxicol. Sci. 2016, 153, 55–69. [Google Scholar] [CrossRef] [Green Version]
- Adhami, N.; Chen, Y.; Martins-Green, M. Biomarkers of disease can be detected in mice as early as 4 weeks after initiation of exposure to third-hand smoke levels equivalent to those found in homes of smokers. Clin. Sci. 2017, 131, 2409–2426. [Google Scholar] [CrossRef] [Green Version]
- Tondera, D.; Grandemange, S.; Jourdain, A.; Karbowski, M.; Mattenberger, Y.; Herzig, S.; Da Cruz, S.; Clerc, P.; Raschke, I.; Merkwirth, C.; et al. SLP-2 is required for stress-induced mitochondrial hyperfusion. EMBO J. 2009, 28, 1589–1600. [Google Scholar] [CrossRef] [Green Version]
- West, R. Tobacco smoking: Health impact, prevalence, correlates and interventions. Psychol. Health 2017, 32, 1018–1036. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Michaeloudes, C.; Zhang, Y.; Wiegman, C.H.; Adcock, I.M.; Lian, Q.; Mak, J.C.W.; Bhavsar, P.K.; Chung, K.F. Mesenchymal stem cells alleviate oxidative stress-induced mitochondrial dysfunction in the airways. J. Allergy Clin. Immunol. 2018, 141, 1634–1645.e1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kanithi, M.; Junapudi, S.; Shah, S.I.; Matta Reddy, A.; Ullah, G.; Chidipi, B. Alterations of Mitochondrial Network by Cigarette Smoking and E-Cigarette Vaping. Cells 2022, 11, 1688. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11101688
Kanithi M, Junapudi S, Shah SI, Matta Reddy A, Ullah G, Chidipi B. Alterations of Mitochondrial Network by Cigarette Smoking and E-Cigarette Vaping. Cells. 2022; 11(10):1688. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11101688
Chicago/Turabian StyleKanithi, Manasa, Sunil Junapudi, Syed Islamuddin Shah, Alavala Matta Reddy, Ghanim Ullah, and Bojjibabu Chidipi. 2022. "Alterations of Mitochondrial Network by Cigarette Smoking and E-Cigarette Vaping" Cells 11, no. 10: 1688. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11101688