
Interferon-γ Preferentially Promotes Necroptosis of Lung Epithelial Cells by Upregulating MLKL


Department of Cellular and Molecular Biology, The University of Texas Health Science Center at Tyler, Tyler, TX 75708, USA
*
Author to whom correspondence should be addressed.
Cells 2022, 11(3), 563; https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030563
Submission received: 30 December 2021
/
Revised: 30 January 2022
/
Accepted: 4 February 2022
/
Published: 6 February 2022
(This article belongs to the Section Cellular Pathology)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Necroptosis, a form of programmed lytic cell death, has emerged as a driving factor in the pathogenesis of acute lung injury (ALI). As ALI is often associated with a cytokine storm, we determined whether pro-inflammatory cytokines modulate the susceptibility of lung cells to necroptosis and which mediators dominate to control necroptosis. In this study, we pretreated/primed mouse primary lung epithelial and endothelial cells with various inflammatory mediators and assessed cell type-dependent responses to different necroptosis inducers and their underlying mechanisms. We found that interferon-γ (IFNγ) as low as 1 ng/mL preferentially promoted necroptosis and accelerated the release of damage-associated molecular patterns from primary alveolar and airway epithelial cells but not lung microvascular endothelial cells. Type-I IFNα was about fifty-fold less effective than IFNγ. Conversely, TNFα or agonists of Toll-like receptor-3 (TLR3), TLR4, TLR7 and TLR9 had a minor effect. The enhanced necroptosis in IFNγ-activated lung epithelial cells was dependent on IFNγ signaling and receptor-interacting protein kinase-3. We further showed that necroptosis effector mixed lineage kinase domain-like protein (MLKL) was predominantly induced by IFNγ, contributing to the enhanced necroptosis in lung epithelial cells. Collectively, our findings indicate that IFNγ is a potent enhancer of lung epithelial cell susceptibility to necroptosis.
1. Introduction
Acute respiratory distress syndrome (ARDS) is a form of life-threatening respiratory failure that is associated with high morbidity and mortality [1,2]. ARDS can develop following various types of acute lung injuries (ALI) resulting from pulmonary (e.g., pneumonia, aspiration) or nonpulmonary (e.g., sepsis, pancreatitis, trauma) insults [2]. Globally, ARDS affects approximately 3 million patients annually and accounts for 10% of intensive care unit admissions [3]. Due to the ongoing coronavirus disease 2019 (COVID-19) pandemic, the number of patients diagnosed with ARDS/ALI increased substantially [4,5]. Clinically, ALI/ARDS manifests as hypoxemia, lung edema, decreased lung compliance and diffuse pulmonary infiltrates [1,2,4]. Despite decades of efforts on research, the pathogenesis of ALI/ARDS is not fully elucidated and treatment options are limited as a result. Supportive care with mechanical ventilation remains the mainstay of management for ALI/ARDS [2,4].
The pathological findings of ALI in the acute phase include the disruption of the alveolar epithelial and endothelial barrier, recruitment of leukocytes and protein-rich fluid in the alveolar space [1]. Inflammation is an important component to the pathogenesis of ALI/ARDS. Previous studies indicate that inhibition of the chemokine CXC–CXCR receptor axis prevents neutrophil recruitment and tissue injury in several sterile inflammation-induced ALI animal models [1]. A recent study shows that palmitoylethanolamide inhibits ALI in mice induced by endotoxin lipopolysaccharide (LPS) and may represent a potential adjuvant anti-inflammatory therapeutic for treating lung injury [6]. Besides the involvement of inflammation, necroptosis, a form of programmed lytic cell death, has emerged as a driving factor in the pathogenesis of ALI [7,8,9,10]. Necroptosis causes cell membrane rupture, and triggers and amplifies inflammation via the release of damage-associated molecular patterns (DAMPs), through which cell death and inflammation promote exaggerated tissue injury and organ dysfunction [11,12]. Necroptosis can be induced by the TNF superfamily, Toll-like receptor 3 (TLR3) or TLR4 ligands when caspases are inhibited and/or when the ubiquitin ligases cIAP1/2 (cellular inhibitor of apoptosis protein) are degraded [13,14]. Necroptosis is executed by receptor-interacting protein kinase-3 (RIPK3) and the downstream effector mixed lineage kinase domain-like protein (MLKL). It has been shown that serum levels of RIPK3 are elevated in critically ill patients and are associated with the development of ALI/ARDs in sepsis, trauma and COVID-19 patients [15,16,17]. Recent experimental studies have demonstrated that inhibition of necroptosis confers protection against ALI induced by LPS [18,19,20], mechanical ventilation [21], sepsis/systemic inflammatory response syndrome [22,23,24], respiratory syncytial virus infection [25], bacterial pneumonia [26,27,28], trauma [29], and blood transfusion [30]. Hence, the targeting of necroptosis or its modulators holds significant promise for the treatment of ALI/ARDS.
The intracellular signaling pathways associated with necroptosis have been well studied [13,14]. However, the extracellular modulators of necroptosis including enhancers and suppressors are not known. As ALI is often associated with a cytokine storm, it remains unclear as to whether the pro-inflammatory cytokines control the susceptibility of lung cells to necroptosis. The critical cytokines that modulate the necroptosis of different types of lung parenchymally derived cells are likewise poorly understood. To address these gaps in current knowledge, we pretreated/primed mouse primary lung epithelial and endothelial cells with various inflammatory mediators and investigated the cell type-dependent responses to necroptosis and the underlying mechanisms. We provide compelling evidence that IFNγ preferentially among various inflammatory mediators promotes necroptosis in primary alveolar and airway epithelial cells but not in lung microvascular endothelial cells. This process involves the upregulation of necroptosis effector MLKL in lung epithelial cells. Our findings provide a mechanistic link and a novel interpretation of the contribution of IFNγ to the pathogenesis of ALI.
2. Materials and Methods
2.1. Antibodies and Reagents
GSK872 (no. 530389) and JAK kinase inhibitor-1 (no. 420099) were from EMD Millipore (Burlington, MA, USA). GSK843 (HY-125402) and GW806742X (no. HY-112292) were from MedChemExpress (Monmouth Junction, NJ, USA). Vinculin (no. V9131) antibody and LPS (Escherichia coli 0111:B4) were from Sigma (St. Louis, MO, USA). Mouse specific MLKL antibody (no. 28640) was from Cell Signaling Technology (Beverly, MA, USA). Recombinant mouse IL-4 (no. 574304), IL-10 (no. 575804) and M-CSF (no. 576404) were from Biolegend (San Diego, CA, USA). Recombinant mouse IFNα2 (no. 14-8312) and IFNγ (no. 34-8311 and no. 14-8311) were from Thermo Fisher Scientific (San Diego, CA, USA). Recombinant mouse TNFα (no. 410-MT) was from R & D Systems (Minneapolis, MN, USA). Polyinosinic-polycytidylic acid (poly(I:C)) (no. tlrl-pic), CL075 (no. tlrl-c75), and ODN 1585 (no. tlrl-1585) were from InvivoGen (San Diego, CA, USA). Z-VAD-FMK (no. A1902) was from ApexBio (Houston, TX, USA). Smac mimetic AZD5582 (no. CT-A5582) was from Chemietek (Indianapolis, IN, USA). Myristoylated autocamtide-2-related inhibitory peptide (Myr-AIP, no. BML-P212) was from Enzo (Farmingdale, NY, USA). Clarity Western ECL substrate (no. 1705060) was from Bio-Rad (Hercules, CA, USA).
2.2. Cell Cultures
Primary alveolar epithelial cells (AECs, no. C57-6053), tracheal and bronchial epithelial cells (TBECs, no. C57-6033) and lung microvascular endothelial cells (no. C57-6011) from wild type C57BL/6 mice were obtained from Cell Biologics (Chicago, IL, USA). These primary lung cells were cultured in epithelial cell growth medium (M6621, Cell Biologics) or endothelial cell growth medium (M1168, Cell Biologics) and used for experiments within 3 passages. RAW264.7 murine macrophages (no. TIB-71) were from American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured in DMEM medium supplemented with 10% fetal bovine serum (FBS).
2.3. Cell Viability, Lactate Dehydrogenase Release and Real Time Necroptosis Assays
Primary cells were seeded into 96-well plates, grown overnight to subconfluence and incubated with control PBS, IFNγ (1–50 ng/mL), IFNα2 (5–50 ng/mL), TNFα (25 ng/mL), LPS (100 ng/mL), poly(I:C) (0.5 μg/mL), CL075 (1 μg/mL), ODN1585 (1 μM), IL-4 (15 ng/mL), or IL-10 (15 ng/mL) for 24 h in the complete growth medium. The media were then aspirated, and the cells were washed once with PBC containing 1% FBS and treated in a 0–10 h period with mouse TNFα (15 ng/mL), poly(I:C) (0.2 µg/mL) or LPS (200 ng/mL) plus Smac mimetic AZD5582 (100 nM) and z-VAD-FMK (25 or 40 µM) to induce necroptosis in the phenol-free epithelial cell growth medium (no. PCS-300-040, ATCC, Manassas, VA, USA) (for lung epithelial cells) or in RPMI-1640 medium supplemented with 2.5% FBS (for endothelial cells). In some experiments, JAK kinase inhibitor-1 (1 µM), GSK872 (5 µM), GSK843 (2 μM), GW806742X (1–2 µM) or Myr-AIP (1 and 5 µM) were added to the medium. All the cells were incubated with the same amount of DMSO. Cell supernatants were saved, and the cell viability was assessed by MTS assay using CellTiter AQueous one solution reagent (no. G3580) according to the manufacturer’s instructions (Promega, Madison, WI, USA). Cell survival rates were calculated by comparison to individual control groups without treatment of necroptosis inducers and are presented as means ± SE from at least three independent experiments.
Lactate dehydrogenase (LDH) activities in cell supernatants were determined by using the LDH-Cytox™ assay kit (no. 426401, Biolegend, San Diego, CA, USA) according to the manufacturer’s protocol. Relative changes in the release of LDH are presented as fold ± SE of individual control groups without treatment of necroptosis inducers from at least three independent experiments.
Real time necroptosis is monitored by a 0–10 h period using the CellTox™ green cytotoxicity assay (no. G8742, Promega, Madison, WI, USA) according to the manufacturer’s protocol. The assay system uses an asymmetric cyanine dye that is excluded from viable cells but preferentially stains the dead cells’ DNA. The percentage of lytic cell death (green dye+ cells) was calculated by comparison to the maximal fluorescence induced by a lysis buffer in each group and is presented as mean ± SE from at least three independent experiments.
2.4. DNA and High-Mobility Group Protein B1 Release Assays
After treatment with necroptosis inducers for 6 h, cell supernatants were saved, and the contents of released double-stranded DNA (dsDNA) were determined by a sensitive fluorescence dsDNA quantitation kit (no. 31007-T, Biotium, Fremont, CA, USA) according to the manufacturer’s protocol. The amounts of high-mobility group protein B1 (HMGB1) in cell supernatants were determined by using ELISA kits (no. MBS2701751, MyBioSource, San Diego, CA, USA).
2.5. Real Time Measurement of ATP Release
Kinetic monitoring of ATP released from lytic cells was performed by using RealTime-Glo™ extracellular ATP assay kits (no. GA5010, Promega, Madison, WI, USA) according to the manufacturer’s protocol. The assay uses an optimized, bioluminescence detection chemistry to measure ATP that has been released into the cultured cell environment as a result of cell death and stress. The nonlytic, homogeneous ATP assay substrate was added directly to cells at the time of treatment with necroptosis inducers for a period of 0–10 h to assess continuous ATP release.
2.6. Western Blot Analysis
Western blot analysis was performed as we described previously [31]. Briefly, cells were washed twice with ice-cold PBS and then lysed on ice in Nonidet P-40 lysis buffer (25 mM Tris-HCl, pH 7.5, 1% Nonidet P-40, 150 mM NaCl, 10 mM NaF, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 10 mg/mL each leupeptin and aprotinin). Equal amounts of whole cell lysates were subjected to SDS-PAGE and then transferred to polyvinylidene difluoride membranes. The membranes were blocked 1 h at room temperature with 5% low fat milk in Tris-buffered saline plus 0.05% Tween 20, and then incubated 1 h at room temperature with primary antibodies against mouse MLKL (1:500, no. 28640, Cell Signaling Technology, Danvers, MA, USA) or vinculin (1:3000, no. V9131, Sigma, St. Louis, MO, USA), followed by 1 h incubation with anti-mouse (no. 7076) or anti-rabbit (no. 7074) HRP-conjugated secondary antibodies (Cell Signaling Technology, Beverly, MA, USA). Labelled proteins were detected with Bio-Rad Clarity Western ECL substrate (no. 1705061, Hercules, CA, USA). Relative changes in the expression levels of the proteins of interest were measured by densitometric analysis using ImageJ 1.47 software and normalized to vinculin and are represented as fold or percentage of control.
2.7. RNA Isolation and Reverse Transcription (RT)-PCR
Total RNA was isolated using the RNeasy RNA isolation kit (no. 74104) according to the manufacturer’s protocol (Qiagen, Valencia, CA, USA). Primers were designed by using Oligo6 software (Molecular Biology Insights) as follows: MLKL (expected product of 269 bp), 5′-CCG AAA GTG TTG GAA TAG TGA-3′ (forward) and 5Ȳ-GAG TGT TTC CGA ATG GTG TAG-3′ (reverse); and internal control glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (608 bp), 5′-CGC TGA GTA CGT CGT GGA G-3′ (forward) and 5′-GAG GAG TGG GTG TCG CTG TTG-3′ (reverse). RT-PCR conditions were 50 °C for 30 min and 95 °C for 15 min, followed by 30 cycles of 94 °C for 30 s, 55 °C for 1 min, and 72 °C for 1 min, followed by 72 °C for 10 min. The relative changes in mRNA levels were measured by densitometric analysis and normalized to GAPDH.
2.8. SiRNAs and siRNA Transfection
AllStars non-targeting negative control siRNA (no. 1027280) and mouse MLKL siRNA (no. SI00868812) were from Qiagen (Valencia, CA, USA). For siRNA transfection, cells were seeded into 96-well plates for 24 h to reach 50–70% confluence, and then siRNA was transfected into the cells in a final concentration of 20 nM by using Lipofectamine 2000 and Opti-MEM I reduced serum medium according to the manufacturer’s protocol (Invitrogen, Carlsbad, CA, USA). The silencing effects of siRNAs were confirmed by Western blot analysis.
2.9. Statistical Analysis
Data are expressed as mean ± SE. Statistical analyses were performed using GraphPad Prism 9 (GraphPad Software, La Jolla, CA, USA). Data were analyzed by using an unpaired, Student’s t test between means of two groups and one-way ANOVA for differences among means with three or more groups. Two-way ANOVA followed by Sidak multiple comparisons were performed to compare a response with 2 factors. Differences with p < 0.05 were considered statistically significant.
3. Results
3.1. IFNγ among Inflammatory Mediators Preferentially Promotes Necroptosis in Primary Alveolar Epithelial Cells
We determined whether lung epithelial cells that are pre-activated by pro-inflammatory mediators preferentially undergo necroptosis. As shown in Figure 1A, primary alveolar epithelial cells (AECs) from C57BL/6 mice were incubated with control PBS, IFNα, IFNγ, agonists of TLR3 (poly(I:C), TLR4 (LPS), TLR7 (CL075) and TLR9 (ODN1585), IL-4 or IL-10 for 24 h, and then treated for 6 h with control vehicle DMSO or three different necroptosis inducers including TNFα plus Smac mimetic and a pan-caspase inhibitor z-VAD-FMK (TSZ), poly(I:C) plus Smac mimetic and z-VAD-FMK (PSZ), or LPS plus Smac mimetic and z-VAD-FMK (LSZ) [32,33,34,35,36]. Cell viability was assessed by an MTS assay that determines the number of viable cells. We found that IFNγ remarkably enhanced cell death induced by TSZ, PSZ and LSZ (Figure 1A). Type-I IFNα also augmented cell death by TSZ and PSZ, but the effect was less than IFNγ. In contrast, TLR agonists, IL-4 or IL-10 demonstrated relatively minor effects (Figure 1A).
The release of LDH is a hallmark of lytic cell death. Consistent with the cell survival data (Figure 1A), the release of LDH from primary AECs by necroptosis inducers TSZ, PSZ or LSZ was greatly enhanced by IFNγ pretreatment (Figure 1B). Type-I IFNα also enhanced the release of LDH induced by TSZ and PSZ, but the effect was much less than IFNγ. In contrast, TLR agonists, IL-4 or IL-10 had a minor effect. To verify the effect of IFNγ, AECs were pretreated with IFNγ in the presence of DMSO or a selective JAK kinase inhibitor [37] and then exposed to necroptosis inducers TSZ or PSZ for 6 h. We found that the enhanced release of LDH by TSZ or PSZ from IFNγ-treated AECs was essentially abolished by the selective JAK kinase inhibitor, indicating a requirement of IFNγ signaling (Figure 1C). The selective JAK kinase inhibitor did not significantly affect the release of LDH from control AECs in response to TSZ or PSZ. Noticeably, the releases of LDH from both control and IFNγ-treated AECs were blocked by a selective RIPK3 inhibitor GSK872 [36,38] (Figure 1C), indicating a key involvement of the RIPK3 kinase activity-dependent signaling pathway.
We next utilized the CellTox™ green cytotoxicity assay to monitor lytic cell death in real time. The assay system uses an asymmetric cyanine dye that is excluded from viable cells, so the viable cells produce no appreciable increases in fluorescence. When the dye binds DNA of compromised cells with impaired membrane integrity, its fluorescent properties are substantially enhanced. Therefore, the fluorescent signal produced by the dye binding to the dead-cell DNA is proportional to cytotoxicity and can be used to accurately monitor real-time lytic cell death. We found that IFNγ greatly accelerated mouse AEC necroptosis in real time by TSZ, PSZ and LSZ (Figure 2). The IFNγ-pretreated AECs had the most susceptibility to TSZ with ~80% of the cells being lysed 5h after TSZ treatment and all the cells lysed 6 h after TSZ treatment (Figure 2A). The IFNγ-promoted real-time necroptosis by TSZ was blocked by a selective JAK kinase inhibitor [37] or two structurally distinct specific RIPK3 inhibitors GSK872 or GSK843 [36,38], confirming the involvement of IFNγ-mediated and RIPK3 kinase activity-dependent signaling pathways (Figure 2B). Moreover, almost all the IFNγ-activated AECs underwent lysis after 10 h treatment with PSZ or LSZ (Figure 2C,D). Type-I IFNα also enhanced real-time necroptosis by TSZ and PSZ, but the effect was much less than IFNγ (Figure 2A,C). Conversely, TNFα, TLR-3, -4, -7, or -9 agonists had a minor effect on necroptosis in primary AECs (Figure 2).
We determined the dose-dependent effects of type-II IFNγ and type-I IFNα on real-time necroptosis in primary AECs. We found that IFNγ at as low as 1 ng/mL markedly promoted real-time necroptosis induced by TSZ or PSZ (Figure 3A,C). IFNα generally had less of an effect. IFNα at 50 ng/mL enhanced TSZ-induced necroptosis (Figure 3A), while at 10 and 50 ng/mL, it significantly promoted PSZ-induced necroptosis (Figure 3C). The dose-dependent effects of IFNγ on necroptosis were verified by measuring the release of LDH from AECs (Figure 3B,D). Moreover, the LSZ-induced real-time necroptosis was dose-dependently accelerated by IFNγ, while IFNα essentially did not affect the necroptosis (Figure 3E). Taken together, our findings indicate that IFNγ preferentially promotes necroptosis in primary AECs.
3.2. IFNγ Preferentially Promotes Necroptosis in Primary Airway Epithelial Cells
We next assessed if IFNγ also promotes necroptosis in primary airway epithelial cells. Tracheal and bronchial epithelial cells (TBECs) from wild type C57BL/6 mice were utilized for this study. We found that IFNγ greatly accelerated TBEC necroptosis induced by TSZ, PSZ and LSZ (Figure 4). IFNγ-activated TBECs had the most susceptibility to TSZ with all cells lysed 5 h after TSZ treatment (Figure 4A). Almost all the IFNγ-activated TBECs underwent lysis after 9 h of treatment with PSZ or LSZ (Figure 4C,E). Dose-dependent real-time necroptosis studies indicate that IFNγ at 1 ng/mL is sufficient to achieve 100% cell lysis after 8 h treatment with TSZ or PSZ (Figure 4B,D). Although type-I IFNα at 50 ng/mL could enhance necroptosis by TSZ and PSZ, the effect was less than that of IFNγ at 1 ng/mL (Figure 4B,D). In contrast, TNFα, TLR-3, -4, -7, or -9 agonists generally had a relatively minor effect on the necroptosis in primary airway epithelial cells. Collectively, these findings indicate that IFNγ also preferentially promotes necroptosis in primary airway epithelial cells.
In contrast to lung epithelial cells, we found that IFNα, IFNγ, TNFα, as well as TLR agonists did not significantly promote necroptosis in response to TSZ, PSZ or LSZ in mouse primary lung microvascular endothelial cells (Figure S1).
3.3. IFNγ Greatly Accelerates the Release of DAMPs from AECs
Necroptosis causes cell membrane rupture, which triggers and amplifies inflammation through the release of DAMPs, such as ATP, the high-mobility group protein B1 (HMGB1), nucleic acids, IL-1 family cytokines and S100 proteins [11,12]. We monitored kinetic ATP release from lytic cells by using RealTime-Glo™ extracellular ATP assay. We found that IFNγ greatly accelerated the release of ATP from AECs in response to TSZ, with a peak at 3 h after TSZ treatment (Figure 5A). The basal level of HMGB1 was detectable in cell supernatants from resting AECs. At 6 h of TSZ treatment, the levels of HMGB1 were not significantly increased in control AECs but were markedly increased in IFNγ-activated AECs (Figure 5B). We further found that IFNγ also accelerated the release of dsDNA from AECs in response to TSZ (Figure 5C). The level of dsDNA was not detectable in cell supernatants from resting AECs and control AECs treated with TSZ for 6 h (Figure 5C). Collectively, these findings demonstrate that IFNγ greatly accelerates the release of DAMPs from primary AECs.
3.4. MLKL Is Predominantly Induced by IFNγ in Primary Lung Epithelial Cells and MLKL Silencing or Inhibition Largely Suppresses the IFNγ-Promoted Necroptosis
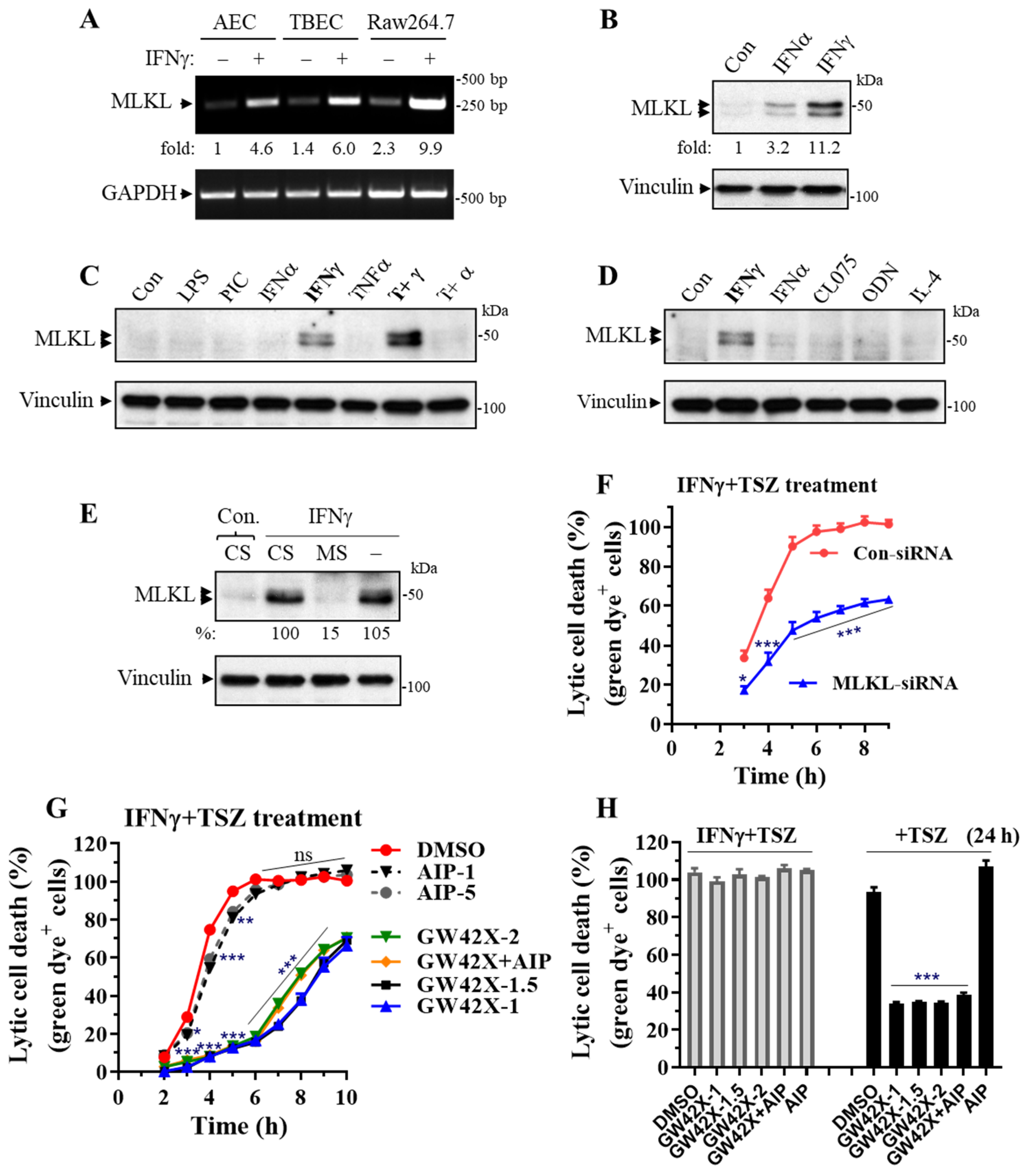
MLKL is a central effector downstream of RIPK3 kinase [13,14]. We found that MLKL mRNAs were readily induced by IFNγ in primary AECs, TBECs and Raw264.7 macrophages (Figure 6A). We further found that MLKL protein levels were predominantly upregulated by IFNγ among the tested inflammatory mediators, although type-I IFNα had an effect (Figure 6B–D). In contrast, TNFα, IL-4, TLR-3, -4, -7 or -9 agonists had a minor effect. Interestingly, MLKL expression could be synergistically induced by IFNγ plus TNFα (Figure 6C).
We knocked down MLKL and determined the effect on the IFNγ-promoted necroptosis in primary AECs. Real-time necroptosis analysis revealed that the enhanced necroptosis in IFNγ-activated AECs was significantly and markedly inhibited by MLKL silencing, although the inhibitory effect was reduced at a later time when necroptosis occurred (after 6 h of TSZ treatment) (Figure 6E,F). We next determined the effect of a selective murine MLKL inhibitor GW806742X on the IFNγ-promoted necroptosis. GW806742X inhibits necroptosis by preventing MLKL membrane translocation [39]. We found that the enhanced necroptosis in IFNγ-activated AECs was inhibited 98%, 92%, 90%, 87%, or 85% from 2 to 6 h during TSZ treatments by GW806742X (1 or 1.5 µM), respectively (Figure 6G). However, GW806742X became less effective, especially in the later stages of necroptosis (after 7 h of TSZ treatment). GW806742X at concentrations of 1 and 1.5 μM appeared to have slightly better inhibitory effects than that of 2 µM (Figure 6G). Interestingly, necroptosis in IFNγ-activated AECs was no longer inhibited by GW806742X at three different doses (1, 1.5 or 2 µM) after 24 h of TSZ treatment, whereas necroptosis in control AECs was sustainably and markedly suppressed by GW806742X at the time point (Figure 6H).
Ca2+-calmodulin-dependent protein kinase-II (CaMKII) has been identified as a new RIPK3 effector mediating myocardial necroptosis [40]. We treated IFNγ-activated AECs with a specific CaMKII myristoylated peptide inhibitor (Myr-AIP) at two different concentrations (1 and 5 µM) that have been shown to effectively inhibit CaMKII activities in vitro and in cultured cells [41,42] and assessed the effects on TSZ-induced necroptosis in primary AECs. We found that inhibition of CaMKII activity with Myr-AIP led to a small but statistically significant reduction in the IFNγ-promoted necroptosis at 3 h (30% inhibition), 4 h (25% inhibition) or 5 h (14% inhibition) of TSZ treatment (Figure 6G). Myr-AIP did not significantly affect necroptosis after 6 h of TSZ treatment. Interestingly, Myr-AIP plus GW806742X did not lead to an additive inhibitory effect on necroptosis compared with GW806742X alone (Figure 6G), suggesting that MLKL and CaMKII may signal and function through the same necroptosis pathway in AECs.
4. Discussion
The present study provides compelling evidence that IFNγ preferentially among various inflammatory mediators promotes necroptosis in primary alveolar and airway epithelial cells but not in lung microvascular endothelial cells. This process involves the IFNγ-mediated upregulation of necroptosis effector MLKL. Unlike apoptosis, necroptosis causes cell membrane rupture, which triggers and amplifies inflammation through the release of DAMPs, such as ATP, HMGB1, nucleic acids, IL-1 family cytokines and S100 proteins [11,12]. Our findings also demonstrate that IFNγ greatly accelerates the release of ATP, HMGB1 and dsDNA from primary lung epithelial cells undergoing necroptosis. DAMPs are detected by pattern recognition receptors that activate immune responses by inducing the expression of cytokines and chemokines. HMGB1 is a highly conserved ubiquitous protein present in the nucleus and cytoplasm of nearly all cell types and is the prototypic DAMP molecule [43]. The inflammatory functions of HMGB1 are mediated through the receptor for advanced glycation end products (RAGE), TLR2, TLR4, and TLR9 [44,45]. ATP activates P2 purinergic receptors, through which it is involved in multiple pulmonary disorders [46,47], while dsDNA including mitochondrial DNA triggers an innate immune response through DNA sensors [48,49]. On the basis of these results, we posit that enhanced necroptosis in IFNγ-activated lung epithelial cells not only promotes cell death but also accelerates the release of immune-stimulatory DAMPs, through which they may contribute to the exaggerated tissue injury, inflammation and lung dysfunction.
IFNγ can be produced by innate (NK, NKT, antigen-presenting cells, neutrophils) and adaptive (CD4+, CD8+, B) immune cells [50,51] and is often elevated in patients with autoimmune diseases [50,52,53] and infections with respiratory viruses such as SARS-CoV-2 [54,55,56,57,58]. A prospective cohort study identified IFNγ, angiopoietin1/2, IL-6, and plasminogen activator inhibitor-1 as four critical biomarkers for the “reactive” phenotype group of ARDS, which has higher mortality than a less “inflamed” phenotypic group [59]. Prior studies demonstrate that neutralization of IFNγ or deficiency of IFNγ receptor confer protection against ALI and/or lethality induced by LPS [60,61,62], sepsis/systemic inflammatory response syndrome [62,63,64,65], hyperoxia/mechanical ventilation [66,67], and respiratory viral infection [68,69,70,71]. Neutralization of both IFNγ and TNFα additionally protects against mortality by SARS-CoV2 infection in mice [72]. Administration of recombinant IFNγ or local IFNγ induction greatly accelerates ALI following LPS challenge in mice [61]. It has been demonstrated that necrosis (including necroptosis) rather than apoptosis is the dominant form of AEC death in LPS-induced ALI in mice [73]. However, IFNγ alone is unable to induce ALI in mice [61]. Collectively, these data indicate that IFNγ appears to play an important role in ALI, but the underlying molecular mechanisms by which this mediator regulates lung injury remain unclear. The present study has now identified a mechanistic link between IFNγ and the enhanced necroptosis of activated lung epithelial cells and expanded understanding of the contribution of IFNγ to the pathogenesis of ALI. High levels of pulmonary IFNγ may cause a rapid initiation and fast progression of ALI that is often observed in ARDS [2,74] where necroptosis signals are triggered by mechanical ventilation, endotoxin inhalation, infections and trauma [7,8,10].
It has been shown that extended incubation (24 to 72 h) of IFNγ induces necroptosis in murine embryo fibroblasts (MEFs) deficient of NF-κB signaling (rela-/-) or Fas-associated death domain (fadd-/-) or when caspases are inhibited [75,76]. NF-κB, FADD and caspases are negative intracellular regulators of necroptosis [13,14], thus IFNγ facilitates necroptosis in the sensitized MEFs. However, IFNγ had a minor effect on the cell survival of primary normal MEFs [75,76]. Conversely, Lee et al. reported that IFNγ slightly promoted a reduction in MLKL and inhibited the phosphorylation of MLKL in mouse splenocytes treated with zVAD and TNFα, in which the mechanism was unknown [77]. Consistent with the effect on primary normal MEFs [75,76], we found that 24 h incubation of IFNγ alone had no effect on cell survival but slightly reduced LDH release from primary normal mouse AECs (Figure S2). We further found that IFNγ markedly promoted the susceptibility of lung epithelial cells to various necroptosis inducers. Hence, we posit that IFNγ has a minor effect on cell survival in primary normal cells but, in a cell type-dependent manner, can greatly promote necrotic cell death in necroptosis-sensitized cells and cells receiving necroptosis signals. The IFNγ-promoted necroptosis of lung epithelial cells was blocked by a selective JAK kinase inhibitor [37] or two structurally distinct specific RIPK3 kinase inhibitors GSK872 or GSK843 [36,38]. These results indicate a requirement of IFNγ signaling and support a key role of RIPK3 kinase activity in the initiation of necroptosis [35]. We further found that the mRNA and protein levels of necroptosis effector MLKL were predominantly upregulated by IFNγ in lung epithelial cells and that the enhanced necroptosis in the cells was significantly and markedly inhibited by MLKL silencing. Moreover, the enhanced necroptosis in IFNγ-activated AECs was almost completely abolished by preventing MLKL membrane translocation with a selective murine MLKL inhibitor GW806742X [39] during the early stages of necroptosis (from 2 to 6 h of TSZ treatments). These findings indicate that the induction of MLKL is a critical determinant of enhanced necrotic cell death that occurs in IFNγ-activated lung epithelial cells. We recently showed that IFNγ, LPS, or poly(I:C) induced MLKL expression and promoted necroptosis in mouse bone marrow-derived macrophages [78]. However, MLKL was barely induced by LPS and poly(I:C) in primary lung epithelial cells and neither LPS nor poly(I:C) promoted lung epithelial cell necroptosis. Similarly, we found that IFNγ did not promote necroptosis in lung microvascular endothelial cells, which may be attributed to the inability of IFNγ to induce MLKL expression. Cell type-dependent regulation of MLKL expression by IFNγ and the underlying mechanism merit further investigation. Interestingly, we found that GW806742X became less effective in suppressing necroptosis during the later stages of necroptosis (after 7 h of TSZ treatment). GW806742X inhibits necroptosis by retarding MLKL translocation to membranes [39]. Our observation could be interpreted as that membrane translocation of MLKL in IFNγ-activated lung epithelial cells may not be effectively blocked by GW806742X, possibly due to a modification of MLKL. Other mechanisms beyond the participation of MLKL could likewise contribute to the enhanced necroptosis at the later stages in IFNγ-activated lung epithelial cells.
CaMKII is another necroptosis effector downstream of RIPK3, mediating myocardial necroptosis [40]. We found that inhibition of CaMKII activity with a specific myristoylated peptide inhibitor (Myr-AIP) [41,42] led to a small but statistically significant reduction in the IFNγ-promoted necroptosis at 3 h (30% inhibition) and 4 h (25% inhibition) of TSZ treatment, where the enhanced necroptosis was suppressed more than 90% by the MLKL inhibitor GW806742X [39]. Furthermore, Myr-AIP plus GW806742X did not lead to an additive inhibitory effect on necroptosis compared with GW806742X alone. A recent report shows that CaMKII regulates necroptosis downstream of MLKL in smooth muscle cells [41]. Hence, our findings indicate that CaMKII contributes to a small proportion of necroptosis early after induction of the process in IFNγ-activated lung epithelial cells and may likely mediate necroptosis downstream of MLKL as well.
In conclusion, we provide compelling evidence showing that IFNγ preferentially among various inflammatory mediators promotes necroptosis and accelerates the release of DAMPs in primary alveolar and airway epithelial cells but not in lung microvascular endothelial cells. We further show that MLKL is predominantly induced by IFNγ, contributing to the enhanced necroptosis in lung epithelial cells. Our findings provide a mechanistic link and a novel interpretation of the contribution of IFNγ to the pathogenesis of cellular perturbations that may contribute to ALI.
Supplementary Materials
The following supporting information can be downloaded at: https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/cells11030563/s1, Figure S1: IFNγ and other agonists do not promote necroptosis in lung microvascular endothelial cells. Figure S2: IFNγ alone has no effect on cell survival and a minor effect on LDH release in primary AECs.
Author Contributions
Conceptualization and design, Q.H. and H.T.; Methodology, Q.H. and H.T.; Data analysis, Q.H. and H.T.; Writing—original draft preparation, H.T.; Writing—review and editing, H.T., S.S., T.A.T. and S.I. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported in part by the National Institute of Health (R21AI135569 to H.T.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All datasets generated for this study are included in the manuscript and the supplementary files.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Manicone, A.M. Role of the pulmonary epithelium and inflammatory signals in acute lung injury. Expert Rev. Clin. Immunol. 2009, 5, 63–75. [Google Scholar] [CrossRef] [Green Version]
- Fan, E.; Brodie, D.; Slutsky, A.S. Acute Respiratory Distress Syndrome: Advances in Diagnosis and Treatment. JAMA 2018, 319, 698–710. [Google Scholar] [CrossRef]
- Bellani, G.; Laffey, J.G.; Pham, T.; Fan, E.; Brochard, L.; Esteban, A.; Gattinoni, L.; van Haren, F.; Larsson, A.; McAuley, D.F.; et al. Epidemiology, Patterns of Care, and Mortality for Patients with Acute Respiratory Distress Syndrome in Intensive Care Units in 50 Countries. JAMA 2016, 315, 788–800. [Google Scholar] [CrossRef]
- Li, L.; Huang, Q.; Wang, D.C.; Ingbar, D.H.; Wang, X. Acute lung injury in patients with COVID-19 infection. Clin. Transl. Med. 2020, 10, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Camporota, L.; Cronin, J.N.; Busana, M.; Gattinoni, L.; Formenti, F. Pathophysiology of coronavirus-19 disease acute lung injury. Curr. Opin. Crit. Care 2022, 28, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Peritore, A.F.; D’Amico, R.; Siracusa, R.; Cordaro, M.; Fusco, R.; Gugliandolo, E.; Genovese, T.; Crupi, R.; Di Paola, R.; Cuzzocrea, S.; et al. Management of Acute Lung Injury: Palmitoylethanolamide as a New Approach. Int. J. Mol. Sci. 2021, 22, 5533. [Google Scholar] [CrossRef]
- Faust, H.; Mangalmurti, N.S. Collateral damage: Necroptosis in the development of lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L215–L225. [Google Scholar] [CrossRef] [PubMed]
- Mizumura, K.; Maruoka, S.; Gon, Y.; Choi, A.M.; Hashimoto, S. The role of necroptosis in pulmonary diseases. Respir. Investig. 2016, 54, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Fan, E.K.Y.; Fan, J. Regulation of alveolar macrophage death in acute lung inflammation. Respir. Res. 2018, 19, 50. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M.K. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, e128834. [Google Scholar] [CrossRef]
- Kaczmarek, A.; Vandenabeele, P.; Krysko, D.V. Necroptosis: The release of damage-associated molecular patterns and its physiological relevance. Immunity 2013, 38, 209–223. [Google Scholar] [CrossRef] [Green Version]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Wallach, D.; Kang, T.B.; Dillon, C.P.; Green, D.R. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science 2016, 352, aaf2154. [Google Scholar] [CrossRef]
- He, S.; Wang, X. RIP kinases as modulators of inflammation and immunity. Nat. Immunol. 2018, 19, 912–922. [Google Scholar] [CrossRef]
- Shashaty, M.G.S.; Reilly, J.P.; Faust, H.E.; Forker, C.M.; Ittner, C.A.G.; Zhang, P.X.; Hotz, M.J.; Fitzgerald, D.; Yang, W.; Anderson, B.J.; et al. Plasma receptor interacting protein kinase-3 levels are associated with acute respiratory distress syndrome in sepsis and trauma: A cohort study. Crit. Care 2019, 23, 235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, H.; Kinjo, T.; Arakaki, W.; Miyagi, K.; Tateyama, M.; Fujita, J. Serum levels of receptor-interacting protein kinase-3 in patients with COVID-19. Crit. Care 2020, 24, 484. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.C.; Schenck, E.J.; Siempos, I.I.; Cloonan, S.M.; Finkelsztein, E.J.; Pabon, M.A.; Oromendia, C.; Ballman, K.V.; Baron, R.M.; Fredenburgh, L.E.; et al. Circulating RIPK3 levels are associated with mortality and organ failure during critical illness. JCI Insight 2018, 3, e99692. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Wang, T.; Li, H.; Liu, Q.; Zhang, Z.; Xie, W.; Feng, Y.; Socorburam, T.; Wu, G.; Xia, Z.; et al. Receptor Interacting Protein 3-Mediated Necroptosis Promotes Lipopolysaccharide-Induced Inflammation and Acute Respiratory Distress Syndrome in Mice. PLoS ONE 2016, 11, e0155723. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, S.; Fu, R.; Zhou, M.; Zhang, T.; Pan, W.; Yang, N.; Huang, Y. RIP3 dependent NLRP3 inflammasome activation is implicated in acute lung injury in mice. J. Transl. Med. 2018, 16, 233. [Google Scholar] [CrossRef]
- Lin, B.; Jin, Z.; Chen, X.; Zhao, L.; Weng, C.; Chen, B.; Tang, Y.; Lin, L. Necrostatin1 protects mice from acute lung injury by suppressing necroptosis and reactive oxygen species. Mol. Med. Rep. 2020, 21, 2171–2181. [Google Scholar]
- Siempos, I.I.; Ma, K.C.; Imamura, M.; Baron, R.M.; Fredenburgh, L.E.; Huh, J.W.; Moon, J.S.; Finkelsztein, E.J.; Jones, D.S.; Lizardi, M.T.; et al. RIPK3 mediates pathogenesis of experimental ventilator-induced lung injury. JCI Insight 2018, 3, e97102. [Google Scholar] [CrossRef]
- Hansen, L.W.; Jacob, A.; Yang, W.L.; Bolognese, A.C.; Prince, J.; Nicastro, J.M.; Coppa, G.F.; Wang, P. Deficiency of receptor-interacting protein kinase 3 (RIPK3) attenuates inflammation and organ injury in neonatal sepsis. J. Pediatr. Surg. 2018, 53, 1699–1705. [Google Scholar] [CrossRef]
- Sharma, A.; Matsuo, S.; Yang, W.L.; Wang, Z.; Wang, P. Receptor-interacting protein kinase 3 deficiency inhibits immune cell infiltration and attenuates organ injury in sepsis. Crit. Care 2014, 18, R142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolognese, A.C.; Yang, W.L.; Hansen, L.W.; Denning, N.L.; Nicastro, J.M.; Coppa, G.F.; Wang, P. Inhibition of necroptosis attenuates lung injury and improves survival in neonatal sepsis. Surgery 2018, 164, 110–116. [Google Scholar] [CrossRef]
- Santos, L.D.; Antunes, K.H.; Muraro, S.P.; de Souza, G.F.; da Silva, A.G.; de Souza Felipe, J.; Zanetti, L.C.; Czepielewski, R.S.; Magnus, K.; Scotta, M.; et al. TNF-mediated alveolar macrophage necroptosis drives disease pathogenesis during Respiratory Syncytial Virus infection. Eur. Respir. J. 2020, 57, 2003764. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Juarbe, N.; Bradley, K.M.; Shenoy, A.T.; Gilley, R.P.; Reyes, L.F.; Hinojosa, C.A.; Restrepo, M.I.; Dube, P.H.; Bergman, M.A.; Orihuela, C.J. Pore-forming toxin-mediated ion dysregulation leads to death receptor-independent necroptosis of lung epithelial cells during bacterial pneumonia. Cell Death Differ. 2017, 24, 917–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Juarbe, N.; Gilley, R.P.; Hinojosa, C.A.; Bradley, K.M.; Kamei, A.; Gao, G.; Dube, P.H.; Bergman, M.A.; Orihuela, C.J. Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PLoS Pathog. 2015, 11, e1005337. [Google Scholar] [CrossRef]
- Gonzalez-Juarbe, N.; Riegler, A.N.; Jureka, A.S.; Gilley, R.P.; Brand, J.D.; Trombley, J.E.; Scott, N.R.; Platt, M.P.; Dube, P.H.; Petit, C.M.; et al. Influenza-Induced Oxidative Stress Sensitizes Lung Cells to Bacterial-Toxin-Mediated Necroptosis. Cell Rep. 2020, 32, 108062. [Google Scholar] [CrossRef]
- Cui, Y.L.; Qiu, L.H.; Zhou, S.Y.; Li, L.F.; Qian, Z.Z.; Liu, X.M.; Zhang, H.L.; Ren, X.B.; Wang, Y.Q. Necroptosis as a potential therapeutic target in multiple organ dysfunction syndrome. Oncotarget 2017, 8, 56980–56990. [Google Scholar] [CrossRef]
- Qing, D.Y.; Conegliano, D.; Shashaty, M.G.; Seo, J.; Reilly, J.P.; Worthen, G.S.; Huh, D.; Meyer, N.J.; Mangalmurti, N.S. Red blood cells induce necroptosis of lung endothelial cells and increase susceptibility to lung inflammation. Am. J. Respir. Crit. Care Med. 2014, 190, 1243–1254. [Google Scholar] [CrossRef]
- Tang, H.; Zhao, Z.J.; Landon, E.J.; Inagami, T. Regulation of calcium-sensitive tyrosine kinase Pyk2 by angiotensin II in endothelial cells. Roles of Yes tyrosine kinase and tyrosine phosphatase SHP-2. J. Biol. Chem. 2000, 275, 8389–8396. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 5322–5327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Wang, L.; Miao, L.; Wang, T.; Du, F.; Zhao, L.; Wang, X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 2009, 137, 1100–1111. [Google Scholar] [CrossRef] [Green Version]
- Kaiser, W.J.; Sridharan, H.; Huang, C.; Mandal, P.; Upton, J.W.; Gough, P.J.; Sehon, C.A.; Marquis, R.W.; Bertin, J.; Mocarski, E.S. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem. 2013, 288, 31268–31279. [Google Scholar] [CrossRef] [Green Version]
- Pedranzini, L.; Dechow, T.; Berishaj, M.; Comenzo, R.; Zhou, P.; Azare, J.; Bornmann, W.; Bromberg, J. Pyridone 6, a pan-Janus-activated kinase inhibitor, induces growth inhibition of multiple myeloma cells. Cancer Res. 2006, 66, 9714–9721. [Google Scholar] [CrossRef] [Green Version]
- Mandal, P.; Berger, S.B.; Pillay, S.; Moriwaki, K.; Huang, C.; Guo, H.; Lich, J.D.; Finger, J.; Kasparcova, V.; Votta, B.; et al. RIP3 induces apoptosis independent of pronecrotic kinase activity. Mol. Cell 2014, 56, 481–495. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, J.M.; Tanzer, M.C.; Lucet, I.S.; Young, S.N.; Spall, S.K.; Sharma, P.; Pierotti, C.; Garnier, J.M.; Dobson, R.C.; Webb, A.I.; et al. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. USA 2014, 111, 15072–15077. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Zhang, Y.; Cui, M.; Jin, L.; Wang, Y.; Lv, F.; Liu, Y.; Zheng, W.; Shang, H.; Zhang, J.; et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016, 22, 175–182. [Google Scholar] [CrossRef]
- Zhou, T.; DeRoo, E.; Yang, H.; Stranz, A.; Wang, Q.; Ginnan, R.; Singer, H.A.; Liu, B. MLKL and CaMKII Are Involved in RIPK3-Mediated Smooth Muscle Cell Necroptosis. Cells 2021, 10, 2397. [Google Scholar] [CrossRef]
- Ishida, A.; Kameshita, I.; Okuno, S.; Kitani, T.; Fujisawa, H. A novel highly specific and potent inhibitor of calmodulin-dependent protein kinase II. Biochem. Biophys. Res. Commun. 1995, 212, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bloom, O.; Zhang, M.; Vishnubhakat, J.M.; Ombrellino, M.; Che, J.; Frazier, A.; Yang, H.; Ivanova, S.; Borovikova, L.; et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science 1999, 285, 248–251. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yang, H.; Czura, C.J.; Sama, A.E.; Tracey, K.J. HMGB1 as a late mediator of lethal systemic inflammation. Am. J. Respir. Crit. Care Med. 2001, 164, 1768–1773. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Czura, C.J.; Tracey, K.J. The cytokine activity of HMGB1. J. Leukoc. Biol. 2005, 78, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Klaver, D.; Thurnher, M. Control of Macrophage Inflammation by P2Y Purinergic Receptors. Cells 2021, 10, 1098. [Google Scholar] [CrossRef]
- Pelleg, A. Extracellular adenosine 5′-triphosphate in pulmonary disorders. Biochem. Pharmacol. 2021, 187, 114319. [Google Scholar] [CrossRef]
- Maelfait, J.; Liverpool, L.; Rehwinkel, J. Nucleic Acid Sensors and Programmed Cell Death. J. Mol. Biol. 2020, 432, 552–568. [Google Scholar] [CrossRef]
- West, A.P.; Shadel, G.S. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 2017, 17, 363–375. [Google Scholar] [CrossRef]
- Fenimore, J. Regulation of IFN-gamma Expression. Adv. Exp. Med. Biol. 2016, 941, 1–19. [Google Scholar] [PubMed]
- Gomez, J.C.; Yamada, M.; Martin, J.R.; Dang, H.; Brickey, W.J.; Bergmeier, W.; Dinauer, M.C.; Doerschuk, C.M. Mechanisms of interferon-gamma production by neutrophils and its function during Streptococcus pneumoniae pneumonia. Am. J. Respir. Cell Mol. Biol. 2015, 52, 349–364. [Google Scholar] [CrossRef] [Green Version]
- Schoenborn, J.R.; Wilson, C.B. Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 2007, 96, 41–101. [Google Scholar]
- Zheng, C.; Yin, S.; Yang, Y.; Yu, Y.; Xie, X. CD24 aggravates acute liver injury in autoimmune hepatitis by promoting IFN-gamma production by CD4(+) T cells. Cell. Mol. Immunol. 2018, 15, 260–271. [Google Scholar] [CrossRef]
- Gadotti, A.C.; de Castro Deus, M.; Telles, J.P.; Wind, R.; Goes, M.; Garcia Charello Ossoski, R.; de Padua, A.M.; de Noronha, L.; Moreno-Amaral, A.; Baena, C.P.; et al. IFN-gamma is an independent risk factor associated with mortality in patients with moderate and severe COVID-19 infection. Virus Res. 2020, 289, 198171. [Google Scholar] [CrossRef]
- Ghazavi, A.; Ganji, A.; Keshavarzian, N.; Rabiemajd, S.; Mosayebi, G. Cytokine profile and disease severity in patients with COVID-19. Cytokine 2021, 137, 155323. [Google Scholar] [CrossRef]
- Theron, M.; Huang, K.J.; Chen, Y.W.; Liu, C.C.; Lei, H.Y. A probable role for IFN-gamma in the development of a lung immunopathology in SARS. Cytokine 2005, 32, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Montalvo Villalba, M.C.; Valdes Ramirez, O.; Mune Jimenez, M.; Arencibia Garcia, A.; Martinez Alfonso, J.; Gonzalez Baez, G.; Roque Arrieta, R.; Rosell Simon, D.; Alvarez Gainza, D.; Sierra Vazquez, B.; et al. Interferon gamma, TGF-beta1 and RANTES expression in upper airway samples from SARS-CoV-2 infected patients. Clin. Immunol. 2020, 220, 108576. [Google Scholar] [CrossRef] [PubMed]
- Akbari, H.; Tabrizi, R.; Lankarani, K.B.; Aria, H.; Vakili, S.; Asadian, F.; Noroozi, S.; Keshavarz, P.; Faramarz, S. The role of cytokine profile and lymphocyte subsets in the severity of coronavirus disease 2019 (COVID-19): A systematic review and meta-analysis. Life Sci. 2020, 258, 118167. [Google Scholar] [CrossRef] [PubMed]
- Bos, L.D.; Schouten, L.R.; van Vught, L.A.; Wiewel, M.A.; Ong, D.S.Y.; Cremer, O.; Artigas, A.; Martin-Loeches, I.; Hoogendijk, A.J.; van der Poll, T.; et al. Identification and validation of distinct biological phenotypes in patients with acute respiratory distress syndrome by cluster analysis. Thorax 2017, 72, 876–883. [Google Scholar] [CrossRef]
- Mock, J.R.; Tune, M.K.; Dial, C.F.; Torres-Castillo, J.; Hagan, R.S.; Doerschuk, C.M. Effects of IFN-gamma on immune cell kinetics during the resolution of acute lung injury. Physiol. Rep. 2020, 8, e14368. [Google Scholar] [CrossRef]
- Aoyagi, T.; Yamamoto, N.; Hatta, M.; Tanno, D.; Miyazato, A.; Ishii, K.; Suzuki, K.; Nakayama, T.; Taniguchi, M.; Kunishima, H.; et al. Activation of pulmonary invariant NKT cells leads to exacerbation of acute lung injury caused by LPS through local production of IFN-gamma and TNF-alpha by Gr-1+ monocytes. Int. Immunol. 2011, 23, 97–108. [Google Scholar] [CrossRef]
- Doherty, G.M.; Lange, J.R.; Langstein, H.N.; Alexander, H.R.; Buresh, C.M.; Norton, J.A. Evidence for IFN-gamma as a mediator of the lethality of endotoxin and tumor necrosis factor-alpha. J. Immunol. 1992, 149, 1666–1670. [Google Scholar] [PubMed]
- Heinzel, F.P. The role of IFN-gamma in the pathology of experimental endotoxemia. J. Immunol. 1990, 145, 2920–2924. [Google Scholar] [PubMed]
- Car, B.D.; Eng, V.M.; Schnyder, B.; Ozmen, L.; Huang, S.; Gallay, P.; Heumann, D.; Aguet, M.; Ryffel, B. Interferon gamma receptor deficient mice are resistant to endotoxic shock. J. Exp. Med. 1994, 179, 1437–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohler, J.; Heumann, D.; Garotta, G.; LeRoy, D.; Bailat, S.; Barras, C.; Baumgartner, J.D.; Glauser, M.P. IFN-gamma involvement in the severity of gram-negative infections in mice. J. Immunol. 1993, 151, 916–921. [Google Scholar] [PubMed]
- Liu, Q.; Xie, W.; Wang, Y.; Chen, S.; Han, J.; Wang, L.; Gui, P.; Wu, Q. JAK2/STAT1-mediated HMGB1 translocation increases inflammation and cell death in a ventilator-induced lung injury model. Lab. Investig. 2019, 99, 1810–1821. [Google Scholar] [CrossRef]
- Yamada, M.; Kubo, H.; Kobayashi, S.; Ishizawa, K.; Sasaki, H. Interferon-gamma: A key contributor to hyperoxia-induced lung injury in mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L1042–L1047. [Google Scholar] [CrossRef]
- Moskophidis, D.; Kioussis, D. Contribution of virus-specific CD8+ cytotoxic T cells to virus clearance or pathologic manifestations of influenza virus infection in a T cell receptor transgenic mouse model. J. Exp. Med. 1998, 188, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Ostler, T.; Davidson, W.; Ehl, S. Virus clearance and immunopathology by CD8(+) T cells during infection with respiratory syncytial virus are mediated by IFN-gamma. Eur. J. Immunol. 2002, 32, 2117–2123. [Google Scholar] [CrossRef]
- Ramana, C.V.; DeBerge, M.P.; Kumar, A.; Alia, C.S.; Durbin, J.E.; Enelow, R.I. Inflammatory impact of IFN-gamma in CD8+ T cell-mediated lung injury is mediated by both Stat1-dependent and -independent pathways. Am. J. Physiol. Lung Cell Mol. Physiol. 2015, 308, L650–L657. [Google Scholar] [CrossRef] [Green Version]
- Wlodarczyk, M.F.; Kraft, A.R.; Chen, H.D.; Kenney, L.L.; Selin, L.K. Anti-IFN-gamma and peptide-tolerization therapies inhibit acute lung injury induced by cross-reactive influenza A-specific memory T cells. J. Immunol. 2013, 190, 2736–2746. [Google Scholar] [CrossRef]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.K.S.; et al. Synergism of TNF-alpha and IFN-gamma Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell 2021, 184, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Tamada, N.; Tojo, K.; Yazawa, T.; Goto, T. Necrosis Rather Than Apoptosis is the Dominant form of Alveolar Epithelial Cell Death in Lipopolysaccharide-Induced Experimental Acute Respiratory Distress Syndrome Model. Shock 2020, 54, 128–139. [Google Scholar] [CrossRef]
- Aeffner, F.; Bolon, B.; Davis, I.C. Mouse Models of Acute Respiratory Distress Syndrome: A Review of Analytical Approaches, Pathologic Features, and Common Measurements. Toxicol. Pathol. 2015, 43, 1074–1092. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.J.; Basagoudanavar, S.H.; Nogusa, S.; Irrinki, K.; Mallilankaraman, K.; Slifker, M.J.; Beg, A.A.; Madesh, M.; Balachandran, S. NF-kappaB protects cells from gamma interferon-induced RIP1-dependent necroptosis. Mol. Cell Biol. 2011, 31, 2934–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thapa, R.J.; Nogusa, S.; Chen, P.; Maki, J.L.; Lerro, A.; Andrake, M.; Rall, G.F.; Degterev, A.; Balachandran, S. Interferon-induced RIP1/RIP3-mediated necrosis requires PKR and is licensed by FADD and caspases. Proc. Natl. Acad. Sci. USA 2013, 110, E3109–E3118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.H.; Kwon, J.Y.; Kim, S.Y.; Jung, K.; Cho, M.L. Interferon-gamma regulates inflammatory cell death by targeting necroptosis in experimental autoimmune arthritis. Sci. Rep. 2017, 7, 10133. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Kundu, S.; Kleam, J.; Zhao, Z.J.; Idell, S.; Tang, H. Enhanced RIPK3 kinase activity-dependent lytic cell death in M1 but not M2 macrophages. Mol. Immunol. 2021, 129, 86–93. [Google Scholar] [CrossRef]
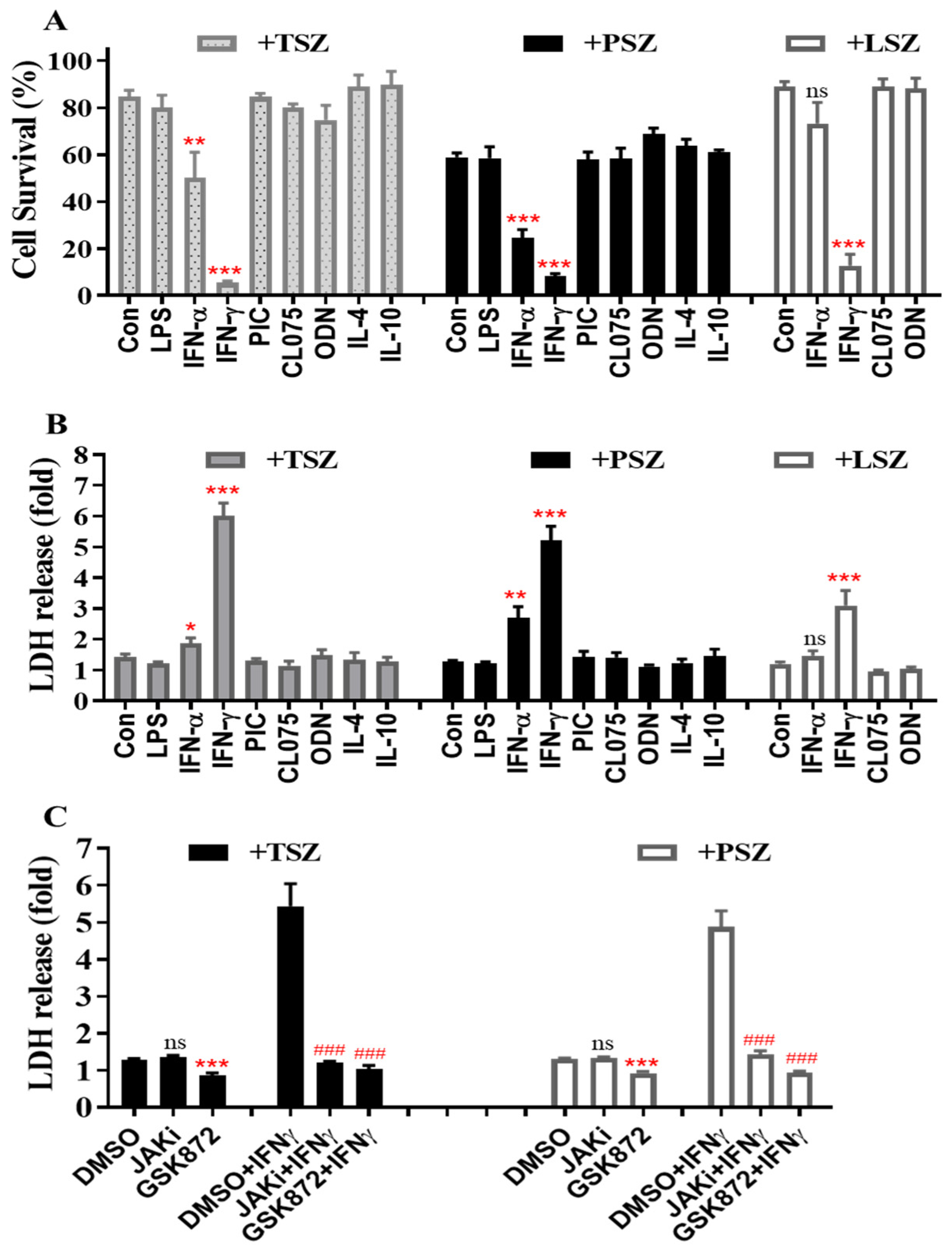
Figure 1.
IFNγ preferentially promotes cell death and the release of LDH induced by various necroptosis inducers in primary AECs. Mouse primary AECs were incubated with LPS (100 ng/mL), IFNα2 (50 ng/mL), IFNγ (50 ng/mL), poly(I:C) (PIC, 0.5 μg/mL), CL075 (1 μg/mL), ODN1585 (1 μM), IL-4 (15 ng/mL), or IL-10 (15 ng/mL) for 24 h, then treated for 6 h with TNFα (15 ng/mL) plus 100 nM Smac mimetic AZD5582 and 25 µM z-VAD-FMK (TSZ), poly(I:C) (0.2 µg/mL) plus 100 nM Smac mimetic AZD5582 and 40 µM z-VAD-FMK (PSZ), or LPS (200 ng/mL) plus 100 nM Smac mimetic AZD5582 and 25 µM z-VAD-FMK (LSZ). (A) AEC viability was assessed by MTS assay using CellTiter AQueous reagent and cell survival rates were calculated by comparison to individual control groups without treatment of necroptosis inducers and are presented as means ± SE. Con.+TSZ or PSZ, n = 12; IFNγ+TSZ or PSZ, n = 10; all other treatments, n = 4–8. ** p < 0.01; *** p < 0.001 versus control (Con.) in each treatment group. NS, no significance. (B) LDH activities in cell supernatant were determined. Relative changes in LDH are presented as fold ± SE of individual control groups without treatment of necroptosis inducers. Con. or IFNγ+TSZ, n = 10; Con. or IFNγ+PSZ, n = 12; all other treatments, n = 4–7. * p < 0.05; ** p < 0.01; *** p < 0.001 versus control (Con.) in each group. NS, no significance. (C) Mouse AECs were incubated without or with IFNγ (50 ng/mL) in the presence of DMSO or JAK kinase inhibitor-1 (JAKi, 1 μM) for 24 h, washed and then treated for 6 h with necroptosis inducers TSZ or PSZ in the presence of DMSO, GSK872 (5 μM) or JAKi (1 μM). LDH activities in cell supernatant were determined and the relative changes are presented as fold ± SE of individual control groups without treatment of necroptosis inducers. TSZ group: DMSO or DMSO+IFNγ, n = 7; others, n = 3–4. PSZ group: DMSO or DMSO+IFNγ, n = 9; others, n = 5. *** p < 0.001 versus DMSO in each group. ### p < 0.001 versus DMSO+IFNγ in each group. NS, no significance. One-way ANOVA was performed in (A–C).
Figure 1.
IFNγ preferentially promotes cell death and the release of LDH induced by various necroptosis inducers in primary AECs. Mouse primary AECs were incubated with LPS (100 ng/mL), IFNα2 (50 ng/mL), IFNγ (50 ng/mL), poly(I:C) (PIC, 0.5 μg/mL), CL075 (1 μg/mL), ODN1585 (1 μM), IL-4 (15 ng/mL), or IL-10 (15 ng/mL) for 24 h, then treated for 6 h with TNFα (15 ng/mL) plus 100 nM Smac mimetic AZD5582 and 25 µM z-VAD-FMK (TSZ), poly(I:C) (0.2 µg/mL) plus 100 nM Smac mimetic AZD5582 and 40 µM z-VAD-FMK (PSZ), or LPS (200 ng/mL) plus 100 nM Smac mimetic AZD5582 and 25 µM z-VAD-FMK (LSZ). (A) AEC viability was assessed by MTS assay using CellTiter AQueous reagent and cell survival rates were calculated by comparison to individual control groups without treatment of necroptosis inducers and are presented as means ± SE. Con.+TSZ or PSZ, n = 12; IFNγ+TSZ or PSZ, n = 10; all other treatments, n = 4–8. ** p < 0.01; *** p < 0.001 versus control (Con.) in each treatment group. NS, no significance. (B) LDH activities in cell supernatant were determined. Relative changes in LDH are presented as fold ± SE of individual control groups without treatment of necroptosis inducers. Con. or IFNγ+TSZ, n = 10; Con. or IFNγ+PSZ, n = 12; all other treatments, n = 4–7. * p < 0.05; ** p < 0.01; *** p < 0.001 versus control (Con.) in each group. NS, no significance. (C) Mouse AECs were incubated without or with IFNγ (50 ng/mL) in the presence of DMSO or JAK kinase inhibitor-1 (JAKi, 1 μM) for 24 h, washed and then treated for 6 h with necroptosis inducers TSZ or PSZ in the presence of DMSO, GSK872 (5 μM) or JAKi (1 μM). LDH activities in cell supernatant were determined and the relative changes are presented as fold ± SE of individual control groups without treatment of necroptosis inducers. TSZ group: DMSO or DMSO+IFNγ, n = 7; others, n = 3–4. PSZ group: DMSO or DMSO+IFNγ, n = 9; others, n = 5. *** p < 0.001 versus DMSO in each group. ### p < 0.001 versus DMSO+IFNγ in each group. NS, no significance. One-way ANOVA was performed in (A–C).

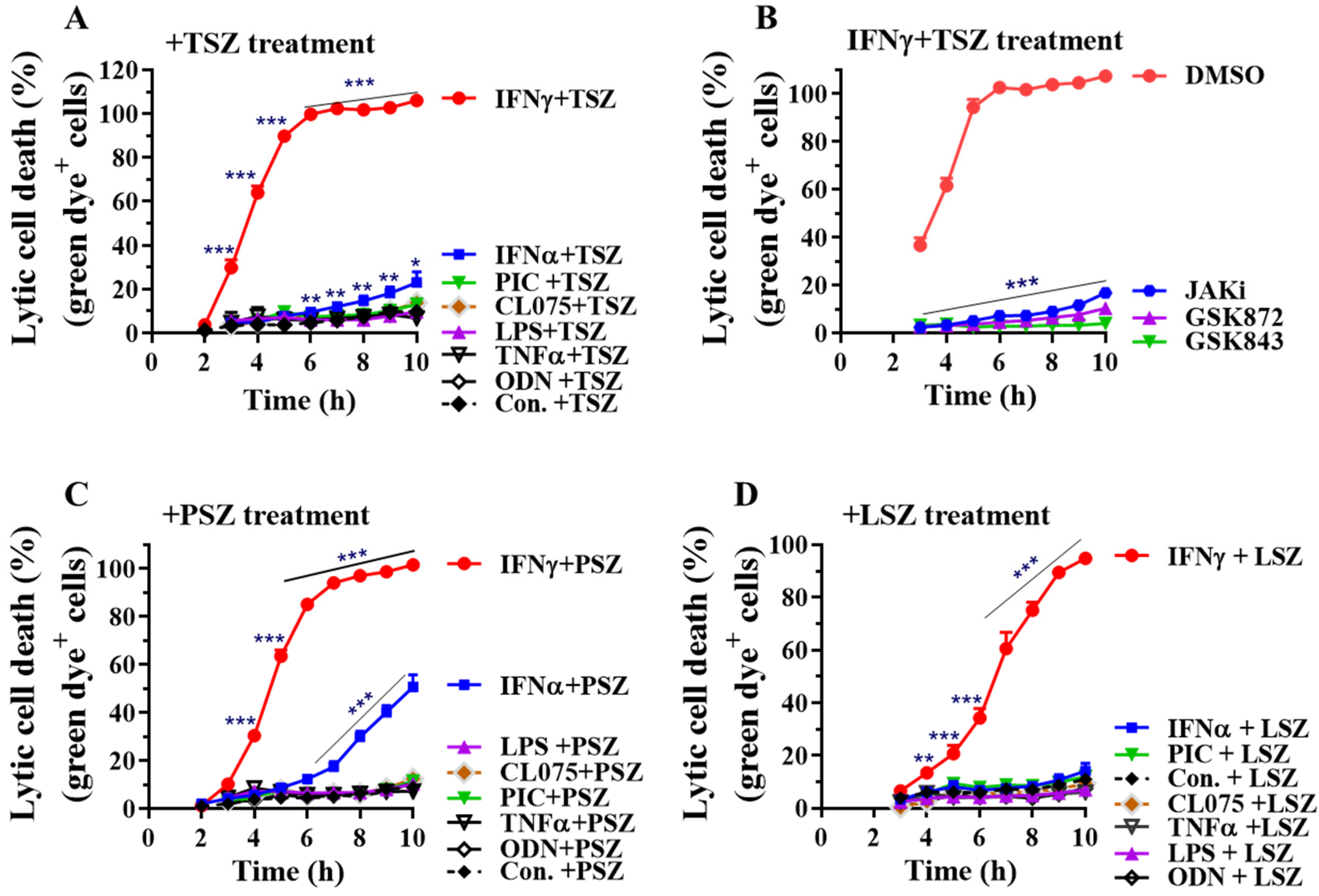
Figure 2.
IFNγ preferentially promotes AEC necroptosis by real-time lytic cell death analysis. Mouse primary AECs were incubated with control PBS (Con.), IFNα2 (50 ng/mL), IFNγ (50 ng/mL), TNFα (25 ng/mL), LPS (100 ng/mL), poly(I:C) (PIC, 0.5 μg/mL), CL075 (1 μg/mL) or ODN1585 (1 μM) for 24 h, then treated in a 0–10 h period with DMSO, or TNFα (15 ng/mL) plus 100 nM Smac mimetic AZD5582 and 25 µM z-VAD-FMK (A, +TSZ), poly(I:C) (0.2 µg/mL) plus 100 nM Smac mimetic and 40 µM z-VAD-FMK (C, +PSZ) or LPS (200 ng/mL) plus 100 nM Smac mimetic AZD5582 and 25 µM z-VAD-FMK (D, +LSZ) in the presence of an asymmetric cyanine dye. (B) Mouse AECs were incubated without or with IFNγ (50 ng/mL) in the presence of DMSO or JAK kinase inhibitor-1 (JAKi, 1 μM) for 24 h, washed and then subjected to TSZ-induced real-time necroptosis assay as described above in the presence of DMSO, GSK872 (5 μM), GSK843 (2 μM) or JAKi (1 μM). The percentage of lytic cell death (green dye+ cells) was calculated by comparison to the maximal fluorescence induced by a lysis buffer in each group and presented as mean ± SE. In panel (A): Con.+TSZ, n = 16; IFNγ+TSZ, n = 15; IFNα+TSZ, n = 11; other treatments, n = 4–6. In panel (B): DMSO, n = 10; others, n = 5–7. In panel (C): Con.+PSZ, n = 15; IFNγ+PSZ, n = 14; IFNα+PSZ, n = 10; other treatments, n = 4–5. In panel (D): Con.+LSZ, n = 8; IFNγ or IFNα+LSZ, n = 6; other treatments, n = 4–5. Two-way ANOVA and unpaired Student’s t test were performed in (A–D). * p < 0.05; ** p < 0.01; *** p < 0.001 versus control (Con.) or DMSO in each group. The orders of each agonist-induced percentage of lytic cell death were listed on the right in panels (A,C,D).
Figure 2.
IFNγ preferentially promotes AEC necroptosis by real-time lytic cell death analysis. Mouse primary AECs were incubated with control PBS (Con.), IFNα2 (50 ng/mL), IFNγ (50 ng/mL), TNFα (25 ng/mL), LPS (100 ng/mL), poly(I:C) (PIC, 0.5 μg/mL), CL075 (1 μg/mL) or ODN1585 (1 μM) for 24 h, then treated in a 0–10 h period with DMSO, or TNFα (15 ng/mL) plus 100 nM Smac mimetic AZD5582 and 25 µM z-VAD-FMK (A, +TSZ), poly(I:C) (0.2 µg/mL) plus 100 nM Smac mimetic and 40 µM z-VAD-FMK (C, +PSZ) or LPS (200 ng/mL) plus 100 nM Smac mimetic AZD5582 and 25 µM z-VAD-FMK (D, +LSZ) in the presence of an asymmetric cyanine dye. (B) Mouse AECs were incubated without or with IFNγ (50 ng/mL) in the presence of DMSO or JAK kinase inhibitor-1 (JAKi, 1 μM) for 24 h, washed and then subjected to TSZ-induced real-time necroptosis assay as described above in the presence of DMSO, GSK872 (5 μM), GSK843 (2 μM) or JAKi (1 μM). The percentage of lytic cell death (green dye+ cells) was calculated by comparison to the maximal fluorescence induced by a lysis buffer in each group and presented as mean ± SE. In panel (A): Con.+TSZ, n = 16; IFNγ+TSZ, n = 15; IFNα+TSZ, n = 11; other treatments, n = 4–6. In panel (B): DMSO, n = 10; others, n = 5–7. In panel (C): Con.+PSZ, n = 15; IFNγ+PSZ, n = 14; IFNα+PSZ, n = 10; other treatments, n = 4–5. In panel (D): Con.+LSZ, n = 8; IFNγ or IFNα+LSZ, n = 6; other treatments, n = 4–5. Two-way ANOVA and unpaired Student’s t test were performed in (A–D). * p < 0.05; ** p < 0.01; *** p < 0.001 versus control (Con.) or DMSO in each group. The orders of each agonist-induced percentage of lytic cell death were listed on the right in panels (A,C,D).

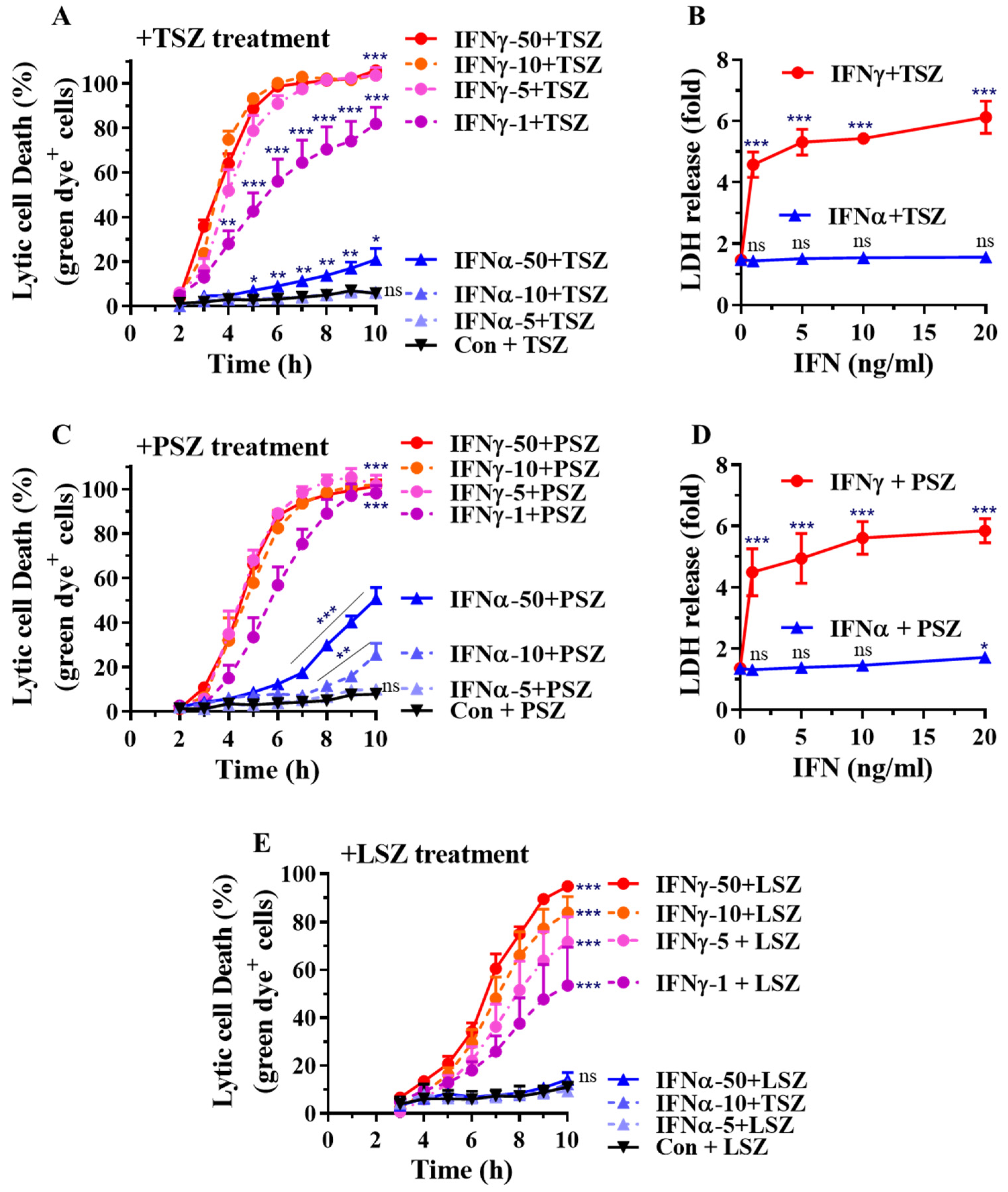
Figure 3.
Dose-dependent effects of IFNγ and IFNα on AEC necroptosis. (A,C,E) Mouse primary AECs were incubated with control PBS (Con.), IFNα2 at 5, 10, or 50 ng/mL, or IFNγ at 1, 5, 10, or 50 ng/mL for 24 h. The cells were then subjected to real-time necroptosis assay as described in Figure 2 in response to TSZ (A), PSZ (C) or LSZ (E). The percentage of lytic cell death (green dye+ cells) was calculated by comparison to the maximal fluorescence induced by a lysis buffer in each group and is presented as mean ± SE. In panel (A): Con., IFNγ-1, IFNγ-10, IFNγ-50, or IFNα-50+TSZ, n = 10; other treatments, n = 4–7. In panel (C): Con., IFNγ-50, or IFNα-50+PSZ, n = 10; other treatments, n = 4–7. In panel (E): Con.+LSZ, n = 8; IFNα-5+LSZ, n = 3; all other treatments, n = 6. Two-way ANOVA and unpaired Student’s t test were performed in (A,C,E). * p < 0.05; ** p < 0.01; *** p < 0.001 versus control (Con.) in each treatment group. NS, no significance. The orders of each agonist-induced percentage of lytic cell death were listed on the right in each panel. (B,D) Mouse primary AECs were incubated with IFNα2 or IFNγ at 0, 1, 5, 10, or 20 ng/mL for 24 h, then treated for 6 h with necroptosis inducers TSZ (B) or PSZ (D). LDH activities in cell supernatant were determined and the relative changes are presented as fold ± SE of individual control groups without treatment of necroptosis inducers. Panel (B), n = 4–7; panel (D), n = 3–6. One-way ANOVA was performed in (B,D). *** p < 0.001 versus control (0 IFN) in each treatment group. NS, no significance.
Figure 3.
Dose-dependent effects of IFNγ and IFNα on AEC necroptosis. (A,C,E) Mouse primary AECs were incubated with control PBS (Con.), IFNα2 at 5, 10, or 50 ng/mL, or IFNγ at 1, 5, 10, or 50 ng/mL for 24 h. The cells were then subjected to real-time necroptosis assay as described in Figure 2 in response to TSZ (A), PSZ (C) or LSZ (E). The percentage of lytic cell death (green dye+ cells) was calculated by comparison to the maximal fluorescence induced by a lysis buffer in each group and is presented as mean ± SE. In panel (A): Con., IFNγ-1, IFNγ-10, IFNγ-50, or IFNα-50+TSZ, n = 10; other treatments, n = 4–7. In panel (C): Con., IFNγ-50, or IFNα-50+PSZ, n = 10; other treatments, n = 4–7. In panel (E): Con.+LSZ, n = 8; IFNα-5+LSZ, n = 3; all other treatments, n = 6. Two-way ANOVA and unpaired Student’s t test were performed in (A,C,E). * p < 0.05; ** p < 0.01; *** p < 0.001 versus control (Con.) in each treatment group. NS, no significance. The orders of each agonist-induced percentage of lytic cell death were listed on the right in each panel. (B,D) Mouse primary AECs were incubated with IFNα2 or IFNγ at 0, 1, 5, 10, or 20 ng/mL for 24 h, then treated for 6 h with necroptosis inducers TSZ (B) or PSZ (D). LDH activities in cell supernatant were determined and the relative changes are presented as fold ± SE of individual control groups without treatment of necroptosis inducers. Panel (B), n = 4–7; panel (D), n = 3–6. One-way ANOVA was performed in (B,D). *** p < 0.001 versus control (0 IFN) in each treatment group. NS, no significance.

Figure 4.
IFNγ predominantly promotes TBEC necroptosis by real time lytic cell death analysis. (A,C,E) Mouse primary TBECs were incubated with PBS (Con.), IFNα2 (50 ng/mL), IFNγ (50 ng/mL), TNFα (25 ng/mL), LPS (100 ng/mL), poly(I:C) (PIC, 0.5 μg/mL), CL075 (1 μg/mL) or ODN1585 (1 μM) for 24 h. (B,D) Mouse primary AECs were incubated with control PBS (Con.), IFNα2 at 5, 10, or 50 ng/mL, or IFNγ at 1, 5, 10, or 50 ng/mL for 24 h. The cells were then treated for 0 to 10 h with necroptosis inducers TSZ (A,B), PSZ (C,D) or LSZ (E) in the presence of an asymmetric cyanine dye. The percentage of lytic cell death (green dye+ cells) was calculated by comparison to the maximal fluorescence induced by a lysis buffer in each group and is presented as mean ± SE. In panel (A): Con. or IFNγ+TSZ, n = 7–8; all other treatments, n = 3. In panel (B): Con. or IFNγ-50+TSZ, n = 7–8; IFNγ-1 or IFNγ-5+TSZ, n = 5; other treatments, n = 3–4. In panel (C): Con. or IFNγ+PSZ, n = 7–8; all other treatments, n = 3. In panel (D): Con. or IFNγ-50+TSZ, n = 7–8; all other treatments, n = 3–4. In panel (E): n = 4–5. Two-way ANOVA was performed in (A–E). * p < 0.05; ** p < 0.01; *** p < 0.001 versus control (Con.) in each treatment group. NS, no significance. The orders of each agonist-induced percentage of lytic cell death were listed on the right in each panel.
Figure 4.
IFNγ predominantly promotes TBEC necroptosis by real time lytic cell death analysis. (A,C,E) Mouse primary TBECs were incubated with PBS (Con.), IFNα2 (50 ng/mL), IFNγ (50 ng/mL), TNFα (25 ng/mL), LPS (100 ng/mL), poly(I:C) (PIC, 0.5 μg/mL), CL075 (1 μg/mL) or ODN1585 (1 μM) for 24 h. (B,D) Mouse primary AECs were incubated with control PBS (Con.), IFNα2 at 5, 10, or 50 ng/mL, or IFNγ at 1, 5, 10, or 50 ng/mL for 24 h. The cells were then treated for 0 to 10 h with necroptosis inducers TSZ (A,B), PSZ (C,D) or LSZ (E) in the presence of an asymmetric cyanine dye. The percentage of lytic cell death (green dye+ cells) was calculated by comparison to the maximal fluorescence induced by a lysis buffer in each group and is presented as mean ± SE. In panel (A): Con. or IFNγ+TSZ, n = 7–8; all other treatments, n = 3. In panel (B): Con. or IFNγ-50+TSZ, n = 7–8; IFNγ-1 or IFNγ-5+TSZ, n = 5; other treatments, n = 3–4. In panel (C): Con. or IFNγ+PSZ, n = 7–8; all other treatments, n = 3. In panel (D): Con. or IFNγ-50+TSZ, n = 7–8; all other treatments, n = 3–4. In panel (E): n = 4–5. Two-way ANOVA was performed in (A–E). * p < 0.05; ** p < 0.01; *** p < 0.001 versus control (Con.) in each treatment group. NS, no significance. The orders of each agonist-induced percentage of lytic cell death were listed on the right in each panel.

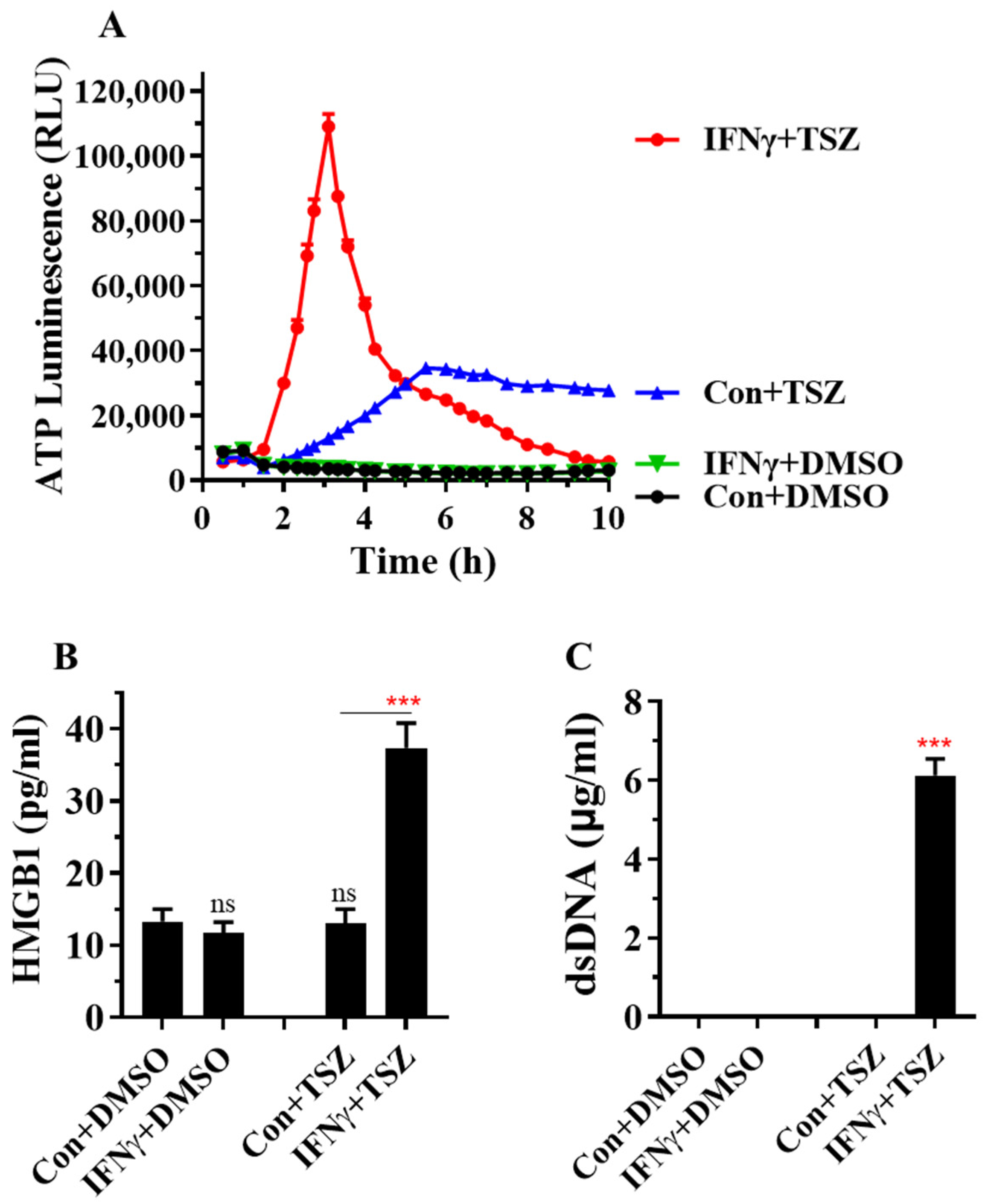
Figure 5.
IFNγ greatly accelerates the release of DAMPs from AECs. (A) Mouse primary AECs were incubated with PBS (Con.) or IFNγ (50 ng/mL) for 24 h, then treated for 0 to 10 h with DMSO or necroptosis inducer TSZ. The release of ATP from AECs was continuously monitored by using RealTime-Glo™ extracellular ATP assay kits, and the levels of ATP luminescence are presented as means ± SE (n = 8). (B,C) Mouse primary AECs were incubated with PBS (Con.) or IFNγ (50 ng/mL) for 24 h, then treated for 6 h with DMSO or necroptosis inducer TSZ. The levels of HMGB1 and dsDNA in cell supernatant were determined and are presented as means ± SE (n = 4). Student’s t test was performed in (B,C). *** p < 0.001 versus control (Con.) in each treatment group. NS, no significance.
Figure 5.
IFNγ greatly accelerates the release of DAMPs from AECs. (A) Mouse primary AECs were incubated with PBS (Con.) or IFNγ (50 ng/mL) for 24 h, then treated for 0 to 10 h with DMSO or necroptosis inducer TSZ. The release of ATP from AECs was continuously monitored by using RealTime-Glo™ extracellular ATP assay kits, and the levels of ATP luminescence are presented as means ± SE (n = 8). (B,C) Mouse primary AECs were incubated with PBS (Con.) or IFNγ (50 ng/mL) for 24 h, then treated for 6 h with DMSO or necroptosis inducer TSZ. The levels of HMGB1 and dsDNA in cell supernatant were determined and are presented as means ± SE (n = 4). Student’s t test was performed in (B,C). *** p < 0.001 versus control (Con.) in each treatment group. NS, no significance.

Figure 6.
MLKL is predominantly induced by IFNγ in primary lung epithelial cells, and MLKL silencing or inhibition largely suppresses the IFNγ-promoted necroptosis. (A) RT-PCR showing the induction of MLKL by IFNγ. Mouse primary AECs and TBECs or Raw264.7 macrophages were treated with IFNγ (50 ng/mL) for 24 h, and mRNA levels of MLKL and GAPDH are shown in a representative RT-PCR. (B–D) Western blots showing the expression of MLKL. Primary mouse AECs were incubated with control PBS (Con.), IFNα2 (50 ng/mL), IFNγ (50 ng/mL), LPS (100 ng/mL), poly(I:C) (PIC, 0.5 μg/mL), TNFα (25 ng/mL), TNFα (25 ng/mL) plus IFNγ (50 ng/mL) (T+γ), TNFα (25 ng/mL) plus IFNα (50 ng/mL) (T+α), CL075 (1 μg/mL), ODN1585 (1 μM) or IL-4 (15 ng/mL) for 24 h. Equal amounts of cell lysates were subjected to Western blot analysis. Results represent the findings of 3 independent experiments. (E) MLKL knockdown by siRNA. Primary mouse AECs were transfected with 20 nM control siRNA (CS) or MLKL siRNA (MS) for 6 h, then treated with control PBS (Con) or IFNγ (25 ng/mL) for 24 h. Equal amounts of cell lysates were subjected to Western blots. Results represent the findings of 3 independent experiments. (F) MLKL silencing largely suppresses the IFNγ-promoted necroptosis. Primary mouse AECs were transfected with control siRNA or MLKL siRNA for 6 h, incubated with IFNγ (25 ng/mL) for 24 h, then subjected to TSZ-induced real-time necroptosis assay as described in Figure 2. * p < 0.05; ** p < 0.01; *** p < 0.001 versus control siRNA in each time point (n = 4). (G,H) Effects of MLKL or CaMKII inhibitors on necroptosis. Mouse AECs were incubated without or with IFNγ (25 ng/mL) for 24 h, then subjected to TSZ-induced real-time necroptosis assay as described above in the presence of DMSO, GW806742X at 1 μM (GW42X-1), 1.5 μM (GW42X-1.5), 2 μM (GW42X-2) or 2 μM plus 1 μM Myr-AIP (GW42X+AIP), or Myr-AIP at 1 μM (AIP-1) or 5 μM (AIP-5) over a 0–10 h period (G) or at 24 h of TSZ treatment (H). In panel (G): DMSO, n = 12; GW42X-1, n = 9; GW42X-1.5, n = 6; all others, n = 4. In panel (H): DMSO, n = 8; GW42X-1 or GW42X-1.5, n = 6; all others, n = 4. * p < 0.05; ** p < 0.01; *** p < 0.001 versus DMSO in each group. NS, no significance. Two-way ANOVA was performed in (F,G) and one-way ANOVA was performed in (H).
Figure 6.
MLKL is predominantly induced by IFNγ in primary lung epithelial cells, and MLKL silencing or inhibition largely suppresses the IFNγ-promoted necroptosis. (A) RT-PCR showing the induction of MLKL by IFNγ. Mouse primary AECs and TBECs or Raw264.7 macrophages were treated with IFNγ (50 ng/mL) for 24 h, and mRNA levels of MLKL and GAPDH are shown in a representative RT-PCR. (B–D) Western blots showing the expression of MLKL. Primary mouse AECs were incubated with control PBS (Con.), IFNα2 (50 ng/mL), IFNγ (50 ng/mL), LPS (100 ng/mL), poly(I:C) (PIC, 0.5 μg/mL), TNFα (25 ng/mL), TNFα (25 ng/mL) plus IFNγ (50 ng/mL) (T+γ), TNFα (25 ng/mL) plus IFNα (50 ng/mL) (T+α), CL075 (1 μg/mL), ODN1585 (1 μM) or IL-4 (15 ng/mL) for 24 h. Equal amounts of cell lysates were subjected to Western blot analysis. Results represent the findings of 3 independent experiments. (E) MLKL knockdown by siRNA. Primary mouse AECs were transfected with 20 nM control siRNA (CS) or MLKL siRNA (MS) for 6 h, then treated with control PBS (Con) or IFNγ (25 ng/mL) for 24 h. Equal amounts of cell lysates were subjected to Western blots. Results represent the findings of 3 independent experiments. (F) MLKL silencing largely suppresses the IFNγ-promoted necroptosis. Primary mouse AECs were transfected with control siRNA or MLKL siRNA for 6 h, incubated with IFNγ (25 ng/mL) for 24 h, then subjected to TSZ-induced real-time necroptosis assay as described in Figure 2. * p < 0.05; ** p < 0.01; *** p < 0.001 versus control siRNA in each time point (n = 4). (G,H) Effects of MLKL or CaMKII inhibitors on necroptosis. Mouse AECs were incubated without or with IFNγ (25 ng/mL) for 24 h, then subjected to TSZ-induced real-time necroptosis assay as described above in the presence of DMSO, GW806742X at 1 μM (GW42X-1), 1.5 μM (GW42X-1.5), 2 μM (GW42X-2) or 2 μM plus 1 μM Myr-AIP (GW42X+AIP), or Myr-AIP at 1 μM (AIP-1) or 5 μM (AIP-5) over a 0–10 h period (G) or at 24 h of TSZ treatment (H). In panel (G): DMSO, n = 12; GW42X-1, n = 9; GW42X-1.5, n = 6; all others, n = 4. In panel (H): DMSO, n = 8; GW42X-1 or GW42X-1.5, n = 6; all others, n = 4. * p < 0.05; ** p < 0.01; *** p < 0.001 versus DMSO in each group. NS, no significance. Two-way ANOVA was performed in (F,G) and one-way ANOVA was performed in (H).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hao, Q.; Shetty, S.; Tucker, T.A.; Idell, S.; Tang, H. Interferon-γ Preferentially Promotes Necroptosis of Lung Epithelial Cells by Upregulating MLKL. Cells 2022, 11, 563. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030563
AMA Style
Hao Q, Shetty S, Tucker TA, Idell S, Tang H. Interferon-γ Preferentially Promotes Necroptosis of Lung Epithelial Cells by Upregulating MLKL. Cells. 2022; 11(3):563. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030563
Chicago/Turabian StyleHao, Qin, Sreerama Shetty, Torry A. Tucker, Steven Idell, and Hua Tang. 2022. "Interferon-γ Preferentially Promotes Necroptosis of Lung Epithelial Cells by Upregulating MLKL" Cells 11, no. 3: 563. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030563
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.