
Mediating EGFR-TKI Resistance by VEGF/VEGFR Autocrine Pathway in Non-Small Cell Lung Cancer

 ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. The Tyrosine Kinase Inhibitors and Growth Factor Ligands
2.2. Antibodies
2.3. Cell Lines
2.4. Quantitative Real-Time PCR
2.5. Immunoblotting
2.6. Immunofluorescence
2.7. Flow Cytometry
2.8. MTT Cell Viability Assay
2.9. ELISA
2.10. Immunohistochemistry
2.11. Statistical Analysis
3. Results
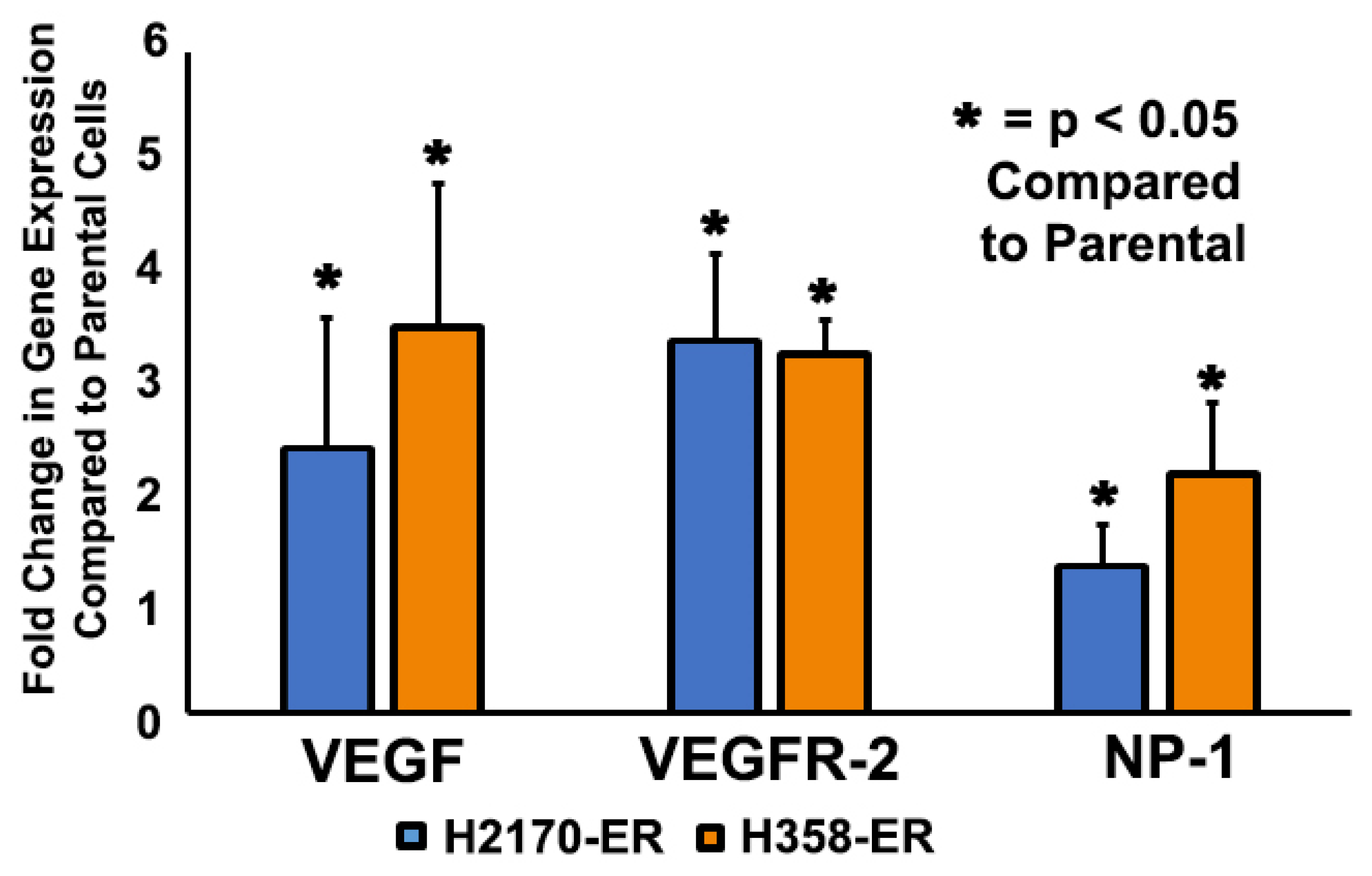
3.1. Increased Gene Expression of VEGF, VEGFR-2, and NP-1 in Erlotinib-Resistant NSCLC Cells
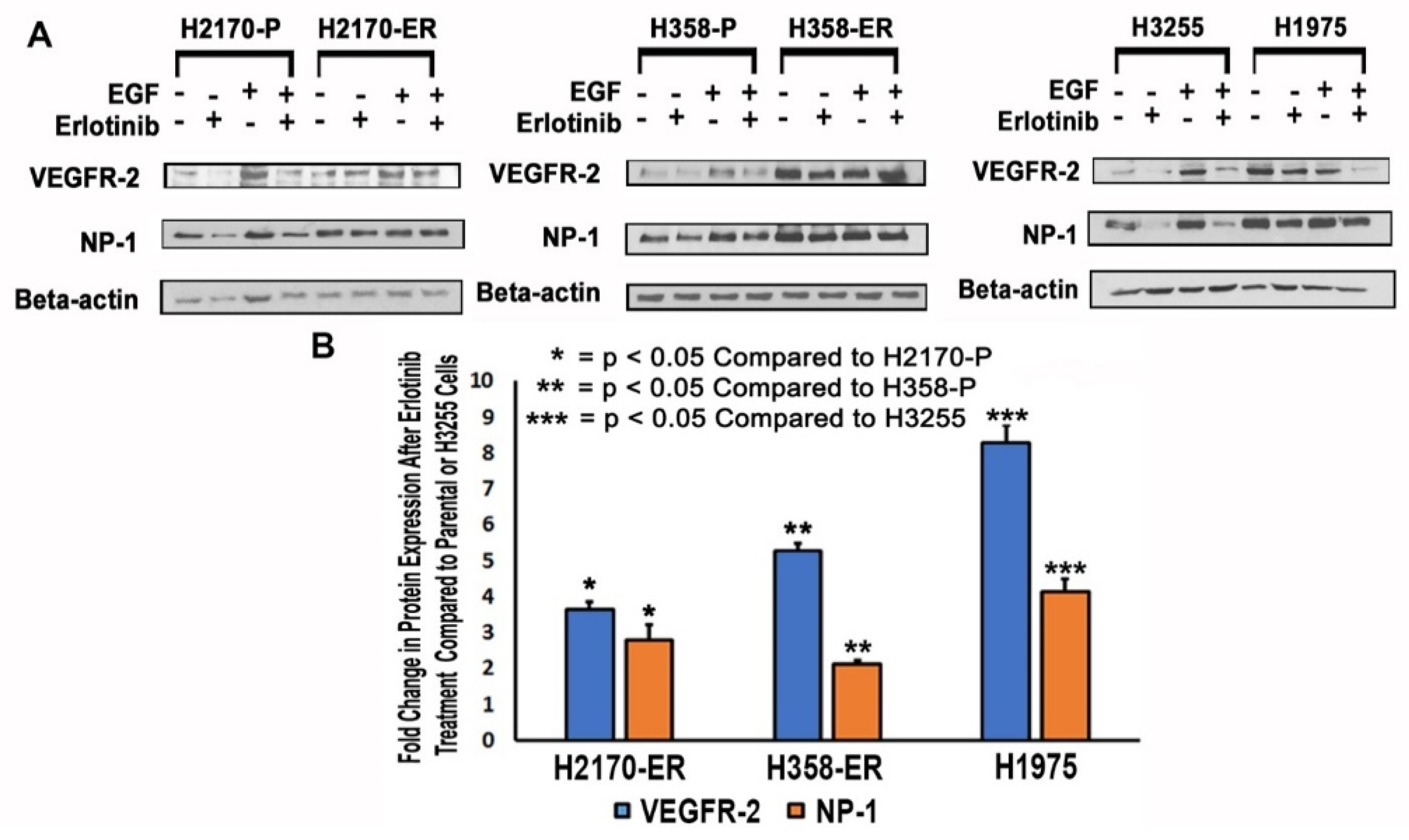
3.2. Modulation of Protein Expression of VEGFR-2 and NP-1 in NSCLC Cell Lines
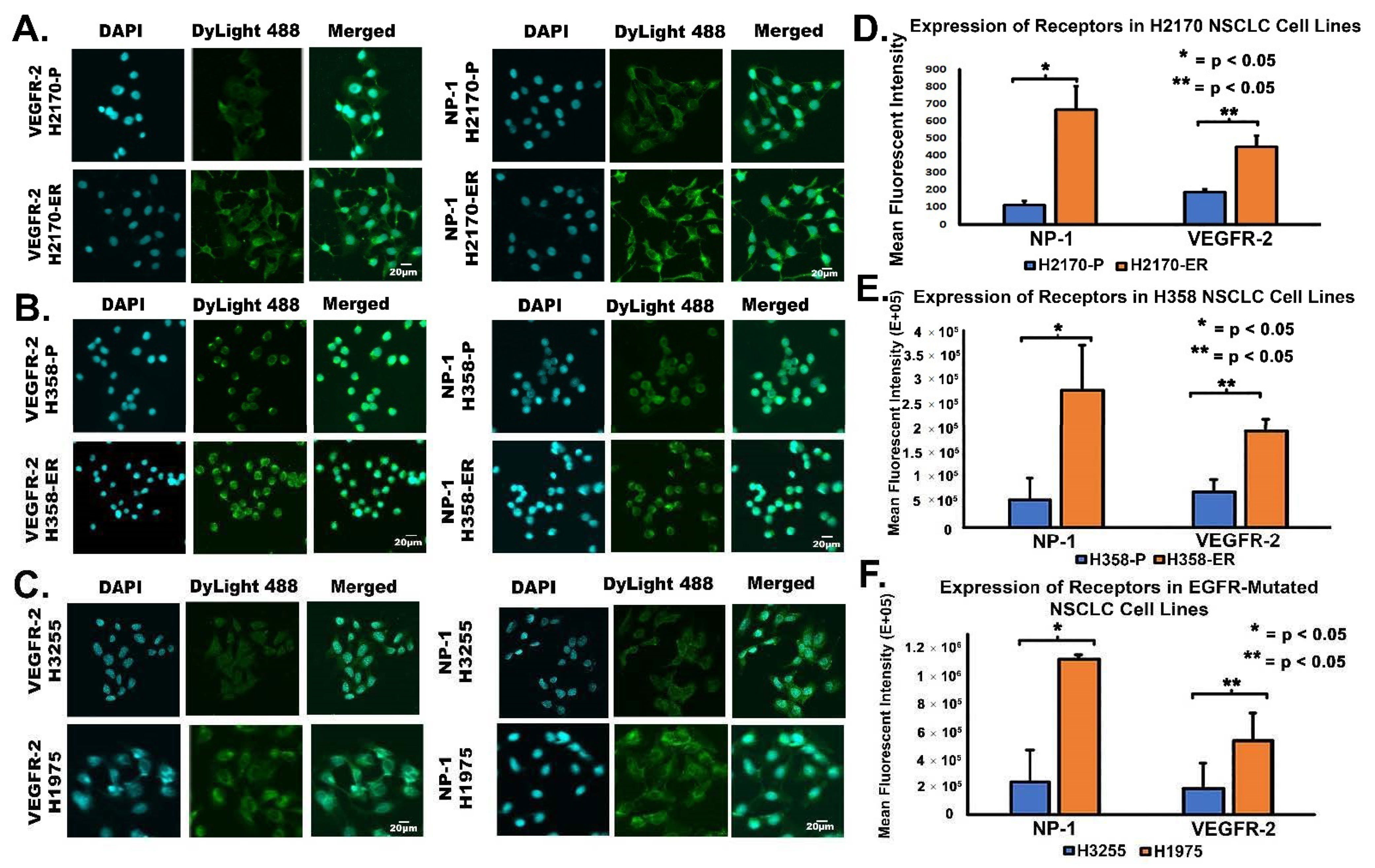
3.3. Increased Cell Surface Expression of VEGFR-2 and NP-1 Receptors in Erlotinib-Resistant NSCLC Cell Lines, as Demonstrated by FACS Analysis
3.4. Increased Cell Surface Expression of VEGFR-2 and NP-1 Receptors in Erlotinib-Resistant NSCLC Cell Lines, as Demonstrated by Immunofluorescence Studies
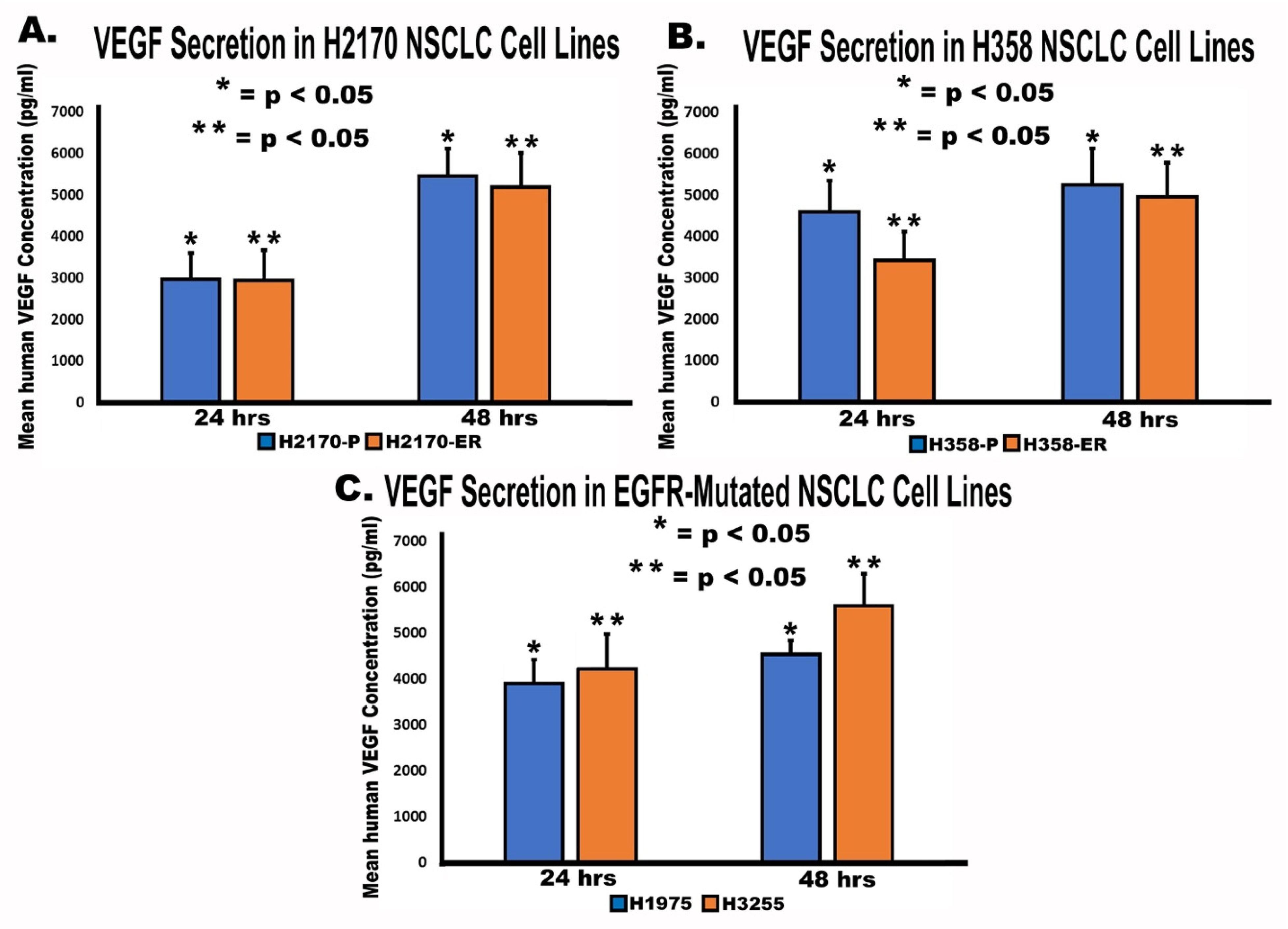
3.5. Secretion of VEGF by NSCLC Cell Lines
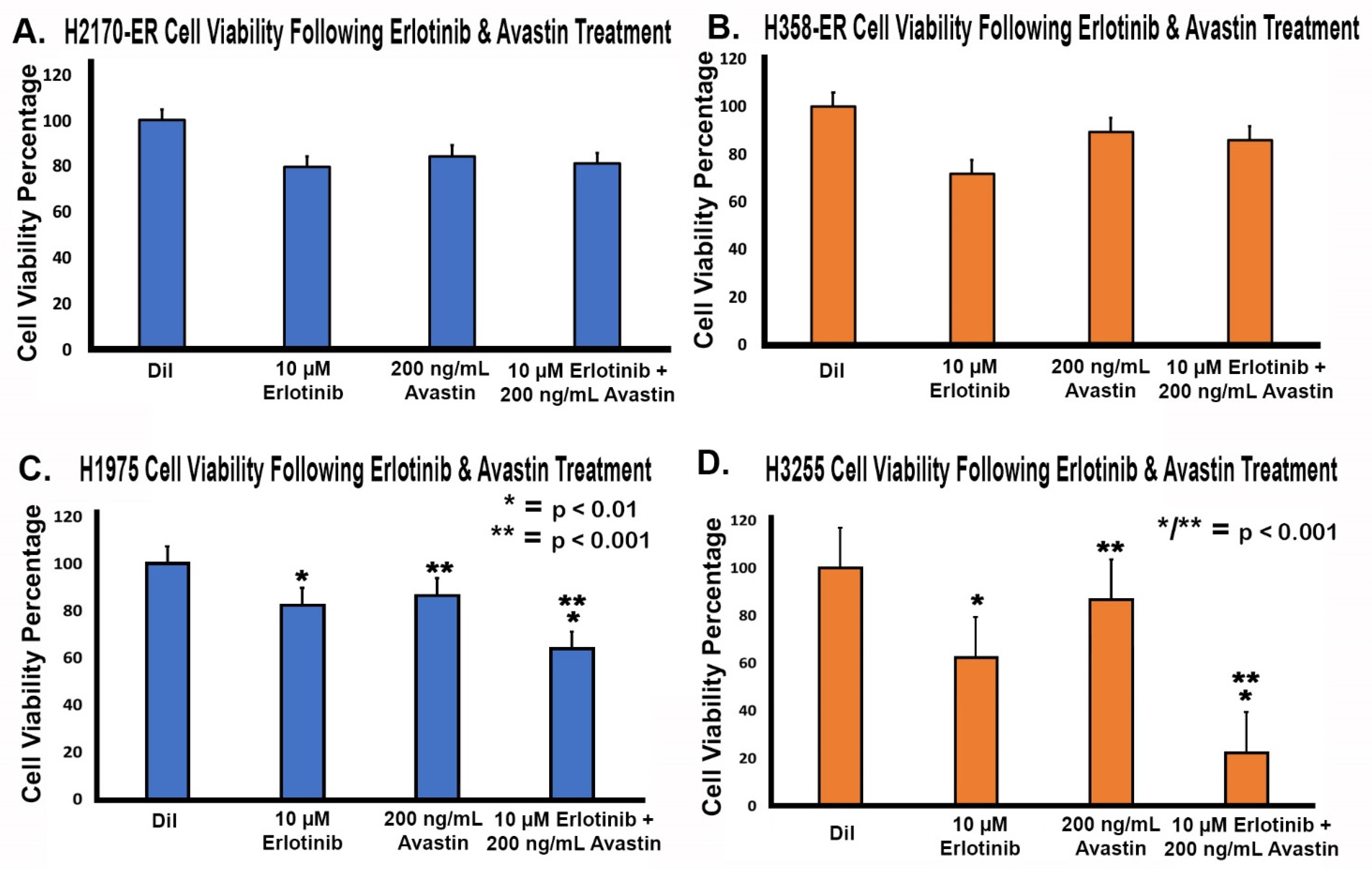
3.6. Effect of Anti-VEGF Treatments on the Proliferation of Erlotinib-Resistant and EGFR-Mutated NSCLC Cell Lines
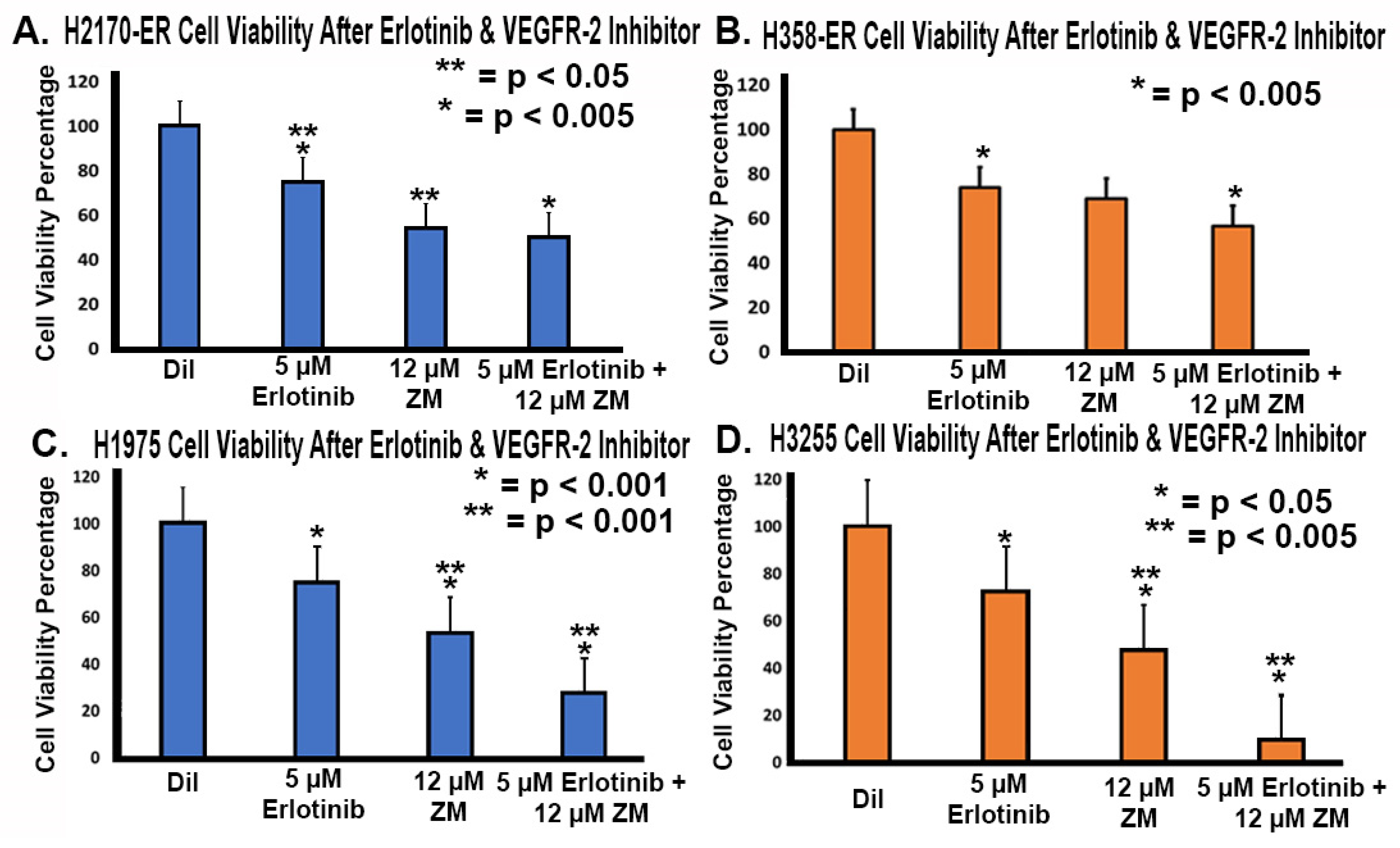
3.7. Effect of Anti-VEGFR-2 Treatments in Erlotinib-Resistant and EGFR-Mutated NSCLC Cell Lines
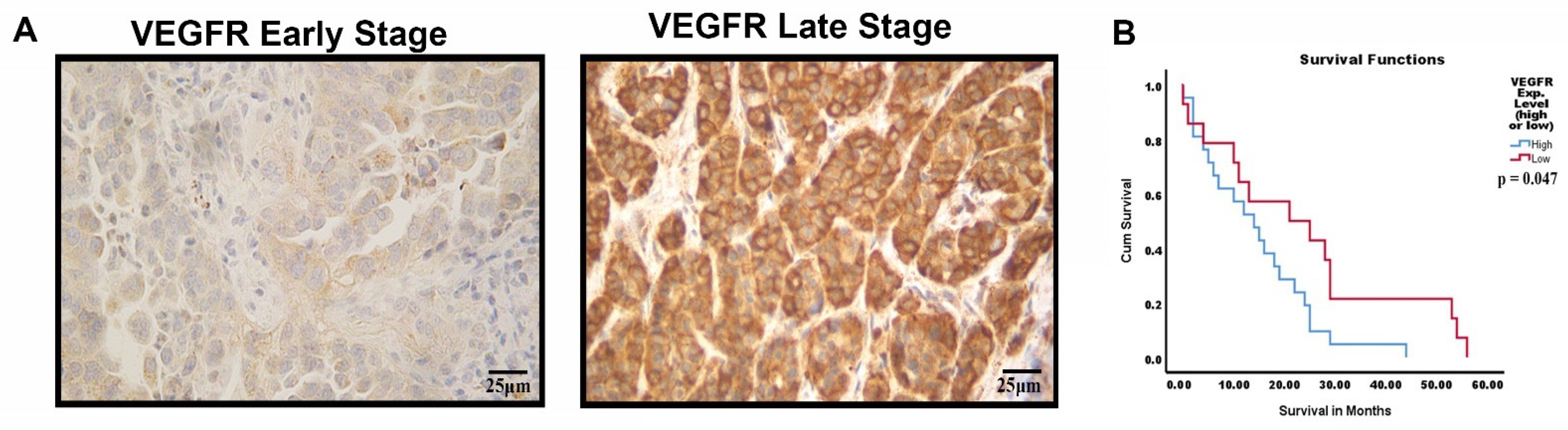
3.8. VEGFR-2 Expression in Late-Stage NSCLC Tumor Samples with Kaplan–Meier Analysis
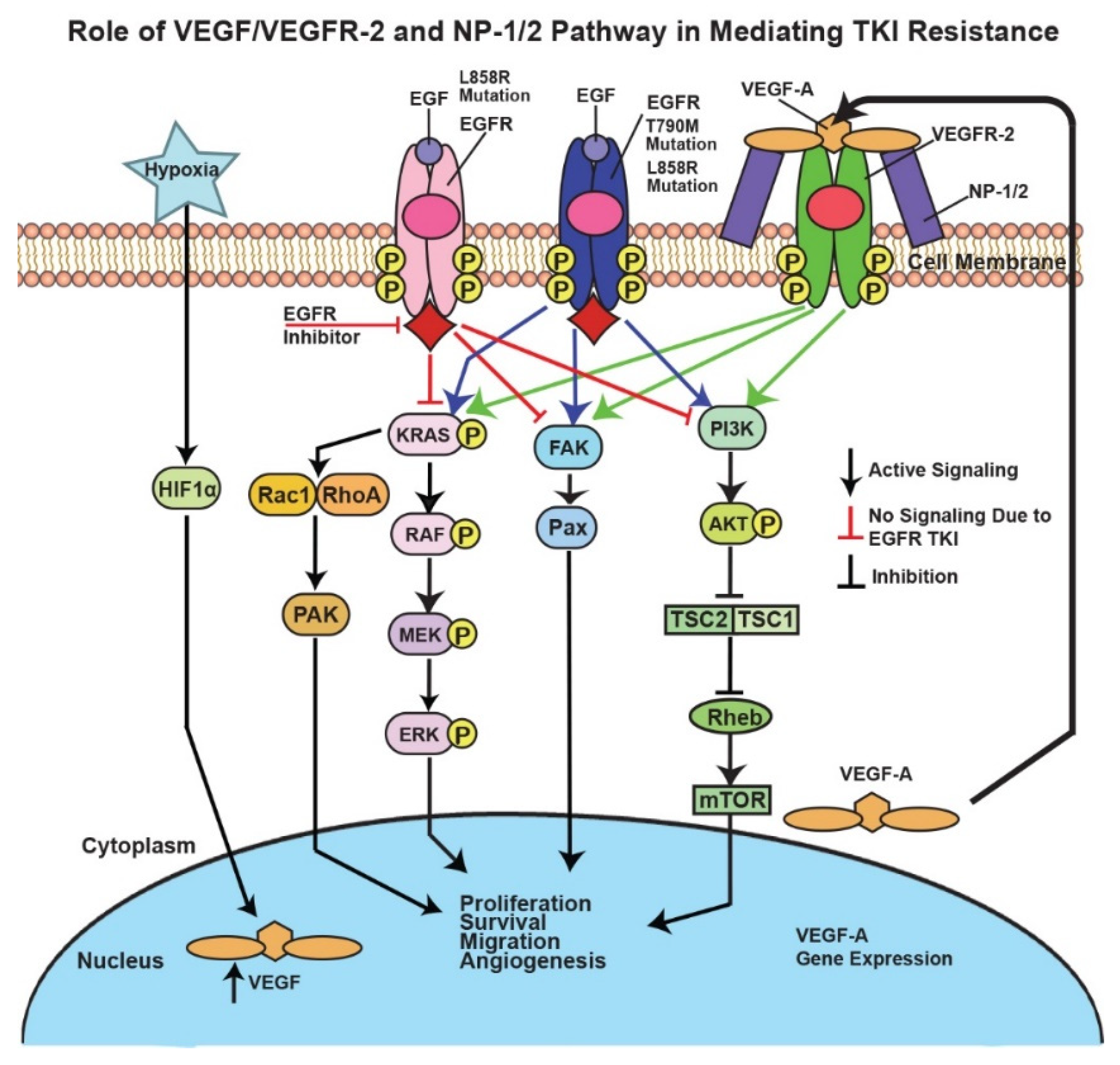
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA A Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Corner, J.; Hopkinson, J.; Fitzsimmons, D.; Barclay, S.; Muers, M. Is late diagnosis of lung cancer inevitable? Interview study of patients’ recollections of symptoms before diagnosis. Thorax 2005, 60, 314–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burki, T.K. Late detection of lung cancer. Lancet Oncol. 2014, 15, e590. [Google Scholar] [CrossRef]
- Schrank, Z.; Chhabra, G.; Lin, L.; Iderzorig, T.; Osude, C.; Khan, N.; Kuckovic, A.; Singh, S.; Miller, R.J.; Puri, N. Current molecular-targeted therapies in nsclc and their mechanism of resistance. Cancers 2018, 10, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, N.; Salgia, R. Synergism of egfr and c-met pathways, cross-talk and inhibition, in non-small cell lung cancer. J. Carcinog. 2008, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Domvri, K.; Zarogoulidis, P.; Darwiche, K.; Browning, R.F.; Li, Q.; Turner, J.F.; Kioumis, I.; Spyratos, D.; Porpodis, K.; Papaiwannou, A.; et al. Molecular targeted drugs and biomarkers in nsclc, the evolving role of individualized therapy. J. Cancer 2013, 4, 736–754. [Google Scholar] [CrossRef] [Green Version]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (egfr) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar]
- da Cunha Santos, G.; Shepherd, F.A.; Tsao, M.S. Egfr mutations and lung cancer. Annu. Rev. Pathol. 2011, 6, 49–69. [Google Scholar] [CrossRef] [Green Version]
- Regad, T. Targeting rtk signaling pathways in cancer. Cancers 2015, 7, 1758–1784. [Google Scholar] [CrossRef]
- Sierra, J.R.; Cepero, V.; Giordano, S. Molecular mechanisms of acquired resistance to tyrosine kinase targeted therapy. Mol. Cancer 2010, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.A.; Sima, C.S.; Huang, J.; Solomon, S.B.; Rimner, A.; Paik, P.; Pietanza, M.C.; Azzoli, C.G.; Rizvi, N.A.; Krug, L.M.; et al. Local therapy with continued egfr tyrosine kinase inhibitor therapy as a treatment strategy in egfr-mutant advanced lung cancers that have developed acquired resistance to egfr tyrosine kinase inhibitors. J. Thorac. Oncol. 2013, 8, 346–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsuka, K.; Hata, A.; Takeshita, J.; Okuda, C.; Kaji, R.; Masago, K.; Fujita, S.; Katakami, N. Egfr-tki rechallenge with bevacizumab in egfr-mutant non-small cell lung cancer. Cancer Chemother. Pharmacol. 2015, 76, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Morgillo, F.; Della Corte, C.M.; Fasano, M.; Ciardiello, F. Mechanisms of resistance to egfr-targeted drugs: Lung cancer. ESMO Open 2016, 1, e000060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdullah, S.E.; Perez-Soler, R. Mechanisms of resistance to vascular endothelial growth factor blockade. Cancer 2012, 118, 3455–3467. [Google Scholar] [CrossRef]
- Tabernero, J. The role of vegf and egfr inhibition: Implications for combining anti-vegf and anti-egfr agents. Mol. Cancer Res. MCR 2007, 5, 203–220. [Google Scholar] [CrossRef] [Green Version]
- Pennell, N.A.; Lynch, T.J., Jr. Combined inhibition of the vegfr and egfr signaling pathways in the treatment of nsclc. Oncologist 2009, 14, 399–411. [Google Scholar] [CrossRef]
- Gille, H.; Kowalski, J.; Li, B.; LeCouter, J.; Moffat, B.; Zioncheck, T.F.; Pelletier, N.; Ferrara, N. Analysis of biological effects and signaling properties of flt-1 (vegfr-1) and kdr (vegfr-2). A reassessment using novel receptor-specific vascular endothelial growth factor mutants. J. Biol. Chem. 2001, 276, 3222–3230. [Google Scholar] [CrossRef] [Green Version]
- Byrne, A.M.; Bouchier-Hayes, D.J.; Harmey, J.H. Angiogenic and cell survival functions of vascular endothelial growth factor (vegf). J. Cell. Mol. Med. 2005, 9, 777–794. [Google Scholar] [CrossRef]
- Alvarez-Aznar, A.; Muhl, L.; Gaengel, K. Vegf receptor tyrosine kinases: Key regulators of vascular function. Curr. Top. Dev. Biol. 2017, 123, 433–482. [Google Scholar]
- Patel, M.; Eckburg, A.; Gantiwala, S.; Hart, Z.; Dein, J.; Lam, K.; Puri, N. Resistance to molecularly targeted therapies in melanoma. Cancers 2021, 13, 1115. [Google Scholar] [CrossRef]
- Bartoli, M.; Gu, X.; Tsai, N.T.; Venema, R.C.; Brooks, S.E.; Marrero, M.B.; Caldwell, R.B. Vascular endothelial growth factor activates stat proteins in aortic endothelial cells. J. Biol. Chem. 2000, 275, 33189–33192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, T.H.; Seng, S.; Sekine, M.; Hinton, C.; Fu, Y.; Avraham, H.K.; Avraham, S. Vascular endothelial growth factor mediates intracrine survival in human breast carcinoma cells through internally expressed vegfr1/flt1. PLoS Med. 2007, 4, e186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, G.G.; Yoon, H.H.; Zerkowski, M.P.; Ghosh, S.; Thomas, L.; Harigopal, M.; Charette, L.A.; Salem, R.R.; Camp, R.L.; Rimm, D.L.; et al. Vascular endothelial growth factor, flt-1, and flk-1 analysis in a pancreatic cancer tissue microarray. Cancer 2006, 106, 1677–1684. [Google Scholar] [CrossRef]
- Silva, S.R.; Bowen, K.A.; Rychahou, P.G.; Jackson, L.N.; Weiss, H.L.; Lee, E.Y.; Townsend, C.M., Jr.; Evers, B.M. Vegfr-2 expression in carcinoid cancer cells and its role in tumor growth and metastasis. Int. J. Cancer 2011, 128, 1045–1056. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.; Heukamp, L.C.; Siobal, M.; Schottle, J.; Wieczorek, C.; Peifer, M.; Frasca, D.; Koker, M.; Konig, K.; Meder, L.; et al. Tumor vegf:Vegfr2 autocrine feed-forward loop triggers angiogenesis in lung cancer. J. Clin. Investig. 2013, 123, 1732–1740. [Google Scholar] [CrossRef] [Green Version]
- Jackson, A.L.; Zhou, B.; Kim, W.Y. Hif, hypoxia and the role of angiogenesis in non-small cell lung cancer. Expert Opin. Ther. Targets 2010, 14, 1047–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, H.; Silverman, J.F.; Santucci, T.S.; Macherey, R.S.; d’Amato, T.A.; Tung, M.Y.; Weyant, R.J.; Landreneau, R.J. Vascular endothelial growth factor expression in stage i non-small cell lung cancer correlates with neoangiogenesis and a poor prognosis. Ann. Surg. Oncol. 2001, 8, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Piperdi, B.; Merla, A.; Perez-Soler, R. Targeting angiogenesis in squamous non-small cell lung cancer. Drugs 2014, 74, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Alevizakos, M.; Kaltsas, S.; Syrigos, K.N. The vegf pathway in lung cancer. Cancer Chemother. Pharmacol. 2013, 72, 1169–1181. [Google Scholar] [CrossRef]
- Wild, J.R.; Staton, C.A.; Chapple, K.; Corfe, B.M. Neuropilins: Expression and roles in the epithelium. Int. J. Exp. Pathol. 2012, 93, 81–103. [Google Scholar] [CrossRef]
- Chaudhary, B.; Khaled, Y.S.; Ammori, B.J.; Elkord, E. Neuropilin 1: Function and therapeutic potential in cancer. Cancer Immunol. Immunother. CII 2014, 63, 81–99. [Google Scholar] [CrossRef] [PubMed]
- Djordjevic, S.; Driscoll, P.C. Targeting vegf signalling via the neuropilin co-receptor. Drug Discov. Today 2013, 18, 447–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial vegf receptor signalling. Nat. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Hong, T.M.; Chen, Y.L.; Wu, Y.Y.; Yuan, A.; Chao, Y.C.; Chung, Y.C.; Wu, M.H.; Yang, S.C.; Pan, S.H.; Shih, J.Y.; et al. Targeting neuropilin 1 as an antitumor strategy in lung cancer. Clin. Cancer Res. 2007, 13, 4759–4768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, X.; Nilsson, M.; Goldman, J.; Reck, M.; Nakagawa, K.; Kato, T.; Ares, L.P.; Frimodt-Moller, B.; Wolff, K.; Visseren-Grul, C.; et al. Dual egfr-vegf pathway inhibition: A promising strategy for patients with egfr-mutant nsclc. J. Thorac. Oncol. 2021, 16, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Whittles, C.E.; Pocock, T.M.; Wedge, S.R.; Kendrew, J.; Hennequin, L.F.; Harper, S.J.; Bates, D.O. Zm323881, a novel inhibitor of vascular endothelial growth factor-receptor-2 tyrosine kinase activity. Microcirculation 2002, 9, 513–522. [Google Scholar] [CrossRef]
- Fong, J.T.; Jacobs, R.J.; Moravec, D.N.; Uppada, S.B.; Botting, G.M.; Nlend, M.; Puri, N. Alternative signaling pathways as potential therapeutic targets for overcoming egfr and c-met inhibitor resistance in non-small cell lung cancer. PLoS ONE 2013, 8, e78398. [Google Scholar] [CrossRef] [Green Version]
- Botting, G.M.; Rastogi, I.; Chhabra, G.; Nlend, M.; Puri, N. Mechanism of resistance and novel targets mediating resistance to egfr and c-met tyrosine kinase inhibitors in non-small cell lung cancer. PLoS ONE 2015, 10, e0136155. [Google Scholar] [CrossRef] [Green Version]
- Ma, P.C.; Jagadeeswaran, R.; Jagadeesh, S.; Tretiakova, M.S.; Nallasura, V.; Fox, E.A.; Hansen, M.; Schaefer, E.; Naoki, K.; Lader, A.; et al. Functional expression and mutations of c-met and its therapeutic inhibition with su11274 and small interfering rna in non-small cell lung cancer. Cancer Res. 2005, 65, 1479–1488. [Google Scholar] [CrossRef] [Green Version]
- Ciardiello, F.; Troiani, T.; Bianco, R.; Orditura, M.; Morgillo, F.; Martinelli, E.; Morelli, M.P.; Cascone, T.; Tortora, G. Interaction between the epidermal growth factor receptor (egfr) and the vascular endothelial growth factor (vegf) pathways: A rational approach for multi-target anticancer therapy. Ann. Oncol. 2006, 17 (Suppl. 7), vii109–vii114. [Google Scholar] [CrossRef]
- Metro, G.; Chiari, R.; Duranti, S.; Siggillino, A.; Fischer, M.J.; Giannarelli, D.; Ludovini, V.; Bennati, C.; Marcomigni, L.; Baldi, A.; et al. Impact of specific mutant kras on clinical outcome of egfr-tki-treated advanced non-small cell lung cancer patients with an egfr wild type genotype. Lung Cancer 2012, 78, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, A.; Milella, M.; Felicioni, L.; Cappuzzo, F.; Irtelli, L.; Del Grammastro, M.; Sciarrotta, M.; Malatesta, S.; Nuzzo, C.; Finocchiaro, G.; et al. Clinical implications of kras mutations in lung cancer patients treated with tyrosine kinase inhibitors: An important role for mutations in minor clones. Neoplasia 2009, 11, 1084–1092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the egfr kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, C.H.; Mengwasser, K.E.; Toms, A.V.; Woo, M.S.; Greulich, H.; Wong, K.K.; Meyerson, M.; Eck, M.J. The t790m mutation in egfr kinase causes drug resistance by increasing the affinity for atp. Proc. Natl. Acad. Sci. USA 2008, 105, 2070–2075. [Google Scholar] [CrossRef] [Green Version]
- Godin-Heymann, N.; Bryant, I.; Rivera, M.N.; Ulkus, L.; Bell, D.W.; Riese, D.J., 2nd; Settleman, J.; Haber, D.A. Oncogenic activity of epidermal growth factor receptor kinase mutant alleles is enhanced by the t790m drug resistance mutation. Cancer Res. 2007, 67, 7319–7326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, K.; Onozato, R.; Yatabe, Y.; Mitsudomi, T. Egfr t790m mutation: A double role in lung cancer cell survival? J. Thorac. Oncol. 2009, 4, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.D.; Wei, S.Q.; Wang, Q.Y. Targeting oncogenic kras in non-small cell lung cancer cells by phenformin inhibits growth and angiogenesis. Am. J. Cancer Res. 2015, 5, 3339–3349. [Google Scholar]
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [Green Version]
- Koch, S.; van Meeteren, L.A.; Morin, E.; Testini, C.; Westrom, S.; Bjorkelund, H.; Le Jan, S.; Adler, J.; Berger, P.; Claesson-Welsh, L. Nrp1 presented in trans to the endothelium arrests vegfr2 endocytosis, preventing angiogenic signaling and tumor initiation. Dev. Cell 2014, 28, 633–646. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, Q.; Ruhrberg, C. Neuropilin, you gotta let me know: Should i stay or should i go? Cell Adhes. Migr. 2010, 4, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Simons, M. An inside view: Vegf receptor trafficking and signaling. Physiology 2012, 27, 213–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basagiannis, D.; Christoforidis, S. Constitutive endocytosis of vegfr2 protects the receptor against shedding. J. Biol. Chem. 2016, 291, 16892–16903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, X.; Yu, J.; Lai, Y.; He, W.; Li, S.; Wang, L.; Ke, Z. L858r-positive lung adenocarcinoma with kras g12v, egfr t790m and egfr l858r mutations: A case report. Oncol. Lett. 2015, 10, 1293–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, C.; Qiu, L.X.; Liao, R.Y.; Du, F.B.; Ding, H.; Yang, W.C.; Li, J.; Chen, Q. Kras mutations and resistance to egfr-tkis treatment in patients with non-small cell lung cancer: A meta-analysis of 22 studies. Lung Cancer 2010, 69, 272–278. [Google Scholar] [CrossRef]
- Lian, L.; Li, X.L.; Xu, M.D.; Li, X.M.; Wu, M.Y.; Zhang, Y.; Tao, M.; Li, W.; Shen, X.M.; Zhou, C.; et al. Vegfr2 promotes tumorigenesis and metastasis in a pro-angiogenic-independent way in gastric cancer. BMC Cancer 2019, 19, 183. [Google Scholar] [CrossRef]
- Ding, M.; Liu, L.; Hu, C.; Liu, Y.; Qiao, Y.; Jiang, X. Expression of vegfr2 and nrp-1 in non-small cell lung cancer and their clinical significance. Chin. J. Cancer Res. 2014, 26, 669–677. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence | Melting Point | DNA Bases |
|---|---|---|---|
| VEGF | F: 5′ CGCAAGCTTAGGAGTACCCTGATGAG 3′ | 60.7 °C | 26 |
| R: 5′ CCGTCTAGAACATTTGTTGTGCTGT 3′ | 57.6 °C | 25 | |
| VEGFR-2 | F: 5′ GCAGGGGACAGAGGGACTTG 3′ | 60.1 °C | 20 |
| R: 5′ GAGGCCATCGCTGCACTCA 3′ | 60.4 °C | 19 | |
| NP-1 | F: 5′ ATGGAGAGGGGGCTGCCG 3′ | 63.0 °C | 18 |
| R: 5′ CTATCGCGCTGTCGGTGTA 3′ | 56.9 °C | 19 | |
| GAPDH | F: 5′ ATGACATCAAGAAGGTGGTG 3′ | 54.4 °C | 20 |
| R: 5′ CAGGAAATGAGCTTGACAAA 3′ | 55.8 °C | 20 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osude, C.; Lin, L.; Patel, M.; Eckburg, A.; Berei, J.; Kuckovic, A.; Dube, N.; Rastogi, A.; Gautam, S.; Smith, T.J.; et al. Mediating EGFR-TKI Resistance by VEGF/VEGFR Autocrine Pathway in Non-Small Cell Lung Cancer. Cells 2022, 11, 1694. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11101694
Osude C, Lin L, Patel M, Eckburg A, Berei J, Kuckovic A, Dube N, Rastogi A, Gautam S, Smith TJ, et al. Mediating EGFR-TKI Resistance by VEGF/VEGFR Autocrine Pathway in Non-Small Cell Lung Cancer. Cells. 2022; 11(10):1694. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11101694
Chicago/Turabian StyleOsude, Chike, Leo Lin, Meet Patel, Adam Eckburg, Joseph Berei, Adijan Kuckovic, Namrata Dube, Aayush Rastogi, Shruti Gautam, Thomas J. Smith, and et al. 2022. "Mediating EGFR-TKI Resistance by VEGF/VEGFR Autocrine Pathway in Non-Small Cell Lung Cancer" Cells 11, no. 10: 1694. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11101694