
The Pivotal Role of the Placenta in Normal and Pathological Pregnancies: A Focus on Preeclampsia, Fetal Growth Restriction, and Maternal Chronic Venous Disease

 , , , ,
, , , ,  , , ,
, , , 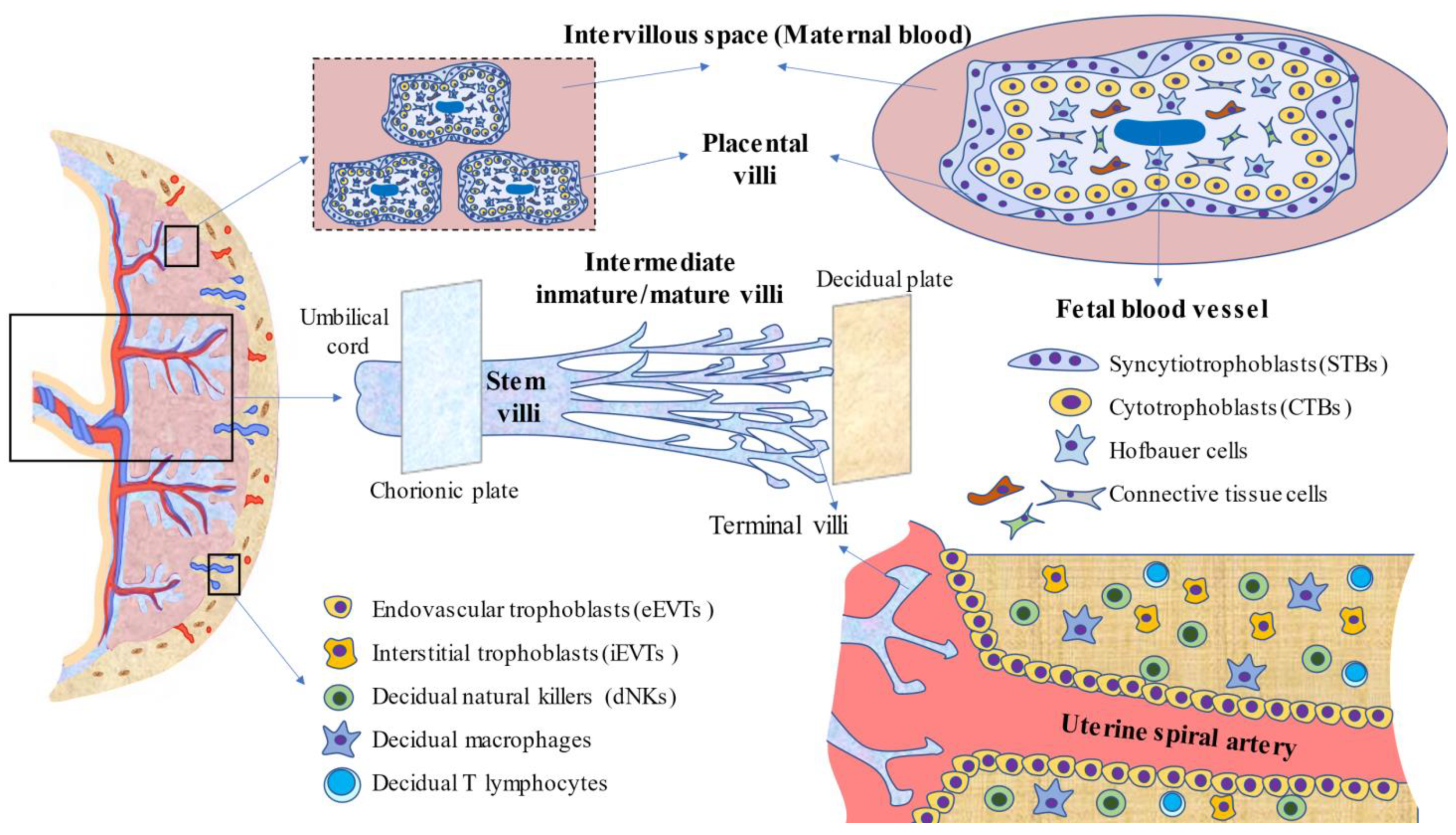{kind=link}
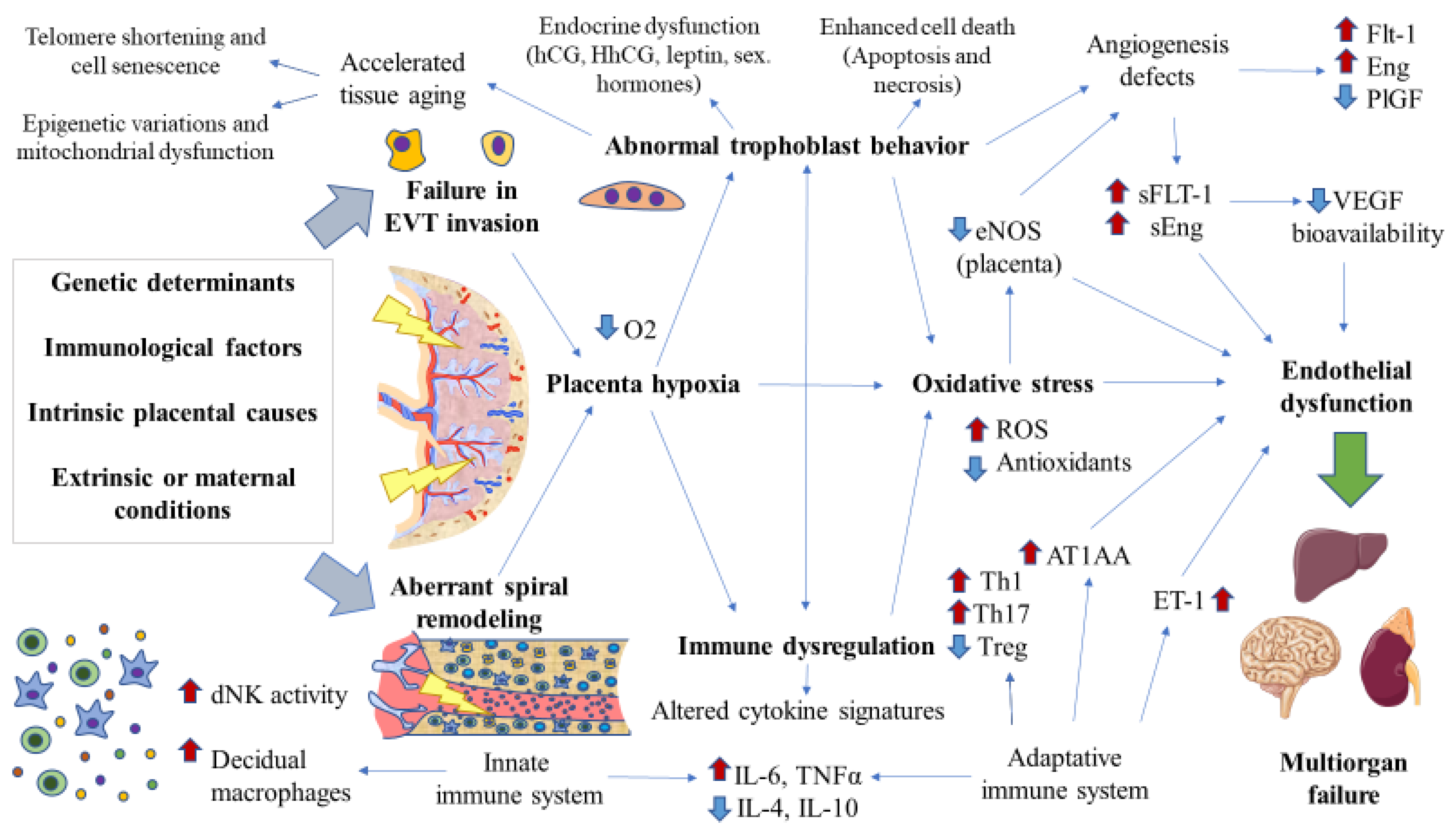{kind=link}
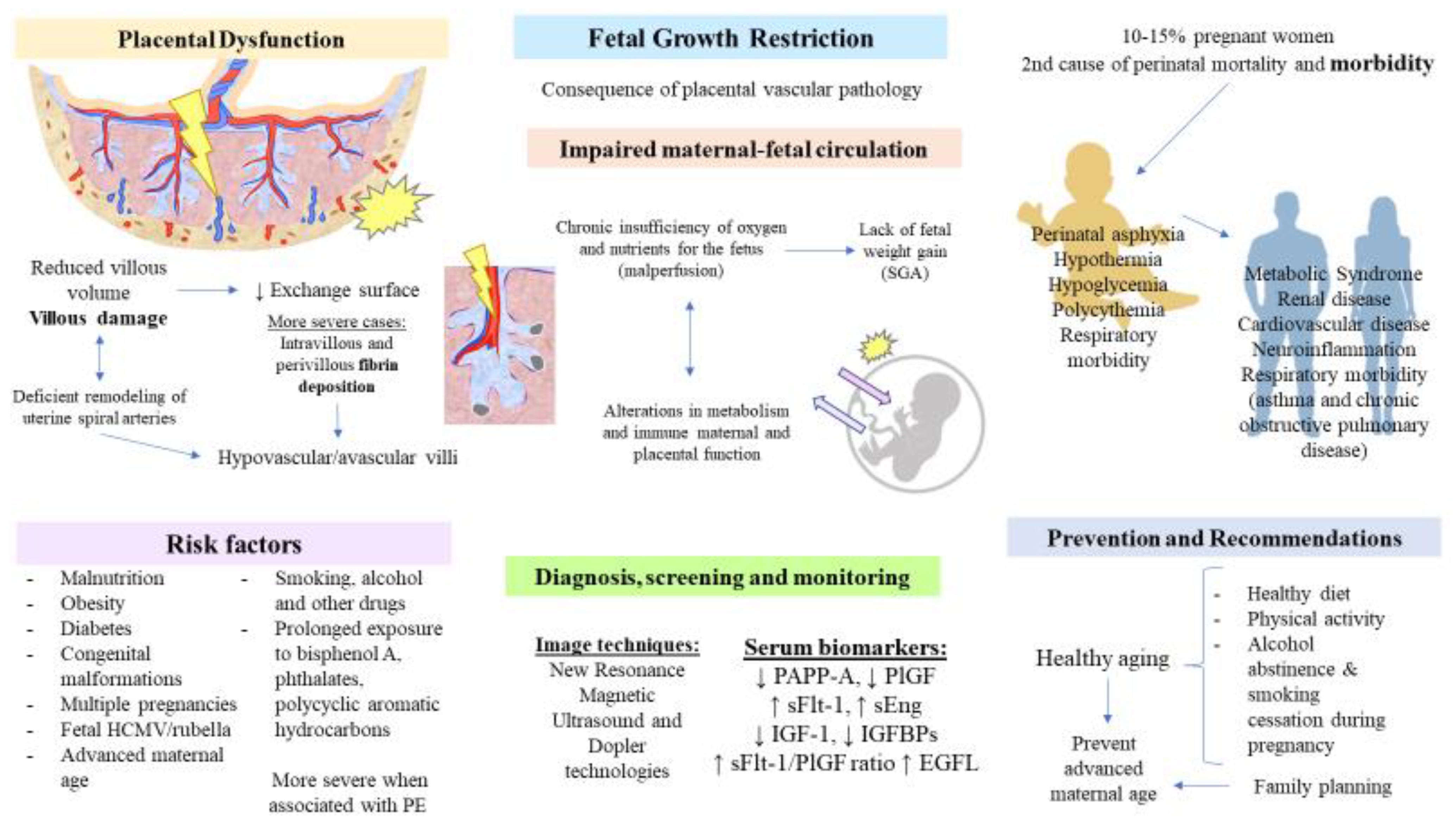{kind=link}
Abstract
:1. Introduction
2. Placental Development, Cytoarchitecture, and Immunology
2.1. Early Development of the Placenta
2.2. Placental Anatomy and Cytoarchitecture
2.3. Immunology of the Placenta
2.4. Function and Activity of the Placenta during Pregnancy
2.4.1. Placenta in Maternofetal Exchange
2.4.2. Endocrine Activity of the Placenta
2.4.3. Placental Barrier
2.4.4. Placenta and Maternofetal Programming
3. Describing the Placenta in Pathological Conditions
3.1. The Role of Placenta in Preeclampsia
3.1.1. Introduction
3.1.2. Preventive and Therapeutic Approaches
3.1.3. Pathophysiology of Early Onset/Placental Preeclampsia
Defective Spiral Artery Remodeling and Trophoblast Invasion
The Antiangiogenic Status
Vascular Inflammation
Oxidative Stress
Endocrine Disruption
Placental Aging and Damage
3.1.4. Pathophysiology of Late-Onset Preeclampsia
3.2. Placenta in Fetal Growth Restriction
3.2.1. Introduction
3.2.2. Pathophysiological Role of the Placenta
3.2.3. Screening, Predictive, and Diagnostic Biomarkers
3.2.4. Preventive and Therapeutic Approaches
3.3. Chronic Venous Disease, Clinical Manifestations, and Repercussions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gude, N.M.; Roberts, C.T.; Kalionis, B.; King, R.G. Growth and function of the normal human placenta. Thromb. Res. 2004, 114, 397–407. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. What is the placenta? Am. J. Obstet. Gynecol. 2015, 213, S6.e1–S6.e4. [Google Scholar] [CrossRef] [Green Version]
- Huppertz, B. Placental pathology in pregnancy complications. Thromb. Res. 2011, 127, S96–S99. [Google Scholar] [CrossRef]
- Konkel, L. Lasting Impact of an Ephemeral Organ: The Role of the Placenta in Fetal Programming. Environ. Health Perspect. 2016, 124, A124–A129. [Google Scholar] [CrossRef] [Green Version]
- Thornburg, K.L.; Marshall, N. The placenta is the center of the chronic disease universe. Am. J. Obstet. Gynecol. 2015, 213, S14–S20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttmacher, A.; Maddox, Y.; Spong, C. The Human Placenta Project: Placental structure, development, and function in real time. Placenta 2014, 35, 303–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.-M.; Kim, J.-S. A Review of Mechanisms of Implantation. Dev. Reprod. 2017, 21, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Staud, F.; Karahoda, R. Trophoblast: The central unit of fetal growth, protection and programming. Int. J. Biochem. Cell Biol. 2018, 105, 35–40. [Google Scholar] [CrossRef]
- Ramathal, C.Y.; Bagchi, I.C.; Taylor, R.N.; Bagchi, M.K. Endometrial Decidualization: Of Mice and Men. Semin. Reprod. Med. 2010, 28, 017–026. [Google Scholar] [CrossRef] [Green Version]
- Riley, J.K. Trophoblast Immune Receptors in Maternal-Fetal Tolerance. Immunol. Investig. 2008, 37, 395–426. [Google Scholar] [CrossRef]
- Huppertz, B. The anatomy of the normal placenta. J. Clin. Pathol. 2008, 61, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- McMaster, M.T.; Fisher, S.J. Placental Development. In Encyclopedia of Hormones; Academic Press: Cambridge, MA, USA, 2003; pp. 213–219. [Google Scholar] [CrossRef]
- Carter, A.M. When is the maternal placental circulation established in man? Placenta 1997, 18, 83–87. [Google Scholar] [CrossRef]
- Jaffe, R.; Jauniaux, E.; Hustin, J. Maternal circulation in the first-trimester human placenta—Myth or reality? Am. J. Obstet. Gynecol. 1997, 176, 695–705. [Google Scholar] [CrossRef]
- Menon, R.; Moore, J.J. Fetal Membranes, Not a Mere Appendage of the Placenta, but a Critical Part of the Fetal-Maternal Interface Controlling Parturition. Obstet. Gynecol. Clin. N. Am. 2020, 47, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, P.; Black, S.; Huppertz, B. Endovascular Trophoblast Invasion: Implications for the Pathogenesis of Intrauterine Growth Retardation and Preeclampsia. Biol. Reprod. 2003, 69, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Castellucci, M.; Kosanke, G.; Verdenelli, F.; Huppertz, B.; Kaufmann, P. Villous sprouting: Fundamental mechanisms of human placental development. Hum. Reprod. Updat. 2000, 6, 485–494. [Google Scholar] [CrossRef] [Green Version]
- Turco, M.Y.; Moffett, A. Development of the human placenta. Development 2019, 146, dev163428. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, K.M.; Yaktine, A.L. Institute of Medicine (US) and National Research Council (US) Committee to Reexamine IOM Pregnancy Weight Guidelines; National Academies Press (US): Washington, DC, USA, 2009. [Google Scholar]
- Fadl, S.; Moshiri, M.; Fligner, C.L.; Katz, D.S.; Dighe, M. Placental Imaging: Normal Appearance with Review of Pathologic Findings. RadioGraphics 2017, 37, 979–998. [Google Scholar] [CrossRef]
- Architecture of Normal Villous Trees. In Pathology of the Human Placenta; Springer: Berlin/Heidelberg, Germany, 2006; pp. 121–173. [CrossRef]
- Huppertz, B. Human Placentation. In Encyclopedia of Reproduction; Academic Press: Cambridge, MA, USA, 2018; pp. 431–439. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, S. Structure of the Placenta. In Vascular Biology of the Placenta; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2010. [Google Scholar]
- Aplin, J.D.; Lewis, R.M.; Jones, C.J. Development of the Human Placental Villus. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar] [CrossRef]
- Barker, D.; Osmond, C.; Grant, S.; Thornburg, K.; Cooper, C.; Ring, S.; Davey-Smith, G. Maternal cotyledons at birth predict blood pressure in childhood. Placenta 2013, 34, 672–675. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, P.; Huppertz, B.; Frank, H.-G. The fibrinoids of the human placenta: Origin, composition and functional relevance. Ann. Anat. Anat. Anz. 1996, 178, 485–501. [Google Scholar] [CrossRef]
- Mor, G.; Cardenas, I. The Immune System in Pregnancy: A Unique Complexity. Am. J. Reprod. Immunol. 2010, 63, 425–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, M.; Abrahams, V.M. Immunology of the Placenta. Obstet. Gynecol. Clin. N. Am. 2020, 47, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Sanguansermsri, D.; Pongcharoen, S. Pregnancy Immunology: Decidual Immune Cells. Asian Pac. J. Allergy Immunol. 2008, 26, 171–181. [Google Scholar] [PubMed]
- Hoo, R.; Nakimuli, A.; Vento-Tormo, R. Innate Immune Mechanisms to Protect Against Infection at the Human Decidual-Placental Interface. Front. Immunol. 2020, 11, 2070. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, S.; Zhao, Y.; Wang, H.; Pan, Q.; Shao, Q. Decidual Natural Killer Cells: A Good Nanny at the Maternal-Fetal Interface During Early Pregnancy. Front. Immunol. 2021, 12, 1684. [Google Scholar] [CrossRef]
- Ander, S.E.; Diamond, M.S.; Coyne, C.B. Immune responses at the maternal-fetal interface. Sci. Immunol. 2019, 4, eaat6114. [Google Scholar] [CrossRef]
- Shmeleva, E.V.; Colucci, F. Maternal natural killer cells at the intersection between reproduction and mucosal immunity. Mucosal Immunol. 2021, 14, 991–1005. [Google Scholar] [CrossRef]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.-E.; Stephenson, E.; Polański, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal–fetal interface in humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Wang, H. Macrophage subsets at the maternal-fetal interface. Cell. Mol. Immunol. 2020, 17, 889–891. [Google Scholar] [CrossRef]
- Ning, F.; Liu, H.; Lash, G.E. The Role of Decidual Macrophages During Normal and Pathological Pregnancy. Am. J. Reprod. Immunol. 2016, 75, 298–309. [Google Scholar] [CrossRef]
- Lissauer, D.; Kilby, M.; Moss, P. Maternal effector T cells within decidua: The adaptive immune response to pregnancy? Placenta 2017, 60, 140–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nancy, P.; Erlebacher, A. T cell behavior at the maternal-fetal interface. Int. J. Dev. Biol. 2014, 58, 189–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilburgs, T.; Strominger, J.L. CD8+ Effector T Cells at the Fetal-Maternal Interface, Balancing Fetal Tolerance and Antiviral Immunity. Am. J. Reprod. Immunol. 2013, 69, 395–407. [Google Scholar] [CrossRef] [Green Version]
- Shima, T.; Sasaki, Y.; Itoh, M.; Nakashima, A.; Ishii, N.; Sugamura, K.; Saito, S. Regulatory T cells are necessary for implantation and maintenance of early pregnancy but not late pregnancy in allogeneic mice. J. Reprod. Immunol. 2010, 85, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Mjösberg, J.; Berg, G.; Jenmalm, M.; Ernerudh, J. FOXP3+ Regulatory T Cells and T Helper 1, T Helper 2, and T Helper 17 Cells in Human Early Pregnancy Decidua1. Biol. Reprod. 2010, 82, 698–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mor, G.; Cardenas, I.; Abrahams, V.; Guller, S. Inflammation and pregnancy: The role of the immune system at the implantation site. Ann. N. Y. Acad. Sci. 2011, 1221, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Burton, G.J.; Fowden, A.L.; Thornburg, K.L. Placental Origins of Chronic Disease. Physiol. Rev. 2016, 96, 1509–1565. [Google Scholar] [CrossRef] [PubMed]
- Sibley, C.P.; Birdsey, T.J.; Brownbill, P.; Clarson, L.H.; Doughty, I.; Glazier, J.D.; Greenwood, S.L.; Hughes, J.; Janssont, T.; Mylona, P.; et al. Mechanisms of maternofetal exchange across the human placenta. Biochem. Soc. Trans. 1998, 26, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Sibley, C.P.; Brownbill, P.; Glazier, J.D.; Greenwood, S.L. Knowledge needed about the exchange physiology of the placenta. Placenta 2018, 64, S9–S15. [Google Scholar] [CrossRef]
- Hay, J.W.W. Placental Transport of Nutrients to the Fetus. Horm. Res. 1994, 42, 215–222. [Google Scholar] [CrossRef]
- Hay, W.W. The Placenta: Not Just a Conduit for Maternal Fuels. Diabetes 1991, 40, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Larqué, E.; Ruiz-Palacios, M.; Koletzko, B. Placental regulation of fetal nutrient supply. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Jansson, T.; Aye, I.; Goberdhan, D. The emerging role of mTORC1 signaling in placental nutrient-sensing. Placenta 2012, 33, e23–e29. [Google Scholar] [CrossRef] [Green Version]
- Carter, A.M. Placental Gas Exchange and the Oxygen Supply to the Fetus. Compr. Physiol. 2015, 5, 1381–1403. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.J. Oxygen delivery and fetal-placental growth: Beyond a question of supply and demand? Placenta 2012, 33, e16–e22. [Google Scholar] [CrossRef]
- Serov, A.S.; Salafia, C.; Grebenkov, D.S.; Filoche, M. The role of morphology in mathematical models of placental gas exchange. J. Appl. Physiol. 2016, 120, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Barapatre, N.; Haeussner, E.; Grynspan, D.; Schmitz, C.; Von Koch, F.E.; Frank, H.-G. The Density of Cell Nuclei at the Materno-Fetal Exchange Barrier is Sexually Dimorphic in Normal Placentas, but not in IUGR. Sci. Rep. 2019, 9, 2359. [Google Scholar] [CrossRef]
- Christians, J.K. The Placenta’s Role in Sexually Dimorphic Fetal Growth Strategies. Reprod. Sci. 2021, 1–13. [Google Scholar] [CrossRef]
- Burton, G. Deportation of syncytial sprouts from the term human placenta. Placenta 2011, 32, 96–98. [Google Scholar] [CrossRef]
- Huppertz, B. Placental Origins of Preeclampsia: Challenging the Current Hypothesis. Hypertension 2008, 51, 970–975. [Google Scholar] [CrossRef]
- Chamley, L.; Holland, O.; Chen, Q.; Viall, C.; Stone, P.; Abumaree, M. Review: Where is the maternofetal interface? Placenta 2013, 35, S74–S80. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Brkić, J.; Liu, M.; Fu, G.; Peng, C.; Wang, Y.-L. Placental trophoblast cell differentiation: Physiological regulation and pathological relevance to preeclampsia. Mol. Asp. Med. 2013, 34, 981–1023. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.J.; Aplin, J.D. A re-examination of the origins of placental bed giant cells. Placenta 2021, 114, 39–41. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.A. The endocrine function of human placenta: An overview. Reprod. Biomed. Online 2016, 32, 14–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomes, J.; Au, F.; Basak, A.; Cakmak, S.; Vincent, R.; Kumarathasan, P. Maternal blood biomarkers and adverse pregnancy outcomes: A systematic review and meta-analysis. Crit. Rev. Toxicol. 2019, 49, 461–478. [Google Scholar] [CrossRef]
- Heerema-McKenney, A. Defense and infection of the human placenta. APMIS 2018, 126, 570–588. [Google Scholar] [CrossRef] [Green Version]
- Ortega, M.A.; Fraile-Martínez, O.; García-Montero, C.; García-Gallego, S.; Sánchez-Trujillo, L.; Torres-Carranza, D.; Álvarez-Mon, M.; Pekarek, L.; García-Honduvilla, N.; Bujan, J.; et al. An integrative look at SARS-CoV-2 (Review). Int. J. Mol. Med. 2020, 47, 415–434. [Google Scholar] [CrossRef]
- Cuñarro-López, Y.; Cano-Valderrama, Ó.; Pintado-Recarte, P.; Cueto-Hernández, I.; González-Garzón, B.; García-Tizón, S.; Bujan, J.; Asúnsolo, Á.; Ortega, M.A.; De León-Luis, J.A. Maternal and Perinatal Outcomes in Patients with Suspected COVID-19 and Their Relationship with a Negative RT-PCR Result. J. Clin. Med. 2020, 9, 3552. [Google Scholar] [CrossRef]
- Zaga-Clavellina, V.; Diaz, L.; Olmos-Ortiz, A.; Godínez-Rubí, M.; Rojas-Mayorquín, A.E.; Ortuño-Sahagún, D. Central role of the placenta during viral infection: Immuno-competences and miRNA defensive responses. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2021, 1867, 166182. [Google Scholar] [CrossRef]
- Kreis, N.-N.; Ritter, A.; Louwen, F.; Yuan, J. A Message from the Human Placenta: Structural and Immunomodulatory Defense against SARS-CoV-2. Cells 2020, 9, 1777. [Google Scholar] [CrossRef]
- Pelzer, E.; Gomez-Arango, L.F.; Barrett, H.L.; Nitert, M.D. Review: Maternal health and the placental microbiome. Placenta 2016, 54, 30–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myllynen, P.; Pasanen, M.; Vähäkangas, K. The fate and effects of xenobiotics in human placenta. Expert Opin. Drug Metab. Toxicol. 2007, 3, 331–346. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.J.; Fowden, A.L. The placenta: A multifaceted, transient organ. Philos. Trans. R. Soc. B Biol. Sci. 2015, 370, 20140066. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Castañeda, R.; Galaviz-Hernández, C.; Souto, P.C.D.S.; Lima, V.V.; Giachini, F.R.; Escudero, C.; Damiano, A.E.; Barragán-Zúñiga, L.J.; Martínez-Aguilar, G.; Sosa-Macías, M. The role of xenobiotic-metabolizing enzymes in the placenta: A growing research field. Expert Rev. Clin. Pharmacol. 2020, 13, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Hakkola, J.; Pelkonen, O.; Pasanen, M.; Raunio, H. Xenobiotic-Metabolizing Cytochrome P450 Enzymes in the Human Feto-Placental Unit: Role in Intrauterine Toxicity. Crit. Rev. Toxicol. 1998, 28, 35–72. [Google Scholar] [CrossRef]
- St-Pierre, M.V.; Serrano, M.A.; Macias, R.I.R.; Dubs, U.; Hoechli, M.; Lauper, U.; Meier, P.J.; Marin, J.J.G. Expression of members of the multidrug resistance protein family in human term placenta. Am. J. Physiol. Integr. Comp. Physiol. 2000, 279, R1495–R1503. [Google Scholar] [CrossRef]
- Ceckova-Novotna, M.; Pavek, P.; Staud, F. P-glycoprotein in the placenta: Expression, localization, regulation and function. Reprod. Toxicol. 2006, 22, 400–410. [Google Scholar] [CrossRef]
- Martineau, M.; Papacleovoulou, G.; Abu-Hayyeh, S.; Dixon, P.; Ji, H.; Powrie, R.; Larson, L.; Chien, E.; Williamson, C. Cholestatic pregnancy is associated with reduced placental 11βHSD2 expression. Placenta 2013, 35, 37–43. [Google Scholar] [CrossRef]
- Stark, M.J.; Wright, I.M.R.; Clifton, V.L. Sex-specific alterations in placental 11β-hydroxysteroid dehydrogenase 2 activity and early postnatal clinical course following antenatal betamethasone. Am. J. Physiol. Integr. Comp. Physiol. 2009, 297, R510–R514. [Google Scholar] [CrossRef]
- Duhig, K.; Chappell, L.C.; Shennan, A.H. Oxidative stress in pregnancy and reproduction. Obstet. Med. 2016, 9, 113–116. [Google Scholar] [CrossRef] [Green Version]
- Poston, L.; Raijmakers, M. Trophoblast Oxidative Stress, Antioxidants and Pregnancy Outcome—A Review. Placenta 2004, 25, S72–S78. [Google Scholar] [CrossRef]
- Jones, M.L.; Mark, P.J.; Lewis, J.L.; Mori, T.A.; Keelan, J.A.; Waddell, B.J. Antioxidant Defenses in the Rat Placenta in Late Gestation: Increased Labyrinthine Expression of Superoxide Dismutases, Glutathione Peroxidase 3, and Uncoupling Protein 21. Biol. Reprod. 2010, 83, 254–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfrey, K.M. The Role of the Placenta in Fetal Programming—A Review. Placenta 2002, 23, S20–S27. [Google Scholar] [CrossRef] [PubMed]
- Myatt, L. Placental adaptive responses and fetal programming. J. Physiol. 2006, 572, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.; Cleal, J.; Hanson, M. Review: Placenta, evolution and lifelong health. Placenta 2012, 33, S28–S32. [Google Scholar] [CrossRef]
- Belkacemi, L.; Nelson, D.M.; Desai, M.; Ross, M.G. Maternal Undernutrition Influences Placental-Fetal Development1. Biol. Reprod. 2010, 83, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Vaag, A.A.; Grunnet, L.G.; Arora, G.P.; Brøns, C. The thrifty phenotype hypothesis revisited. Diabetologia 2012, 55, 2085–2088. [Google Scholar] [CrossRef] [Green Version]
- Şanlı, E.; Kabaran, S. Maternal Obesity, Maternal Overnutrition and Fetal Programming: Effects of Epigenetic Mechanisms on the Development of Metabolic Disorders. Curr. Genom. 2019, 20, 419–427. [Google Scholar] [CrossRef]
- Connor, K.L.; Kibschull, M.; Matysiak-Zablocki, E.; Nguyen, T.T.-T.N.; Matthews, S.G.; Lye, S.J.; Bloise, E. Maternal malnutrition impacts placental morphology and transporter expression: An origin for poor offspring growth. J. Nutr. Biochem. 2020, 78, 108329. [Google Scholar] [CrossRef]
- Ramírez-Vélez, R.; Bustamante, J.; Czerniczyniec, A.; De Plata, A.C.A.; Lores-Arnaiz, S. Effect of Exercise Training on Enos Expression, NO Production and Oxygen Metabolism in Human Placenta. PLoS ONE 2013, 8, e80225. [Google Scholar] [CrossRef] [Green Version]
- Clapp, J. Influence of Endurance Exercise and Diet on Human Placental Development and Fetal Growth. Placenta 2006, 27, 527–534. [Google Scholar] [CrossRef]
- Dahlerup, B.R.; Egsmose, E.L.; Siersma, V.; Mortensen, E.L.; Hedegaard, M.; Knudsen, L.E.; Mathiesen, L. Maternal stress and placental function, a study using questionnaires and biomarkers at birth. PLoS ONE 2018, 13, e0207184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bronson, S.L.; Bale, T.L. The Placenta as a Mediator of Stress Effects on Neurodevelopmental Reprogramming. Neuropsychopharmacology 2015, 41, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, C.; Sanchez, S.E.; Gelaye, B.; Enquobahrie, D.A.; Ananth, C.V.; Williams, M.A. Maternal sleep duration and complaints of vital exhaustion during pregnancy is associated with placental abruption. J. Matern. Neonatal Med. 2014, 28, 350–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soma-Pillay, P.; Nelson-Piercy, C.; Tolppanen, H.; Mebazaa, A. Physiological changes in pregnancy. Cardiovasc. J. Afr. 2016, 27, 89–94. [Google Scholar] [CrossRef] [Green Version]
- Sanghavi, M.; Rutherford, J.D. Cardiovascular Physiology of Pregnancy. Circulation 2014, 130, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Parks, W.T.; Catov, J.M. The Placenta as a Window to Maternal Vascular Health. Obstet. Gynecol. Clin. N. Am. 2019, 47, 17–28. [Google Scholar] [CrossRef]
- Del Gobbo, G.; Konwar, C.; Robinson, W.P. The significance of the placental genome and methylome in fetal and maternal health. Qual. Life Res. 2019, 139, 1183–1196. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.A.; Gallagher, K.; Beck, C.; Kumar, R.; Gernand, A.D. Maternal-Fetal Inflammation in the Placenta and the Developmental Origins of Health and Disease. Front. Immunol. 2020, 11, 531543. [Google Scholar] [CrossRef]
- Yeung, E.; Saha, A.; Zhu, C.; Trinh, M.; Hinkle, S.; Pollack, A.; Grantz, K.; Mills, J.; Mumford, S.; Zhang, C.; et al. Placental characteristics and risks of maternal mortality 50 years after delivery. Placenta 2021, 117, 194–199. [Google Scholar] [CrossRef]
- Hutcheon, J.A.; Lisonkova, S.; Joseph, K. Epidemiology of pre-eclampsia and the other hypertensive disorders of pregnancy. Best Pr. Res. Clin. Obstet. Gynaecol. 2011, 25, 391–403. [Google Scholar] [CrossRef] [PubMed]
- Filipek, A.; Jurewicz, E. Preeclampsia—A Disease of Pregnant Women. Postepy Biochem. 2018, 64, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.A.; Magee, L.A.; Kenny, L.C.; Karumanchi, S.A.; McCarthy, F.; Saito, S.; Hall, D.R.; Warren, C.E.; Adoyi, G.; Ishaku, S. Hypertensive Disorders of Pregnancy. Hypertension 2018, 72, 24–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gestational Hypertension and Preeclampsia: ACOG Practice Bulletin, Number 222. Obstet. Gynecol. 2020, 135, e237–e260. [CrossRef] [PubMed]
- Kenny, L.; English, F.; McCarthy, F. Risk factors and effective management of preeclampsia. Integr. Blood Press. Control 2015, 8, 7–12. [Google Scholar] [CrossRef] [Green Version]
- Paré, E.; Parry, S.; McElrath, T.F.; Pucci, D.; Newton, A.; Lim, K.-H. Clinical Risk Factors for Preeclampsia in the 21st Century. Obstet. Gynecol. 2014, 124, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Tranquilli, A.L. Introduction to ISSHP new classification of preeclampsia. Pregnancy Hypertens. 2013, 3, 58–59. [Google Scholar] [CrossRef]
- Raymond, D.; Peterson, E. A Critical Review of Early-Onset and Late-Onset Preeclampsia. Obstet. Gynecol. Surv. 2011, 66, 497–506. [Google Scholar] [CrossRef]
- Bokslag, A.; van Weissenbruch, M.; Mol, B.W.; de Groot, C.J.M. Preeclampsia; short and long-term consequences for mother and neonate. Early Hum. Dev. 2016, 102, 47–50. [Google Scholar] [CrossRef]
- Wallace, K.; Harris, S.; Addison, A.; Bean, C. HELLP Syndrome: Pathophysiology and Current Therapies. Curr. Pharm. Biotechnol. 2018, 19, 816–826. [Google Scholar] [CrossRef]
- Berhan, Y. No Hypertensive Disorder of Pregnancy; No Preeclampsia-eclampsia; No Gestational Hypertension; No Hellp Syndrome. Vascular Disorder of Pregnancy Speaks for All. Ethiop. J. Health Sci. 2016, 26, 177–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magley, M.; Hinson, M.R. Eclampsia. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Marasciulo, F.; Orabona, R.; Fratelli, N.; Fichera, A.; Valcamonico, A.; Ferrari, F.; Odicino, F.E.; Sartori, E.; Prefumo, F. Pre-eclampsia and late fetal growth restriction. Minerva Obstet. Gynecol. 2021, 73, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Frost, A.L.; Suriano, K.; Aye, C.Y.L.; Leeson, P.; Lewandowski, A.J. The Immediate and Long-Term Impact of Preeclampsia on Offspring Vascular and Cardiac Physiology in the Preterm Infant. Front. Pediatr. 2021, 9, 625726. [Google Scholar] [CrossRef] [PubMed]
- Ives, C.W.; Sinkey, R.; Rajapreyar, I.; Tita, A.T.; Oparil, S. Preeclampsia—Pathophysiology and Clinical Presentations. J. Am. Coll. Cardiol. 2020, 76, 1690–1702. [Google Scholar] [CrossRef]
- Rolnik, D.L.; Wright, D.; Poon, L.C.Y.; Syngelaki, A.; O’Gorman, N.; de Paco Matallana, C.; Akolekar, R.; Cicero, S.; Janga, D.; Singh, M.; et al. ASPRE trial: Performance of screening for preterm pre-eclampsia. Ultrasound Obstet. Gynecol. 2017, 50, 492–495. [Google Scholar] [CrossRef]
- Bujold, E.; Roberge, S.; Lacasse, Y.; Bureau, M.; Audibert, F.; Marcoux, S.; Forest, J.-C.; Giguère, Y. Prevention of Preeclampsia and Intrauterine Growth Restriction With Aspirin Started in Early Pregnancy. Obstet. Gynecol. 2010, 116, 402–414. [Google Scholar] [CrossRef]
- Roberge, S.; Bujold, E.; Nicolaides, K. Aspirin for the prevention of preterm and term preeclampsia: Systematic review and metaanalysis. Am. J. Obstet. Gynecol. 2018, 218, 287–293.e1. [Google Scholar] [CrossRef] [Green Version]
- Hofmeyr, G.J.; Manyame, S.; Medley, N.; Williams, M.J. Calcium supplementation commencing before or early in pregnancy, for preventing hypertensive disorders of pregnancy. Cochrane Database Syst. Rev. 2019, 2019, CD011192. [Google Scholar] [CrossRef]
- Omotayo, M.O.; Dickin, K.; O’Brien, K.O.; Neufeld, L.M.; De Regil, L.M.; Stoltzfus, R.J. Calcium Supplementation to Prevent Preeclampsia: Translating Guidelines into Practice in Low-Income Countries. Adv. Nutr. Int. Rev. J. 2016, 7, 275–278. [Google Scholar] [CrossRef] [Green Version]
- Maia e Holanda Moura, S.B.; Marques Lopes, L.; Murthi, P.; da Silva Costa, F. Prevention of Preeclampsia. J. Pregnancy 2012, 2012, 1–9. [Google Scholar] [CrossRef]
- Sunjaya, A.F.; Med, B.; Sunjaya, A.P. Evaluation of Serum Biomarkers and Other Diagnostic Modalities for Early Diagnosis of Preeclampsia. J. Fam. Reprod. Health 2019, 13, 56. [Google Scholar]
- Park, H.J.; Shim, S.S.; Cha, D.H. Combined Screening for Early Detection of Pre-Eclampsia. Int. J. Mol. Sci. 2015, 16, 17952–17974. [Google Scholar] [CrossRef] [PubMed]
- Chaiworapongsa, T.; Chaemsaithong, P.; Yeo, L.; Romero, R. Pre-eclampsia part 1: Current understanding of its pathophysiology. Nat. Rev. Nephrol. 2014, 10, 466–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gathiram, P.; Moodley, J. Pre-eclampsia: Its pathogenesis and pathophysiolgy. Cardiovasc. J. Afr. 2016, 27, 71–78. [Google Scholar] [CrossRef] [Green Version]
- Roberts, J.; Hubel, C. The Two Stage Model of Preeclampsia: Variations on the Theme. Placenta 2009, 30, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Lyall, F.; Bulmer, J.N.; Duffie, E.; Cousins, F.; Theriault, A.; Robson, S.C. Human Trophoblast Invasion and Spiral Artery Transformation: The Role of PECAM-1 in Normal Pregnancy, Preeclampsia, and Fetal Growth Restriction. Am. J. Pathol. 2001, 158, 1713–1721. [Google Scholar] [CrossRef]
- Burke, S.D.; Karumanchi, S.A. Spiral Artery Remodeling in Preeclampsia Revisited. Hypertension 2013, 62, 1013–1014. [Google Scholar] [CrossRef] [Green Version]
- Bakrania, B.A.; George, E.M.; Granger, J.P. Animal models of preeclampsia: Investigating pathophysiology and therapeutic targets. Am. J. Obstet. Gynecol. 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Staff, A.C.; Johnsen, G.M.; Dechend, R.; Redman, C.W. Preeclampsia and uteroplacental acute atherosis: Immune and inflammatory factors. J. Reprod. Immunol. 2013, 101–102, 120–126. [Google Scholar] [CrossRef]
- Predoi, C.; Grigoriu, C.; Vladescu, R.; Mihart, A. Placental damages in preeclampsia—From ultrasound images to histopathological findings. J. Med. Life 2015, 8, 62–65. [Google Scholar]
- Soleymanlou, N.; Jurisica, I.; Nevo, O.; Ietta, F.; Zhang, X.; Zamudio, S.; Post, M.; Caniggia, I. Molecular Evidence of Placental Hypoxia in Preeclampsia. J. Clin. Endocrinol. Metab. 2005, 90, 4299–4308. [Google Scholar] [CrossRef] [Green Version]
- Redman, C.W.; Sargent, I.L. Latest Advances in Understanding Preeclampsia. Science 2005, 308, 1592–1594. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.S.; Nijland, M.J.; Knoblich, P. Placental ischemia and cardiovascular dysfunction in preeclampsia and beyond: Making the connections. Expert Rev. Cardiovasc. Ther. 2008, 6, 1367–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, M. Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): A dual regulator for angiogenesis. Angiogenesis 2006, 9, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Dijke, P.T.; Goumans, M.-J.; Pardali, E. Endoglin in angiogenesis and vascular diseases. Angiogenesis 2008, 11, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Lewis, D.F.; Wang, Y. Placental Productions and Expressions of Soluble Endoglin, Soluble fms-Like Tyrosine Kinase Receptor-1, and Placental Growth Factor in Normal and Preeclamptic Pregnancies. J. Clin. Endocrinol. Metab. 2008, 93, 260–266. [Google Scholar] [CrossRef] [Green Version]
- Lecarpentier, E.; Tsatsaris, V. Angiogenic balance (sFlt-1/PlGF) and preeclampsia. Ann. Endocrinol. 2016, 77, 97–100. [Google Scholar] [CrossRef]
- Zhou, Q.; Qiao, F.-Y.; Zhao, C.; Liu, H.-Y. Hypoxic trophoblast-derived sFlt-1 may contribute to endothelial dysfunction: An implication for the mechanism of trophoblast-endothelial dysfunction in preeclampsia. Cell Biol. Int. 2010, 35, 61–66. [Google Scholar] [CrossRef]
- Venkatesha, S.; Toporsian, M.; Lam, C.; Hanai, J.-I.; Mammoto, T.; Kim, Y.M.; Bdolah, Y.; Lim, K.-H.; Yuan, H.-T.; Libermann, T.A.; et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat. Med. 2006, 12, 642–649. [Google Scholar] [CrossRef]
- Nikuei, P.; Rajaei, M.; Roozbeh, N.; Mohseni, F.; Poordarvishi, F.; Azad, M.; Haidari, S. Diagnostic accuracy of sFlt1/PlGF ratio as a marker for preeclampsia. BMC Pregnancy Childbirth 2020, 20, 80. [Google Scholar] [CrossRef]
- Zeisler, H.; Llurba, E.; Chantraine, F.; Vatish, M.; Staff, A.C.; Sennström, M.; Olovsson, M.; Brennecke, S.P.; Stepan, H.; Allegranza, D.; et al. Predictive Value of the sFlt-1:PlGF Ratio in Women with Suspected Preeclampsia. N. Engl. J. Med. 2016, 374, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Caillon, H.; Tardif, C.; Dumontet, E.; Winer, N.; Masson, D. Evaluation of sFlt-1/PlGF Ratio for Predicting and Improving Clinical Management of Pre-eclampsia: Experience in a Specialized Perinatal Care Center. Ann. Lab. Med. 2018, 38, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, B.; Amaral, L.M.; Harmon, A.C.; Cornelius, D.C.; Faulkner, J.L.; Cunningham, M.W., Jr. Placental Ischemia and Resultant Phenotype in Animal Models of Preeclampsia. Curr. Hypertens. Rep. 2016, 18, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Laresgoiti-Servitje, E. A leading role for the immune system in the pathophysiology of preeclampsia. J. Leukoc. Biol. 2013, 94, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.M. The role of RAS in the pathogenesis of preeclampsia. Curr. Hypertens. Rep. 2006, 8, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Lumbers, E.R.; Delforce, S.J.; Arthurs, A.; Pringle, K. Causes and Consequences of the Dysregulated Maternal Renin-Angiotensin System in Preeclampsia. Front. Endocrinol. 2019, 10, 563. [Google Scholar] [CrossRef] [PubMed]
- Herse, F.; LaMarca, B. Angiotensin II Type 1 Receptor Autoantibody (AT1-AA)-Mediated Pregnancy Hypertension. Am. J. Reprod. Immunol. 2012, 69, 413–418. [Google Scholar] [CrossRef] [Green Version]
- Saito, S.; Shiozaki, A.; Nakashima, A.; Sakai, M.; Sasaki, Y. The role of the immune system in preeclampsia. Mol. Asp. Med. 2007, 28, 192–209. [Google Scholar] [CrossRef]
- Aggarwal, R.; Jain, A.K.; Mittal, P.; Kohli, M.; Jawanjal, P.; Rath, G. Association of pro- and anti-inflammatory cytokines in preeclampsia. J. Clin. Lab. Anal. 2019, 33, e22834. [Google Scholar] [CrossRef] [Green Version]
- Perez-Sepulveda, A.; Torres, M.J.; Khoury, M.; Illanes, S.E. Innate Immune System and Preeclampsia. Front. Immunol. 2014, 5, 244. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhao, G.; Fan, H.; Zhao, X.; Li, P.; Wang, Z.; Hu, Y.; Hou, Y. Mesenchymal Stem Cells Ameliorate Th1-Induced Pre-Eclampsia-Like Symptoms in Mice via the Suppression of TNF-α Expression. PLoS ONE 2014, 9, e88036. [Google Scholar] [CrossRef] [PubMed]
- Olaniyi, K.S.; Moodley, J.; Mahabeer, Y.; Mackraj, I. Placental Microbial Colonization and Its Association With Pre-eclampsia. Front. Cell. Infect. Microbiol. 2020, 10, 413. [Google Scholar] [CrossRef] [PubMed]
- Lamarca, B. The role of immune activation in contributing to vascular dysfunction and the pathophysiology of hypertension during preeclampsia. Minerva Ginecol. 2010, 62, 105–120. [Google Scholar] [PubMed]
- Saleh, L.; Verdonk, K.; Visser, W.; van den Meiracker, A.H.; Danser, A.H.J. The emerging role of endothelin-1 in the pathogenesis of pre-eclampsia. Ther. Adv. Cardiovasc. Dis. 2016, 10, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, I.A.; Jaleel, A.; Tamimi, W.; Al Kadri, H.M.F. Role of oxidative stress in the pathogenesis of preeclampsia. Arch. Gynecol. Obstet. 2010, 282, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Aouache, R.; Biquard, L.; Vaiman, D.; Miralles, F. Oxidative Stress in Preeclampsia and Placental Diseases. Int. J. Mol. Sci. 2018, 19, 1496. [Google Scholar] [CrossRef] [Green Version]
- Baker, B.C.; Heazell, A.E.P.; Sibley, C.; Wright, R.; Bischof, H.; Beards, F.; Guevara, T.; Girard, S.; Jones, R.L. Hypoxia and oxidative stress induce sterile placental inflammation in vitro. Sci. Rep. 2021, 11, 7281. [Google Scholar] [CrossRef]
- Silvestro, S.; Calcaterra, V.; Pelizzo, G.; Bramanti, P.; Mazzon, E. Prenatal Hypoxia and Placental Oxidative Stress: Insights from Animal Models to Clinical Evidences. Antioxidants 2020, 9, 414. [Google Scholar] [CrossRef]
- Wu, F.; Tian, F.-J.; Lin, Y. Oxidative Stress in Placenta: Health and Diseases. BioMed Res. Int. 2015, 2015, 293271. [Google Scholar] [CrossRef] [Green Version]
- Guerby, P.; Tasta, O.; Swiader, A.; Pont, F.; Bujold, E.; Parant, O.; Vayssiere, C.; Salvayre, R.; Negre-Salvayre, A. Role of oxidative stress in the dysfunction of the placental endothelial nitric oxide synthase in preeclampsia. Redox Biol. 2021, 40, 101861. [Google Scholar] [CrossRef]
- Sandrim, V.C.; Palei, A.C.; Metzger, I.F.; Gomes, V.A.; Cavalli, R.C.; Tanus-Santos, J.E. Nitric Oxide Formation Is Inversely Related to Serum Levels of Antiangiogenic Factors Soluble Fms-Like Tyrosine Kinase-1 and Soluble Endogline in Preeclampsia. Hypertension 2008, 52, 402–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashie, W.; Fondjo, L.A.; Owiredu, W.K.B.A.; Ephraim, R.K.D.; Asare, L.; Adu-Gyamfi, E.A.; Seidu, L. Altered Bioavailability of Nitric Oxide and L-Arginine Is a Key Determinant of Endothelial Dysfunction in Preeclampsia. BioMed Res. Int. 2020, 2020, 3251956. [Google Scholar] [CrossRef] [PubMed]
- Jurado, S.; Saraiva, K.; Marceliano, C.; Souza, V.; Vieira, I. Maternal and Fetal Complications Due to Decreased Nitric Oxide Synthesis during Gestation. Available online: https://www.intechopen.com/chapters/66384 (accessed on 10 December 2021).
- Sanchez-Aranguren, L.C.; Prada, C.E.; Riãno-Medina, C.E.; Lopez, M. Endothelial dysfunction and preeclampsia: Role of oxidative stress. Front. Physiol. 2014, 5, 372. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Sheehan, P.M.; Brennecke, S.; Keogh, R.J. Vessel remodelling, pregnancy hormones and extravillous trophoblast function. Mol. Cell. Endocrinol. 2012, 349, 138–144. [Google Scholar] [CrossRef]
- Handschuh, K.; Guibourdenche, J.; Tsatsaris, V.; Guesnon, M.; Laurendeau, I.; Evain-Brion, D.; Fournier, T. Human Chorionic Gonadotropin Produced by the Invasive Trophoblast But Not the Villous Trophoblast Promotes Cell Invasion and Is Down-Regulated by Peroxisome Proliferator-Activated Receptor-γ. Endocrinology 2007, 148, 5011–5019. [Google Scholar] [CrossRef] [Green Version]
- Atamer, Y.; Erden, A.C.; Demir, B.; Koçyigit, Y.; Atamer, A. The relationship between plasma levels of leptin and androgen in healthy and preeclamptic pregnant women. Acta Obstet. Gynecol. Scand. 2004, 83, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Salustiano, E.; De Pinho, J.C.; Provost, K.; Ruano, R.; Zugaib, M. Maternal Serum Hormonal Factors in the Pathogenesis of Preeclampsia. Obstet. Gynecol. Surv. 2013, 68, 141–150. [Google Scholar] [CrossRef]
- Goswami, D.; Tannetta, D.; Magee, L.; Fuchisawa, A.; Redman, C.; Sargent, I.; von Dadelszen, P. Excess syncytiotrophoblast microparticle shedding is a feature of early-onset pre-eclampsia, but not normotensive intrauterine growth restriction. Placenta 2006, 27, 56–61. [Google Scholar] [CrossRef]
- Raguema, N.; Moustadraf, S.; Bertagnolli, M. Immune and Apoptosis Mechanisms Regulating Placental Development and Vascularization in Preeclampsia. Front. Physiol. 2020, 11, 98. [Google Scholar] [CrossRef]
- Gerasimova, E.M.; Fedotov, S.A.; Kachkin, D.V.; Vashukova, E.S.; Glotov, A.S.; Chernoff, Y.O.; Rubel, A.A. Protein Misfolding during Pregnancy: New Approaches to Preeclampsia Diagnostics. Int. J. Mol. Sci. 2019, 20, 6183. [Google Scholar] [CrossRef] [Green Version]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manna, S.; McCarthy, C.; McCarthy, F. Placental Ageing in Adverse Pregnancy Outcomes: Telomere Shortening, Cell Senescence, and Mitochondrial Dysfunction. Oxidative Med. Cell. Longev. 2019, 2019, 3095383. [Google Scholar] [CrossRef] [PubMed]
- Mayne, B.T.; Leemaqz, S.; Smith, A.K.; Breen, J.; Roberts, C.; Bianco-Miotto, T. Accelerated placental aging in early onset preeclampsia pregnancies identified by DNA methylation. Epigenomics 2017, 9, 279–289. [Google Scholar] [CrossRef] [Green Version]
- Herzog, E.M.; Eggink, A.J.; Reijnierse, A.; Kerkhof, M.A.; de Krijger, R.R.; Roks, A.J.; Reiss, I.K.; Nigg, A.L.; Eilers, P.H.; Steegers, E.A.; et al. Impact of early- and late-onset preeclampsia on features of placental and newborn vascular health. Placenta 2016, 49, 72–79. [Google Scholar] [CrossRef]
- Lisonkova, S.; Sabr, Y.; Mayer, C.; Young, C.; Skoll, A.; Joseph, K. Maternal Morbidity Associated With Early-Onset and Late-Onset Preeclampsia. Obstet. Gynecol. 2014, 124, 771–781. [Google Scholar] [CrossRef]
- Lisonkova, S.; Joseph, K. Incidence of preeclampsia: Risk factors and outcomes associated with early- versus late-onset disease. Am. J. Obstet. Gynecol. 2013, 209, 544.e1–544.e12. [Google Scholar] [CrossRef] [PubMed]
- Staff, A.C. The two-stage placental model of preeclampsia: An update. J. Reprod. Immunol. 2019, 134–135, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Verlohren, S.; Melchiorre, K.; Khalil, A.; Thilaganathan, B. Uterine artery Doppler, birth weight and timing of onset of pre-eclampsia: Providing insights into the dual etiology of late-onset pre-eclampsia. Ultrasound Obstet. Gynecol. 2014, 44, 293–298. [Google Scholar] [CrossRef] [Green Version]
- Thilaganathan, B. Pre-eclampsia and the cardiovascular-placental axis. Ultrasound Obstet. Gynecol. 2018, 51, 714–717. [Google Scholar] [CrossRef] [Green Version]
- Berg, C.V.D.; Chaves, I.; Herzog, E.M.; Willemsen, S.P.; Van Der Horst, G.T.J.; Steegers-Theunissen, R.P.M. Early- and late-onset preeclampsia and the DNA methylation of circadian clock and clock-controlled genes in placental and newborn tissues. Chronobiol. Int. 2017, 34, 921–932. [Google Scholar] [CrossRef]
- Suhag, A.; Berghella, V. Intrauterine Growth Restriction (IUGR): Etiology and Diagnosis. Curr. Obstet. Gynecol. Rep. 2013, 2, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Unterscheider, J.; Daly, S.; Geary, M.P.; Kennelly, M.M.; McAuliffe, F.M.; O’Donoghue, K.; Hunter, A.; Morrison, J.J.; Burke, G.; Dicker, P.; et al. Optimizing the definition of intrauterine growth restriction: The multicenter prospective PORTO Study. Am. J. Obstet. Gynecol. 2013, 208, 290.e1–290.e6. [Google Scholar] [CrossRef] [PubMed]
- Osuchukwu, O.O.; Reed, D.J. Small for Gestational Age. Available online: https://0-www-ncbi-nlm-nih-gov.brum.beds.ac.uk/books/NBK563247/ (accessed on 10 December 2021).
- Beune, I.M.; Bloomfield, F.H.; Ganzevoort, W.; Embleton, N.; Rozance, P.J.; van Wassenaer-Leemhuis, A.G.; Wynia, K.; Gordijn, S.J. Consensus Based Definition of Growth Restriction in the Newborn. J. Pediatr. 2018, 196, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Beune, I.M.; Damhuis, S.E.; Ganzevoort, W.; Hutchinson, J.C.; Khong, T.Y.; Mooney, E.E.; Sebire, N.J.; Gordijn, S.J. Consensus Definition of Fetal Growth Restriction in Intrauterine Fetal Death: A Delphi Procedure. Arch. Pathol. Lab. Med. 2020, 145, 428–436. [Google Scholar] [CrossRef]
- Martín-Estal, I.; de la Garza, R.G.; de Cortázar, I.C. Intrauterine Growth Retardation (IUGR) as a Novel Condition of Insulin-Like Growth Factor-1 (IGF-1) Deficiency. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Cham, Switzerland, 2016; Volume 170. [Google Scholar] [CrossRef]
- Saleem, T.; Sajjad, N.; Fatima, S.; Habib, N.; Ali, S.R.; Qadir, M. Intrauterine growth retardation-small events, big consequences. Ital. J. Pediatr. 2011, 37, 41–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallego, E.M.; Pujol, A.T.; Bartra, A.J.C.; Roig, M.D.G. Fetal Growth Restriction. Available online: https://www.intechopen.com/chapters/70711 (accessed on 10 December 2021).
- Salam, R.A.; Das, J.K.; Ali, A.; Lassi, Z.S.; Bhutta, Z.A. Maternal undernutrition and intrauterine growth restriction. Expert Rev. Obstet. Gynecol. 2013, 8, 559–567. [Google Scholar] [CrossRef]
- Radulescu, L.; Munteanu, O.; Popa, F.; Cirstoiu, M. The implications and consequences of maternal obesity on fetal intrauterine growth restriction. J. Med. Life 2013, 6, 292–298. [Google Scholar] [PubMed]
- Parikh, R.M.; Joshi, S.R.; Menon, P.S.; Shah, N.S. Intensive glycemic control in diabetic pregnancy with intrauterine growth restriction is detrimental to fetus. Med. Hypotheses 2007, 69, 203–205. [Google Scholar] [CrossRef]
- Lean, S.C.; Heazell, A.E.P.; Dilworth, M.; Mills, T.; Jones, R.L. Placental Dysfunction Underlies Increased Risk of Fetal Growth Restriction and Stillbirth in Advanced Maternal Age Women. Sci. Rep. 2017, 7, 9677. [Google Scholar] [CrossRef]
- Reeves, S.; Bernstein, I. Effects of maternal tobacco-smoke exposure on fetal growth and neonatal size. Expert Rev. Obstet. Gynecol. 2008, 3, 719–730. [Google Scholar] [CrossRef] [Green Version]
- Garrison, L.; Leeman, L.; Savich, R.D.; Gutierrez, H.; Rayburn, W.F.; Bakhireva, L.N. Fetal Growth Outcomes in a Cohort of Polydrug- and Opioid-Dependent Patients. J. Reprod. Med. 2016, 61, 311–319. [Google Scholar] [PubMed]
- Romo, A.; Carceller, R.; Tobajas, J. Intrauterine Growth Retardation (IUGR): Epidemiology and Etiology—PubMed. Pediatr. Endocrinol. Rev. 2009, 6, 332–336. [Google Scholar] [PubMed]
- Vrachnis, N.; Loukas, N.; Vrachnis, D.; Antonakopoulos, N.; Zygouris, D.; Kοlialexi, A.; Pergaliotis, V.; Iavazzo, C.; Mastorakos, G.; Iliodromiti, Z. A Systematic Review of Bisphenol A from Dietary and Non-Dietary Sources during Pregnancy and Its Possible Connection with Fetal Growth Restriction: Investigating Its Potential Effects and the Window of Fetal Vulnerability. Nutrients 2021, 13, 2426. [Google Scholar] [CrossRef] [PubMed]
- Armengaud, J.; Yzydorczyk, C.; Siddeek, B.; Peyter, A.; Simeoni, U. Intrauterine growth restriction: Clinical consequences on health and disease at adulthood. Reprod. Toxicol. 2020, 99, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Wang, L.; Lin, X.; Spengler, J.D.; Perera, F.P. Fetal Window of Vulnerability to Airborne Polycyclic Aromatic Hydrocarbons on Proportional Intrauterine Growth Restriction. PLoS ONE 2012, 7, e35464. [Google Scholar] [CrossRef] [Green Version]
- Fleiss, B.; Wong, F.; Brownfoot, F.; Shearer, I.K.; Baud, O.; Walker, D.W.; Gressens, P.; Tolcos, M. Knowledge Gaps and Emerging Research Areas in Intrauterine Growth Restriction-Associated Brain Injury. Front. Endocrinol. 2019, 10, 188. [Google Scholar] [CrossRef] [Green Version]
- Cherian, A.; Vijayaselvi, R. Risk assessment of intrauterine growth restriction. Curr. Med. Issues 2017, 15, 262. [Google Scholar] [CrossRef]
- Silver, K.L.; Conroy, A.L.; Leke, R.G.F.; Leke, R.J.I.; Gwanmesia, P.; Molyneux, M.E.; Wallace, D.T.; Rogerson, S.J.; Kain, K.C. Circulating Soluble Endoglin Levels in Pregnant Women in Cameroon and Malawi—Associations with Placental Malaria and Fetal Growth Restriction. PLoS ONE 2011, 6, e24985. [Google Scholar] [CrossRef]
- Gaccioli, F.; Lager, S. Placental Nutrient Transport and Intrauterine Growth Restriction. Front. Physiol. 2016, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Malhotra, A.; Allison, B.J.; Castillo-Melendez, M.; Jenkin, G.; Polglase, G.R.; Miller, S.L. Neonatal Morbidities of Fetal Growth Restriction: Pathophysiology and Impact. Front. Endocrinol. 2019, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- Sharma, D.; Shastri, S.; Sharma, P. Intrauterine Growth Restriction: Antenatal and Postnatal Aspects. Clin. Med. Insights Pediatr. 2016, 10, 67–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinar, B.; Sert, A.; Gokmen, Z.; Aypar, E.; Aslan, E.; Odabas, D. Left ventricular dimensions, systolic functions, and mass in term neonates with symmetric and asymmetric intrauterine growth restriction. Cardiol. Young 2013, 25, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Czernik, C.; Rhode, S.; Metze, B.; Bührer, C.; Schmitz, L. Comparison of left ventricular cardiac dimensions between small and appropriate for gestational age preterm infants below 30 weeks of gestation. J. Périnat. Med. 2013, 41, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Aburawi, E.H.; Malcus, P.; Thuring, A.; Fellman, V.; Pesonen, E. Coronary Flow in Neonates with Impaired Intrauterine Growth. J. Am. Soc. Echocardiogr. 2012, 25, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Guerineau, L.; Perez-Cruz, M.; Roig, M.D.G.; Cambra, F.J.; Carretero, J.; Prada, F.; Gómez, O.; Crispi, F.; Bartrons, J. Cardiovascular adaptation to extrauterine life after intrauterine growth restriction. Cardiol. Young 2017, 28, 284–291. [Google Scholar] [CrossRef]
- Cosmi, E.; Fanelli, T.; Visentin, S.; Trevisanuto, D.; Zanardo, V. Consequences in Infants That Were Intrauterine Growth Restricted. J. Pregnancy 2011, 2011, 364381. [Google Scholar] [CrossRef]
- Pike, K.; Pillow, J.J.; Lucas, J.S. Long term respiratory consequences of intrauterine growth restriction. Semin. Fetal Neonatal Med. 2012, 17, 92–98. [Google Scholar] [CrossRef]
- Briana, D.D.; Malamitsi-Puchner, A. Perinatal biomarkers implying ‘Developmental Origins of Health and Disease’ consequences in intrauterine growth restriction. Acta Paediatr. 2019, 109, 1317–1322. [Google Scholar] [CrossRef]
- Shetty, S.; Idell, S. Fibrinolysis: Plasminogen Activator and Plasmin. Available online: https://0-www-sciencedirect-com.brum.beds.ac.uk/topics/neuroscience/plasminogen-activator (accessed on 10 December 2021).
- Burton, G.; Jauniaux, E. Pathophysiology of placental-derived fetal growth restriction. Am. J. Obstet. Gynecol. 2018, 218, S745–S761. [Google Scholar] [CrossRef] [Green Version]
- Krishna, U.; Bhalerao, S. Placental Insufficiency and Fetal Growth Restriction. J. Obstet. Gynecol. India 2011, 61, 505–511. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.Y.; Milligan, N.; Keating, S.; Windrim, R.; Keunen, J.; Thakur, V.; Ohman, A.; Portnoy, S.; Sled, J.G.; Kelly, E.; et al. The hemodynamics of late-onset intrauterine growth restriction by MRI. Am. J. Obstet. Gynecol. 2015, 214, 367.e1–367.e17. [Google Scholar] [CrossRef] [PubMed]
- Romero, R.; Whitten, A.; Korzeniewski, S.; Than, N.G.; Chaemsaithong, P.; Miranda, J.; Dong, Z.; Hassan, S.S.; Chaiworapongsa, T. Maternal Floor Infarction/Massive Perivillous Fibrin Deposition: A Manifestation of Maternal Antifetal Rejection? Am. J. Reprod. Immunol. 2013, 70, 285–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.J.; Romero, R.; Chaemsaithong, P.; Kim, J.-S. Chronic inflammation of the placenta: Definition, classification, pathogenesis, and clinical significance. Am. J. Obstet. Gynecol. 2015, 213, S53–S69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martín-Estal, I.; Castilla-Cortázar, I.; Castorena-Torres, F. The Placenta as a Target for Alcohol During Pregnancy: The Close Relation with IGFs Signaling Pathway. Rev. Physiol. Biochem. Pharmacol. 2021, 119–153. [Google Scholar] [CrossRef]
- Burd, L.; Roberts, D.; Olson, M.; Odendaal, H. Ethanol and the placenta: A review. J. Matern. Neonatal Med. 2007, 20, 361–375. [Google Scholar] [CrossRef]
- Steane, S.E.; Young, S.L.; Clifton, V.L.; Gallo, L.A.; Akison, L.K.; Moritz, K.M. Prenatal alcohol consumption and placental outcomes: A systematic review and meta-analysis of clinical studies. Am. J. Obstet. Gynecol. 2021, 225, 607.e1–607.e22. [Google Scholar] [CrossRef]
- Sovio, U.; White, I.; Dacey, A.; Pasupathy, D.; Smith, G. Screening for fetal growth restriction with universal third trimester ultrasonography in nulliparous women in the Pregnancy Outcome Prediction (POP) study: A prospective cohort study. Lancet 2015, 386, 2089–2097. [Google Scholar] [CrossRef] [Green Version]
- Mesdaghi-Nia, E.; Behrashi, M.; Saeidi, A.; Abedzadeh-Kalahroudi, M. Association between PAPP-A and placental thickness. Int. J. Reprod. Biomed. 2016, 14, 421–426. [Google Scholar] [CrossRef] [Green Version]
- Ekin, A.; Gezer, C.; Taner, C.E.; Özeren, M. The association between low PAPP-A levels at first trimester and poor pregnancy outcomes. Périnat. J. 2014, 22, 142–146. [Google Scholar] [CrossRef]
- Caliskan, R.; Atis, A.; Aydin, Y.; Acar, D.; Kiyak, H.; Topbas, F. PAPP-A concentrations change in patients with gestational diabetes. J. Obstet. Gynaecol. 2019, 40, 190–194. [Google Scholar] [CrossRef]
- Antwi, E.; Amoakoh-Coleman, M.; Vieira, D.; Madhavaram, S.; Koram, K.A.; Grobbee, D.E.; Agyepong, I.A.; Klipstein-Grobusch, K. Systematic review of prediction models for gestational hypertension and preeclampsia. PLoS ONE 2020, 15, e0230955. [Google Scholar] [CrossRef] [PubMed]
- Fruscalzo, A.; Cividino, A.; Rossetti, E.; Maurigh, A.; Londero, A.P.; Driul, L. First trimester PAPP-A serum levels and long-term metabolic outcome of mothers and their offspring. Sci. Rep. 2020, 10, 5131. [Google Scholar] [CrossRef] [Green Version]
- Westgren, L.; Anneren, R.; Axelsson, O.; Evald, U.; Leb-lanc, K.; Ringden, O.; Winsor, S.; Hornberger, L.; Johnson, J.; Sinai Hospital Toronto, M. Low First-Trimester PAPP-A Identifies Pregnancies Requiring IUGR Screening. Am. J. Obstet. Gynecol. 2003, 189, S215. [Google Scholar] [CrossRef]
- Antsaklis, P.; Fasoulakis, Z.; Theodora, M.; Diakosavvas, M.; Kontomanolis, E.N. Association of Low Maternal Pregnancy-associated Plasma Protein A with Adverse Perinatal Outcome. Cureus 2019, 11, e4912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pascual-Mancho, J.; Pintado-Recarte, P.; Romero-Román, C.; Morales-Camino, J.; Hernández-Martin, C.; Bujan, J.; Ortega, M.; De León-Luis, J. Influence of Cerebral Vasodilation on Blood Reelin Levels in Growth Restricted Fetuses. Diagnostics 2021, 11, 1036. [Google Scholar] [CrossRef] [PubMed]
- Cetin, I.; Mazzocco, M.I.; Giardini, V.; Cardellicchio, M.; Calabrese, S.; Algeri, P.; Martinelli, A.; Todyrenchuk, L.; Vergani, P. PLGF in a clinical setting of pregnancies at risk of Preeclampsia and/or Intrauterine Growth Restriction. J. Matern. Neonatal Med. 2016, 30, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Birdir, C.; Droste, L.; Fox, L.; Frank, M.; Fryze, J.; Enekwe, A.; Köninger, A.; Kimmig, R.; Schmidt, B.; Gellhaus, A. Predictive value of sFlt-1, PlGF, sFlt-1/PlGF ratio and PAPP-A for late-onset preeclampsia and IUGR between 32 and 37 weeks of pregnancy. Pregnancy Hypertens. 2018, 12, 124–128. [Google Scholar] [CrossRef]
- Schoofs, K.; Grittner, U.; Engels, T.; Pape, J.; Denk, B.; Henrich, W.; Verlohren, S. The importance of repeated measurements of the sFlt-1/PlGF ratio for the prediction of preeclampsia and intrauterine growth restriction. J. Périnat. Med. 2014, 42, 61–68. [Google Scholar] [CrossRef]
- Raia-Barjat, T.; Prieux, C.; Gris, J.-C.; Chapelle, C.; Laporte, S.; Chauleur, C. Angiogenic factors for prediction of preeclampsia and intrauterine growth restriction onset in high-risk women: AngioPred study. J. Matern. Neonatal Med. 2017, 32, 248–257. [Google Scholar] [CrossRef]
- Sirikunalai, P.; Wanapirak, C.; Sirichotiyakul, S.; Tongprasert, F.; Srisupundit, K.; Luewan, S.; Traisrisilp, K.; Tongsong, T. Associations between maternal serum free beta human chorionic gonadotropin (β-hCG) levels and adverse pregnancy outcomes. J. Obstet. Gynaecol. 2015, 36, 178–182. [Google Scholar] [CrossRef]
- Massimiani, M.; Salvi, S.; Tiralongo, G.M.; Moresi, S.; Stuhlmann, H.; Valensise, H.; Lanzone, A.; Campagnolo, L. Circulating EGFL7 distinguishes between IUGR and PE: An observational case–control study. Sci. Rep. 2021, 11, 17919. [Google Scholar] [CrossRef] [PubMed]
- Audette, M.C.; Kingdom, J.C. Screening for fetal growth restriction and placental insufficiency. Semin. Fetal Neonatal Med. 2018, 23, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Molvarec, A.; Gullai, N.; Stenczer, B.; Fügedi, G.; Nagy, B.; Jr, J.R. Comparison of placental growth factor and fetal flow Doppler ultrasonography to identify fetal adverse outcomes in women with hypertensive disorders of pregnancy: An observational study. BMC Pregnancy Childbirth 2013, 13, 161. [Google Scholar] [CrossRef] [Green Version]
- Bamfo, J.E.A.K.; Odibo, A.O. Diagnosis and Management of Fetal Growth Restriction. J. Pregnancy 2011, 2011, 640715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantake, M.; Ikeda, N.; Nakaoka, H.; Ohkawa, N.; Tanaka, T.; Miyabayashi, K.; Shoji, H.; Shimizu, T. IGF1 gene is epigenetically activated in preterm infants with intrauterine growth restriction. Clin. Epigenet. 2020, 12, 1–9. [Google Scholar] [CrossRef]
- Merialdi, M.; Carroli, G.; Villar, J.; Abalos, E.; Guülmezoglu, A.M.; Kulier, R.; De Onis, M. Nutritional Interventions during Pregnancy for the Prevention or Treatment of Impaired Fetal Growth: An Overview of Randomized Controlled Trials. J. Nutr. 2003, 133, 1626S–1631S. [Google Scholar] [CrossRef] [Green Version]
- Clarke, P.E.; Gross, H. Women’s behaviour, beliefs and information sources about physical exercise in pregnancy. Midwifery 2004, 20, 133–141. [Google Scholar] [CrossRef]
- Gatford, K.L.; Kaur, G.; Falcão-Tebas, F.; Wadley, G.; Wlodek, M.; Laker, R.C.; Ebeling, P.R.; McConell, G. Exercise as an intervention to improve metabolic outcomes after intrauterine growth restriction. Am. J. Physiol. Metab. 2014, 306, E999–E1012. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, J.; Mohammad, S.; Goudreau, A.D.; Adamo, K.B. Physical activity differentially regulates VEGF, PlGF, and their receptors in the human placenta. Physiol. Rep. 2021, 9, e14710. [Google Scholar] [CrossRef]
- Nascimento, S.L.; Surita, F.; Cecatti, J.G. Physical exercise during pregnancy. Curr. Opin. Obstet. Gynecol. 2012, 24, 387–394. [Google Scholar] [CrossRef]
- Tomić, V.; Sporis, G.; Tomić, J.; Milanović, Z.; Zigmundovac-Klaić, D.; Pantelić, S. The effect of maternal exercise during pregnancy on abnormal fetal growth. Croat. Med. J. 2013, 54, 362–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melamed, N.; Baschat, A.; Yinon, Y.; Athanasiadis, A.; Mecacci, F.; Figueras, F.; Berghella, V.; Nazareth, A.; Tahlak, M.; McIntyre, H.D.; et al. FIGO (International Federation of Gynecology and Obstetrics) initiative on fetal growth: Best practice advice for screening, diagnosis, and management of fetal growth restriction. Int. J. Gynecol. Obstet. 2021, 152, 3–57. [Google Scholar] [CrossRef]
- Groom, K.M.; David, A. The role of aspirin, heparin, and other interventions in the prevention and treatment of fetal growth restriction. Am. J. Obstet. Gynecol. 2018, 218, S829–S840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, L.; Petitt, M.; Fong, A.; Tsuge, M.; Tabata, T.; Fang-Hoover, J.; Maidji, E.; Zydek, M.; Zhou, Y.; Inoue, N.; et al. Intrauterine Growth Restriction Caused by Underlying Congenital Cytomegalovirus Infection. J. Infect. Dis. 2014, 209, 1573–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, S.A.; Wang, D.; Oualim, A.; Diamond, D.J.; Kotton, C.N.; Mossman, S.; Carfi, A.; Anderson, D.; Dormitzer, P.R. The Status of Vaccine Development Against the Human Cytomegalovirus. J. Infect. Dis. 2020, 221, S113–S122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piazza, G. Varicose Veins. Circulation 2014, 130, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Lurie, F.; Passman, M.; Meisner, M.; Dalsing, M.; Masuda, E.; Welch, H.; Bush, R.L.; Blebea, J.; Carpentier, P.H.; De Maeseneer, M.; et al. The 2020 update of the CEAP classification system and reporting standards. J. Vasc. Surg. Venous Lymphat. Disord. 2020, 8, 342–352. [Google Scholar] [CrossRef]
- Youn, Y.J.; Lee, J. Chronic venous insufficiency and varicose veins of the lower extremities. Korean J. Intern. Med. 2019, 34, 269–283. [Google Scholar] [CrossRef]
- Davies, A.H. The Seriousness of Chronic Venous Disease: A Review of Real-World Evidence—PubMed. Adv. Ther. 2019, 36 (Suppl. S1), 5–12. [Google Scholar] [CrossRef]
- Lohr, J.M.; Bush, R.L. Venous disease in women: Epidemiology, manifestations, and treatment. J. Vasc. Surg. 2013, 57, 37S–45S. [Google Scholar] [CrossRef] [Green Version]
- Ortega, M.; Fraile-Martínez, O.; García-Montero, C.; Álvarez-Mon, M.; Chaowen, C.; Ruiz-Grande, F.; Pekarek, L.; Monserrat, J.; Asúnsolo, A.; García-Honduvilla, N.; et al. Understanding Chronic Venous Disease: A Critical Overview of Its Pathophysiology and Medical Management. J. Clin. Med. 2021, 10, 3239. [Google Scholar] [CrossRef] [PubMed]
- Cornu-Thenard, A.; Boivin, P. Chronic Venous Disease during Pregnancy-Servier-Phlebolymphology Servier-Phlebolymphology. Phlebolymphology 2014, 21, 138–145. [Google Scholar]
- Li, X.; Jiang, X.-Y.; Ge, J.; Wang, J.; Chen, G.-J.; Xu, L.; Xie, D.-Y.; Yuan, T.-Y.; Zhang, D.-S.; Zhang, H.; et al. Aberrantly Expressed lncRNAs in Primary Varicose Great Saphenous Veins. PLoS ONE 2014, 9, e86156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, M.A.; Romero, B.; Asúnsolo, Á.; Sola, M.; Álavrez-Rocha, M.J.; Sainz, F.; Álavrez-Mon, M.; Buján, J.; García-Honduvilla, N. Patients with Incompetent Valves in Chronic Venous Insufficiency Show Increased Systematic Lipid Peroxidation and Cellular Oxidative Stress Markers. Oxidative Med. Cell. Longev. 2019, 2019, 5164576. [Google Scholar] [CrossRef]
- Smith, R.K.; Golledge, J. A systematic review of circulating markers in primary chronic venous insufficiency. Phlebol. J. Venous Dis. 2013, 29, 570–579. [Google Scholar] [CrossRef]
- Guss, L.G.; Javvaji, S.; Case, J.; Bs, B.B.; Schaefer, K.N.; Bs, R.G.; Waalen, J.; Greenway, H.T.; Housman, L.B. Differences in Inflammatory Cytokine Levels between Patients with Varying Severity of Chronic Venous Insufficiency. J. Vasc. Med. Surg. 2018, 6, 1–6. [Google Scholar] [CrossRef]
- Raffetto, J.D.; Qiao, X.; Beauregard, K.G.; Khalil, R.A. Estrogen receptor-mediated enhancement of venous relaxation in female rat: Implications in sex-related differences in varicose veins. J. Vasc. Surg. 2010, 51, 972–981. [Google Scholar] [CrossRef] [Green Version]
- Honduvilla, N.G.; Asúnsolo, Á.; Ortega, M.A.; Sainz, F.; Leal, J.; Lopez-Hervas, P.; Pascual, G.; Buján, J. Increase and Redistribution of Sex Hormone Receptors in Premenopausal Women Are Associated with Varicose Vein Remodelling. Oxidative Med. Cell. Longev. 2018, 2018, 3974026. [Google Scholar] [CrossRef] [Green Version]
- Lenković, M.; Cabrijan, L.; Gruber, F.; Batinac, T.; Manestar-Blazić, T.; Stanić Zgombić, Z.; Stasić, A. Effect of Progesterone and Pregnancy on the Development of Varicose Veins. Acta Derm. Croat. 2009, 17, 263–267. [Google Scholar]
- Ciardullo, A.V.; Panico, S.; Bellati, C.; Rubba, P.; Rinaldi, S.; Iannuzzi, A.; Cioffi, V.; Iannuzzo, G.; Berrino, F. High endogenous estradiol is associated with increased venous distensibility and clinical evidence of varicose veins in menopausal women. J. Vasc. Surg. 2000, 32, 544–549. [Google Scholar] [CrossRef]
- Ortega, M.A.; Romero, B.; Asúnsolo, Á.; Martínez-Vivero, C.; Sainz, F.; Bravo, C.; De León-Luis, J.; Álvarez-Mon, M.; Buján, J.; García-Honduvilla, N. Pregnancy-associated venous insufficiency course with placental and systemic oxidative stress. J. Cell. Mol. Med. 2020, 24, 4157–4170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honduvilla, N.G.; Ortega, M.A.; Asúnsolo, Á.; Álvarez-Rocha, M.J.; Romero, B.; De León-Luis, J.; Álvarez-Mon, M.; Buján, J. Placentas from women with pregnancy-associated venous insufficiency show villi damage with evidence of hypoxic cellular stress. Hum. Pathol. 2018, 77, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.; Sánchez-Trujillo, L.; Bravo, C.; Fraile-Martinez, O.; García-Montero, C.; Saez, M.; Alvarez-Mon, M.; Sainz, F.; Alvarez-Mon, M.; Bujan, J.; et al. Newborns of Mothers with Venous Disease during Pregnancy Show Increased Levels of Lipid Peroxidation and Markers of Oxidative Stress and Hypoxia in the Umbilical Cord. Antioxidants 2021, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Saez, M.A.; Fraile-Martínez, O.; Asúnsolo, Á.; Pekarek, L.; Bravo, C.; Coca, S.; Sainz, F.; Mon, M.; Buján, J.; et al. Increased Angiogenesis and Lymphangiogenesis in the Placental Villi of Women with Chronic Venous Disease during Pregnancy. Int. J. Mol. Sci. 2020, 21, 2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortega, M.A.; Saez, M.A.; Asúnsolo, Á.; Romero, B.; Bravo, C.; Coca, S.; Sainz, F.; Álvarez-Mon, M.; Buján, J.; García-Honduvilla, N. Upregulation of VEGF and PEDF in Placentas of Women with Lower Extremity Venous Insufficiency during Pregnancy and Its Implication in Villous Calcification. BioMed Res. Int. 2019, 2019, 5320902. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fraile-Martínez, O.; Saez, M.A.; Álvarez-Mon, M.A.; Gómez-Lahoz, A.M.; Bravo, C.; Luis, J.A.D.L.; Sainz, F.; Coca, S.; Asúnsolo, Á.; et al. Abnormal proinflammatory and stressor environmental with increased the regulatory cellular IGF-1/PAPP-A/STC and Wnt-1/β-Catenin canonical pathway in placenta of women with Chronic venous Disease during Pregnancy. Int. J. Med. Sci. 2021, 18, 2814–2827. [Google Scholar] [CrossRef]
- Ortega, M.A.; Saez, M.A.; Sainz, F.; Fraile-Martínez, O.; García-Gallego, S.; Pekarek, L.; Bravo, C.; Coca, S.; Mon, M.; Buján, J.; et al. Lipidomic profiling of chorionic villi in the placentas of women with chronic venous disease. Int. J. Med. Sci. 2020, 17, 2790–2798. [Google Scholar] [CrossRef]
- Ortega, M.A.; Asúnsolo, Á.; Álvarez-Rocha, M.J.; Romero, B.; De León-Luis, J.; Álvarez-Mon, M.; Buján, J.; García-Honduvilla, N. Remodelling of collagen fibres in the placentas of women with venous insufficiency during pregnancy. Histol. Histopathol. 2018, 33, 567–576. [Google Scholar] [CrossRef]
- Ortega, M.A.; Asúnsolo, Á.; Fraile-Martínez, O.; Sainz, F.; Saez, M.A.; Bravo, C.; De León-Luis, J.A.; Alvarez-Mon, M.A.; Coca, S.; Álvarez-Mon, M.; et al. An increase in elastogenic components in the placental villi of women with chronic venous disease during pregnancy is associated with decreased EGFL7 expression level. Mol. Med. Rep. 2021, 24, 556. [Google Scholar] [CrossRef]
- Asúnsolo, Á.; Chaowen, C.; Ortega, M.A.; Coca, S.; Borrell, L.N.; De León-Luis, J.; García-Honduvilla, N.; Álvarez-Mon, M.; Buján, J. Association Between Lower Extremity Venous Insufficiency and Intrapartum Fetal Compromise: A Nationwide Cross-Sectional Study. Front. Med. 2021, 8, 577096. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortega, M.A.; Fraile-Martínez, O.; García-Montero, C.; Sáez, M.A.; Álvarez-Mon, M.A.; Torres-Carranza, D.; Álvarez-Mon, M.; Bujan, J.; García-Honduvilla, N.; Bravo, C.; et al. The Pivotal Role of the Placenta in Normal and Pathological Pregnancies: A Focus on Preeclampsia, Fetal Growth Restriction, and Maternal Chronic Venous Disease. Cells 2022, 11, 568. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030568
Ortega MA, Fraile-Martínez O, García-Montero C, Sáez MA, Álvarez-Mon MA, Torres-Carranza D, Álvarez-Mon M, Bujan J, García-Honduvilla N, Bravo C, et al. The Pivotal Role of the Placenta in Normal and Pathological Pregnancies: A Focus on Preeclampsia, Fetal Growth Restriction, and Maternal Chronic Venous Disease. Cells. 2022; 11(3):568. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030568
Chicago/Turabian StyleOrtega, Miguel A., Oscar Fraile-Martínez, Cielo García-Montero, Miguel A. Sáez, Miguel Angel Álvarez-Mon, Diego Torres-Carranza, Melchor Álvarez-Mon, Julia Bujan, Natalio García-Honduvilla, Coral Bravo, and et al. 2022. "The Pivotal Role of the Placenta in Normal and Pathological Pregnancies: A Focus on Preeclampsia, Fetal Growth Restriction, and Maternal Chronic Venous Disease" Cells 11, no. 3: 568. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030568