
Targeting Breast Cancer and Their Stem Cell Population through AMPK Activation: Novel Insights



Laser Research Centre, Faculty of Health Sciences, University of Johannesburg, P.O. Box 17011, Doornfontein 2028, South Africa
*
Author to whom correspondence should be addressed.
Cells 2022, 11(3), 576; https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030576
Submission received: 23 November 2021
/
Revised: 24 January 2022
/
Accepted: 25 January 2022
/
Published: 7 February 2022
(This article belongs to the Special Issue Advances in AMPK Research: Basic and Translational Aspects)
Abstract
:Despite some significant advancements, breast cancer has become the most prevalent cancer in the world. One of the main reasons for failure in treatment and metastasis has been attributed to the presence of cancer initiating cells—cancer stem cells. Consequently, research is now being focussed on targeting cancer cells along with their stem cell population. Non-oncology drugs are gaining increasing attention for their potent anticancer activities. Metformin, a drug commonly used to treat type 2 diabetes, is the best example in this regard. It exerts its therapeutic action by activating 5′ adenosine monophosphate-activated protein kinase (AMPK). Activated AMPK subsequently phosphorylates and targets several cellular pathways involved in cell growth and proliferation and the maintenance of stem-like properties of cancer stem cells. Therefore, AMPK is emerging as a target of choice for developing effective anticancer drugs. Vanadium compounds are well-known PTP inhibitors and AMPK activators. They find extensive applications in treatment of diabetes and obesity via PTP1B inhibition and AMPK-mediated inhibition of adipogenesis. However, their role in targeting cancer stem cells has not been explored yet. This review is an attempt to establish the applications of insulin mimetic vanadium compounds for the treatment of breast cancer by AMPK activation and PTP1B inhibition pathways.
1. Introduction
Breast cancer is the most common cancer in women of all races across the globe. According to the World Health Organization, 2.3 million women were diagnosed with breast cancer in 2020 alone, with approximately 685,000 deaths reported globally. As per the 2021 WHO statistics, breast cancer has become the most prevalent cancer in the world, and it is estimated that it would constitute over 30% of all cancers diagnosed in women [1,2]. However, recent advances in medicine and technology have caused significant improvement in the early detection and treatment of cancer through radiation and targeted chemotherapy using specific surface markers (ALDH1, CD44) on cancer and cancer stem cells [3]. Targeted chemotherapies, such as immunotherapy and metal–drug nanoconjugates, have been developed based on gene expression profiling and cancer subtype as well as hormonal therapies for hormone receptor-positive subtype cancers and many more. However, although these approaches cause an initial positive response, there remains a high risk for tumour relapse and drug resistance. In many cases, the relapsed tumour metastasizes, and currently remains incurable [4]. Recent studies suggest that most of these drawbacks experienced with the current line of treatment are caused by a minute population of cells within cancer cells—cancer stem cells (CSCs). The occurrence of cancer stem cells amongst breast cancer cells (BCCs) was first established by Al-Hajj et al. They discovered that as few as 100 of these cells were potent enough to form tumours when xenotransplanted in mice [5].
Cancer stem cells are capable of asymmetric cell division: self-renewal and the generation of heterogeneous lineages (Figure 1). Breast cancer stem cells have been shown to differentiate into endothelial cells to support vasculogenic mimicry and angiogenesis, which subsequently provides nutritional supplies and oxygen to support propagating tumour cells. They are also known to be more resistant to treatment and programmed cell death (anoikis). These sturdy and strong cells can migrate through blood circulation thus causing metastasis. Therefore, it is imperative to kill breast cancer stem cells along with breast cancer cells to ensure complete treatment and prevention of relapse and metastasis. Consequently, BCSCs have emerged as important therapeutic targets especially in overcoming metastasis and drug resistance [6]. Therefore, it is crucial to develop novel chemotherapeutics that can target and/or kill BCSCs.
Breast Cancer Stem Cells
The term cancer stem cells essentially refer to the relatively smaller cell population within cancer cells that can repopulate the tumour cells over a long period of time as well as retain their ability to regenerate themselves continuously. Thus, cancer stem cells, or tumour-initiating cells, function like normal stem cells but in a diseased manner [7]. Pioneering work on BCSC isolation and characterisation was conducted by Hajj et al. They isolated BCSCs as CD44+CD24−/Lineage− subpopulation within breast cancer cells and demonstrated that their tumorigenic ability resonated with normal stem cells. These cells can undergo self-renewal as well as differentiation. They can multiply extensively, as few as 200 of these unpassaged cells were able to form solid tumours that could be planted in mice. Their multiplication resulted in a heterogeneous population of cells composed of the CD44+CD24−/Lineage− population as well as non-tumorigenic cells that could compose the bulk of the tumour mass [5].
Breast cancer stem cells display increased cell motility and invasion and they overexpress genes that promote metastasis [8]. Most cancer therapies developed and being used today were designed to target cancer cells. Cancer stem cells can easily evade these by virtue of their “stemness”. BCSCs are characterised by an overexpression of the BCL-2 family of proteins and ABC transporters as well as multiple drug resistance. They possess the inherent ability to efflux any administered drug and, thus, can elude all therapies. Therefore, BCSCs are fundamentally more resistant to the currently employed clinical techniques (chemo and radiation) [5,9]. In fact, there are a few reports suggesting that most of these treatments enhance the fraction of CD44+CD24− cells and ALDH+ cells, i.e., BCSCs within the tumour cells. Therefore, it is pertinent to treat breast cancer stem cells along with breast cancer cells to ensure a complete and effective treatment. Consequently, many treatment strategies are being designed to directly target the cancer stem cell population by modulating specific pathways involved in the cancer stem cell phenotype [10,11,12]. Significant progress has been achieved in targeting the Notch pathway, Wnt, and Hedgehog signalling pathway in ongoing clinical trials. In vitro studies using breast cancer stem cell markers reveal that BCSCs are relatively resistant to radiation therapy and chemotherapy. This was also resonated in clinical studies, wherein molecular profiles of tumours treated with chemotherapy closely resembled the gene profiling of CD44+CD24− cells [13]. This resistance is imparted by stem cell specific pathways such as Wnt/β–catenin, Notch, and Hedgehog signalling. These pathways are often dysregulated in breast cancer and, thus, various approaches are being designed to target them for effective treatment [11].
The most noteworthy progress in targeted breast cancer treatment involves the development of HER2 targeted therapies using trastuzumab and lapatinib. This treatment approach is also proposed to act against the ALDH1 cancer stem cell population [14,15]. Apart from immunotherapy, novel treatment strategies are also being developed to target stem cell specific pathways. The most noteworthy of these include a combination of docetaxel and γ-secretase inhibitor MK-0752 against the Notch pathway and is currently in Phase I/II clinical trials. Similarly, clinical trials have also been initiated using combination therapies targeting the canonical Wnt signalling pathway (ClinicalTrials.gov Identifier: NCT01351103) as well as Hedgehog signalling pathways (ClinicalTrials.gov Identifier: NCT02694224).
2. Drug Repurposing: The Anti-Diabetic to Anticancer Action of Metformin
The discovery and understanding of the crucial role played by breast cancer stem cells in cancer propagation and metastasis has now led researchers to develop therapeutic strategies to target and treat cancerous stem cells. Several therapies have been tested in vivo and in vitro to target BCSCs through membrane markers and transporters as well as alteration of the cancer stem cell microenvironment. However, although some of these treatments have been promising in vitro, in vivo toxicity and pharmacokinetics remains a big challenge. Intriguingly, some success has been reported recently, with drugs used clinically to treat other ailments. There are researchers and clinicians all over the globe working towards finding a safe and effective cure for cancer. The average cost for the development of an anticancer drug is over 600 million dollars and takes approximately 10–20 years. Moreover, less than 1% of the drug candidates enter clinical trials, and the proportion of drugs successfully making it to market is even smaller. Therefore, drug repurposing is increasingly being employed particularly in cancer research [16,17,18]. Drug repurposing means exploring the possibility of new therapeutic applications of FDA approved drugs that were not listed in the original drug medication. The major benefit provided by drug repurposing is the fact that the said drugs have already been FDA approved and their safety and biocompatibility, i.e., pharmacokinetics and pharmacodynamics, for humans has already been established. Therefore, these drugs can move fast between preclinical and clinical trials. However, with the plethora of FDA approved drugs available, the discovery of anticancer applications of non-oncology drugs are usually serendipitous [19,20]. Recently, there was also a study reporting a systematic screening of over 4000 FDA approved drugs for over 500 cancer cell lines using PRISM software analysis [19].
Many independent systematic studies reported lower risk and occurrences of cancer in diabetic patients on metformin treatment [21,22]. Consequently, the most noteworthy results for breast cancer using a non-oncology drug have been reported with metformin [23,24,25,26,27].
Metformin acts on mitochondria in the cell, inhibiting the mitochondrial electron transport system by enhancing the AMP-activated protein kinase (AMPK) activity. 5′ adenosine monophosphate-activated protein kinase is an enzyme involved in cellular energy homeostasis, largely to activate glucose and fatty acid uptake and oxidation when cellular energy is low. AMPK is identified as the crucial candidate causing the interaction between metabolism and cancer [28,29]. Activated AMPK induces the resting catabolic state of the cell, thus restoring homeostasis. AMPK activity is also implicated in targeted breast cancer stem cells therapy. There are several reports suggesting that augmented AMPK activity eliminates breast cancer stem cells [30,31,32]. Moreover, there are numerous reports suggesting that metformin can specifically target the cancer stem cell population in breast, pancreatic, glioblastoma, and ovarian cancer models. Metformin treatment significantly reduces the sphere-forming ability and inhibits proliferation in the CD44+CD24− and ALDH+ stem-cell-rich population in breast cancer cells [21,24]. Consequently, metformin is part of several ongoing clinical trials, alone and in combination with clinically used anticancer drugs such as doxorubicin and paclitaxel to target cancer stem cell population along with tumour suppression for breast cancer. The therapeutic actions of metformin are known to proceed via the induction of cellular stress by the inhibition of oxidative phosphorylation in mitochondria, followed by the activation of AMPK [29].
3. AMPK Activation
Mechanism of AMPK Activation
5′ adenosine monophosphate-activated protein kinase (AMPK) is an enzyme complex that undergoes autoactivation under conditions of cellular stress such as exercise, hypoxia, and hypoglycaemia. Some anti-diabetic drugs, such as metformin, and thiazolidinediones, such as pioglitazone, also exert their therapeutic effect by activating AMPK [33]. Structurally, it is a heterotrimeric protein kinase consisting of AMPK α, β, and γ subunits. The catalytic α subunit is primarily involved in the activation of the kinase, while the AMPKβ and AMPKγ subunits are mainly regulatory [34]. The generally accepted mechanism for physiological AMPK activation involves phosphorylation of the Thr-172 residue within the catalytic active site of AMPKα subunit mainly by LKB1 (liver kinase B1) and CaMKKβ (Ca2+/calmodulin-dependent protein kinase β) [35,36]. The LKB1 mediated Thr phosphorylation can also be AMP dependent. Under conditions of cellular stress and a high AMP:ATP ratio, AMP allosterically activates AMPK by binding to the AMPKγ and protects the physiological integrity of the activated AMPK by inhibiting its dephosphorylation. In contrast, the CaMKKβ catalysed activation of AMPK is arbitrated by the concentration of cellular calcium ions. AMPK can also be activated passively by molecules that can induce the accumulation of intracellular AMP or calcium ions. The most well-known examples of indirect activators of AMPK are anti-diabetic drugs such as metformin and pioglitazone. They act on the mitochondria and increase the AMP:ATP ratio, thereby leading to cellular stress and AMPK activation. The various mechanisms of AMPK activation are depicted in Figure 2. After activation, AMPK phosphorylates multiple substrates, initiating a cascade of cellular responses to limit ATP consumption. The overall effect of this regulation is to reduce the synthesis of cholesterol, fatty acids, ribosomal RNAs (rRNAs), and proteins. Consequently, several energy-intense pathways, such as fatty acid synthesis and oxidation, are inhibited. Activated AMPK arrests the G1 phase of the cell cycle along with the expression of apoptotic proteins, p53 and p21, and is known to induce autophagy [37,38,39,40].
4. AMPK Activation for the Treatment of Breast Cancer Stem Cells
AMPK is fast emerging as a target of choice in the treatment of cancer cells and cancer stem cells. It is an energy-sensitive molecule and is activated under conditions of cellular stress, a common phenomenon in cancer cells. However, a few reports argue this activation may actually protect cancer cells [41]; nevertheless, there is ample evidence to suggest the therapeutic action of AMPK activation on cancer stem cells, particularly breast cancer stem cells [31,42,43,44,45,46,47]. Paclitaxel, a well-known and commonly used drug for the treatment of breast and lung cancers, involves the activation of AMPK during its therapeutic pathway [48]. Activated AMPK also influences several cancer stem cell specific pathways, indicating the role of AMPK as a tumour suppressor [49]. Activated AMPK interrupts the G1-S and G2-M phases of mitosis, resulting in cell cycle arrest via downregulation of mTORC1 activity and reduced lipogenesis. AMPK activation can also affect the cell cycle by non-metabolic pathways. Prolonged AMPK activation also results in mitotic spindle misorientation as well as abnormal chromosome segregation [50]. Moreover, AMPK activation has also been shown to target cancer stem cell population by counteracting the epithelial-to-mesenchymal transition (EMT) in breast and prostate cancer cells by activating forkhead box O3 (Foxo3a). EMT in cancer cells is known to generate and maintain stem-like properties in cancer cells and lead to chemoresistance and metastasis [51]. Several direct and indirect AMPK activators have been proposed and tested for their anticancer activities. A-769662, a direct AMPK activator, was shown to delay tumour formation in hypomorphic mice and reduced proliferation in breast, colon and prostate cancer cells. Another AMPK activator, OSU-53, suppresses tumour growth in triple-negative breast cancer models in vitro and in vivo. OSU-53 also constrains EMT in breast and prostate cells by activating Foxo3a, thus influencing the cancer stem cell population as well [52]. Similar results have also been observed in clinical studies, wherein enhanced AMPK expression is associated with relapse-free, longer survival rates in breast cancer patients [53].
An increased AMPK activity could assist in anticancer activity through the interplay of various pathways, some of which are discussed below.
4.1. Activated AMPK Downregulates Cyclin Proteins and Induces Cell Cycle Arrest and Autophagy
About 30–60% of breast cancers are characterised by an overexpression of the cyclin D1 encoding gene, CCND1, and over 50% of breast cancers overexpress cyclin D1 protein. Cyclin D1 plays a crucial role in tumour progression and metastasis by causing abnormal phosphorylation and inhibiting the tumour suppressor protein, retinoblastoma (pRB) [54]. It also serves to enrich the population of cancer stem cells and induce angiogenesis. Cyclin D1 is a vital modulator of mammary and embryonic stem cells and is known to assist transforming growth factor β (TGF-β) in inducing stem cell renewal in triple-negative breast cancer cells [55,56]. Therefore, many promising studies have focussed on downregulating cyclin D1 for enhanced anticancer action [57,58,59,60]. Activated AMPK phosphorylates several substrates and different cyclin(s) leading to autophagy and cell cycle arrest. For instance, AMPK activation is a requirement for cyclin D1 downregulation [61,62]. Activated AMPK phosphorylates phosphoinositide 3-kinase enhancer-activating Akt (PIKE–A) at the Ser 351 and Ser 377 residues, thereby inducing the translocation of PIKE-A into the nucleus. Within the nucleus, PIKE-A complexes with CDK4, destabilizing the CDK4/CyclinD1 complex, thus inhibiting the cyclin D1 activity and inducing cell cycle arrest and decreased cell proliferation [63]. The LKB1–AMPK complex also catalyses the phosphorylation of p27Kip1 at the Thr 198, thereby activating the cyclin-dependent kinase inhibitor [64,65]. Furthermore, activated AMPK induces autophagy by phosphorylating S326 of cyclin Y, leading to the formation of the cyclin Y–CDK16 complex and activation of CDK16 [66]. Although the role of autophagy in cancer can be dual and conflicting, there are shreds of evidence suggesting AMPK activation-mediated autophagy results in decreased cell proliferation and cell death [67,68].
An important consequence of AMPK-mediated cyclin D1 inhibition is the arrest of cell cycle progression in the G0/G1 phase. It is also a commonly encountered pathway of cell death relating to the activation of AMPK. Many studies using metformin on different cancer cell lines as well as cancer stem cells suggest the arresting of the cell cycle at the G1:S boundary, following AMPK-mediated upregulation of CDK-inhibiting p21 and p27 [30,61,69,70,71,72]. The pathways for AMPK-mediated downregulation of cyclin D and induction of autophagy are shown in Figure 3.
4.2. AMPK Activation Inhibits Lipogenic Enzymes
AMPK is the master regulator of energy molecules in the body and is involved in glucose and fat metabolism. Phosphorylation of AMPK is a crucial step involved in the inhibition of adipogenesis and lipogenesis [73,74,75]. Fatty acids play crucial roles in maintaining cellular functions and structure. Normal cells exhibit low activity of fatty acid synthase enzyme; however, the activity is elevated by many folds in fast proliferating cancer cells, making it a suitable therapeutic target. Furthermore, most breast cancers exhibit raised levels of acetyl-CoA carboxylase (ACC), a key enzyme for the synthesis of long-chain fatty acids. AMPK activation downregulates the synthesis of fatty acids, lipids, triglycerides, and cholesterol by acting on multiple substrates. Activated AMPK restrains fatty acid synthesis by deactivating ACC. It affects triglyceride synthesis by inhibiting the activity of glycerol-3-phosphate acyltransferase (GPAT). It also downregulates 3-hydroxy-3-methylglutaryl-CoA reductase (HMG-CoA), a crucial enzyme involved in the synthesis of cholesterol. There are also a few studies suggesting the role of AMPK in deactivating lipogenic gene transcription factors. Therefore, it can be said that activated AMPK acts as the vital controller of lipogenic pathways; and this has direct consequences on oncogenic signalling [76]. Unlike normal cells, cancer cells are commonly characterised by enhanced activity of fatty acid and lipid synthesizing enzymes to compensate for the additional energy and nutrition requirement of fast proliferating cells [77]. These differential adipogenesis and lipogenesis activities have been targeted to develop effective and targeted anticancer therapies. Since AMPK is the master regulator of metabolism, most of these therapies are known to act via direct or indirect activation of AMPK [78,79,80,81,82].
4.3. AMPK Activation Downregulates the Mammalian Target of the Rapamycin (mTOR) Pathway and Insulin Growth Factors (IGFs)
AMPK activation also leads to the downregulation of the mechanistic target of rapamycin complex 1 (mTORC1) to restrict the ATP-expensive anabolic processes. mTORC1 is a nutrition-sensitive protein complex composed of mTOR kinase and the scaffolding protein raptor, catalysing various biosynthetic pathways in the human body [83]. mTOR is a Ser/Thr protein kinase that plays crucial roles in cell growth and survival, autophagy, cell division and proliferation. It promotes tumour growth and progression by providing nutrients to cancer cells and promotes angiogenesis. Thus, mTOR plays a pivotal role in cancer cells, particularly cancer stem cells [84]. There is increasing evidence suggesting the pivotal role played by phosphatidylinositol-3-kinase (PI3K)/Akt and the mammalian target of rapamycin (mTOR) (PI3K/Akt/mTOR) signalling in cancer stem cell biology. mTOR activity is frequently associated with the maintenance of cancer stem cells, and particularly in breast cancer stem cells, mTOR is required for tumorigenicity. mTOR along with IGF also controls cell size, growth, and proliferation in tumours [85]. Many drugs being used clinically today to treat cancers are PI3K/Akt/mTOR suppressors [86,87]. There are many studies reporting a direct link between AMPK activation and mTOR suppression [88]. Activated AMPK suppresses mTOR by phosphorylating and activating the tuberous sclerosis complex (TSC) that negatively regulates mTOR signalling [23,89,90]. It can also directly cause the phosphorylation of raptor at Ser 722 and Ser 792, thereby deactivating the activity of mTORC1 [83]. Deactivation of mTOR subsequently downregulates several downstream substrates, resulting in the loss of cell proliferation, cell shrinkage, loss of translation, and many more effects that result in apoptosis in cancer cells (Figure 4). AMPK activation by metformin also involves downregulation of mTOR for the anticancer effect [91,92]. However, there are a few reports suggesting the metformin-mediated repression of mTOR can be independent of AMPK as well [26].
4.4. AMPK Activity Opposes the Warburg Effect
Cancer cells are fast growing and proliferate at an uncontrolled rate. Therefore, they have higher energy requirements than normal cells. The greater energy demands are met by the Warburg effect—cancer cells that operate at higher rates of glucose uptake and lactate production irrespective of the available oxygen concentration and normal mitochondrial function [93]. The exact role of the Warburg effect on cancer cells is still a topic of research; nevertheless, it is considered an important hallmark of cancer. Furthermore, increased glucose uptake and lactate production might result in the acidosis of the tumour microenvironment. LKB1-mediated AMPK activation has been associated as a negative regulator of the Warburg effect in cancer cells [94,95,96]. Thus, AMPK is identified as the crucial candidate causing the interaction between metabolism and cancer [28]. AMPK activity also exerts anti-inflammatory properties and maintains cellular redox balance. Moreover, AMPK also increases inhibitory phosphorylation of glycogen synthase kinase 3 beta (GSK3β), which contributes to mitochondrial protection against iron-induced oxidant stress [97,98].
4.5. Anticancer Stem Cell Action of AMPK
Apart from the various tumour suppression mechanisms described above, activated AMPK also inhibits several self-renewal and metastasis-related pathways that are often associated with the cancer stem cell population. There are ample reports suggesting the involvement of Wnt/β-catenin, NF-κB, TNFα, interleukin proteins, Notch, and Hedgehog signalling pathways in the differentiation and proliferation of breast cancer stem cells by inducing epithelial-to-mesenchymal transition (EMT) and tumorigenesis [99]. AMPK activation results in a cascade of cellular responses that can affect the cancer stem cell phenotype in different cancers including breast cancer. Activated AMPK is known to inhibit EMT in pancreatic and breast cancer cells by downregulating the sonic Hedgehog signalling pathway and TGFβ cytokines by phosphorylating and destabilizing the transcriptional activator form of Gli-1 [29,100]. AMPK activation also inhibits the canonical Wnt signalling pathway in breast and prostate cancers by downregulating DVL3 [101,102]. The canonical and noncanonical Wnt signalling promotes stemness in breast cancer stem cells. It plays crucial roles in the proliferation, orientation, migration, and resistance in the BCSC subpopulation. Therefore, treatments targeting the Wnt/β-catenin pathway may be potent for removing BCSCs [99]. Furthermore, activated AMPK negatively impacts several inflammatory pathways, such as NF-κB, TNFα, and interleukin proteins, that play crucial roles in populating the cancer stem cell population. There are also studies suggesting the role of AMPK activators in targeting specific microRNA-mediated pathways against cancer stem cells. Metformin upregulates let-7a in MCF7 human breast cancer cells and inhibits TGFβ-induced miR-181a expression subsequently reducing the mammosphere-forming ability in vitro [103].
5. Vanadium Compounds: Insulin Mimetic Agents with Possible Anticancer Action
5.1. Anti-Diabetic Action of Vanadium—PTP1B Inhibition
The anti-diabetic effects of vanadium compounds are well documented in the literature [106,107,108,109]. There are surplus reports of vanadium (IV) and (V) complexes exhibiting moderate to excellent insulin mimetic activity [106,107,110,111,112,113]. The geometrical and physiological similarity between vanadate and phosphate allows for the involvement of the former in phosphate-dependent pathways [114,115]. Vanadium compounds, particularly in oxidation states (IV) and (V), are biocompatible and have shown to improve glucose homeostasis and insulin resistance in both in vitro and in vivo models [110,111,116]. The most successful vanadium compounds to be studied with insulin mimetic properties are BMOV (bis(maltolato)oxovanadium(IV)) and its derivatives such as BEOV (bis(ethylmaltolato)oxovanadium(IV)). They have been shown to induce normoglycemic effects in streptozotocin (STZ)-induced type 1 and type 2 diabetic rats in a dose-dependent manner in the range of 1–10 mg V kg−1 body weight [112]. Despite their exemplary activities, these vanadium compounds have not been successful yet in pharma markets. Nevertheless, a deep understanding of their mechanism of action can be applied to cater to the needs of pharmacological and medicinal research.
Vanadium compounds are known inhibitors of several ATPases and phosphatases with varying potency [111]. The most extensively studied phosphatases in this regard have been tyrosine protein phosphatases (PTPases), alkaline and acid phosphatases. PTPases are a superfamily of enzymes that control signal transduction pathways and regulate cellular functions. They play important roless in several oncogenic and metabolic disorders [117,118]. The process of physiological phosphorylation of proteins is strictly balanced and monitored by two opposing enzymes—protein tyrosine kinases (PTKs) and protein tyrosine phosphatases. PTPases have cysteine residues at the active site, and they catalyse the hydrolysis of phosphate esters. Vanadium compounds can oxidise these cysteine residues via free radical and ROS mechanisms thus inhibiting the biological responses that depend on the thiol-reducing ability. Alkaline phosphatases are membrane-bound glycoproteins that catalyse the phosphorylation of small organic molecules. The vanadium induced inhibition of alkaline phosphatases results from the geometrical similarity between vanadates and phosphates (Figure 6). The greater coordination flexibility of vanadate allows them to mimic and replace the penta-coordinated transition states of phosphates in biological systems, thereby initiating a cascade of cellular responses and inhibition [119]. Moreover, the “hard” nature of vanadium allows for the formation of strong hydrogen bonds with amino acid residues, thereby leading to the stabilisation of negatively charged transition states in physiological processes. This also results in inactivation or inhibition of the respective enzyme and the cellular pathway. Thus, vanadium compounds can inhibit most phosphatases in a non-specific manner [114]. The insulin mimicking action of vanadium results from this inhibition, particularly the inhibition of PTP1B. PTP1B is the key negative regulator of insulin signalling [120]. The insulin receptors are present as a transmembrane protein on all mammalian cells. Insulin binding results in the phosphorylation of the tyrosine residues at the insulin receptors. This tyrosine phosphorylation subsequently activates intracellular signalling, leading to glucose uptake and metabolism. PTP1B negatively impacts the cascade by dephosphorylating the tyrosine residues at the insulin receptors. Therefore, inhibition of PTP1B improves insulin sensitivity and has been widely used to treat diabetes [121]. There are ample reviews on vanadium-mediated PTP1B inhibition [110,114,122,123], and to keep this work concise, it will not be discussed in detail here.
5.2. PTP1B and Breast Cancer
PTP1B is localised in the endoplasmic reticulum in the cells and downregulates leptin receptors and insulin signalling by dephosphorylating the IRS [124]. This dephosphorylation results in the deactivation of the insulin receptor, thereby leading to hyperglycaemic conditions and insulin resistance. Therefore, PTP1B has emerged as a target of choice for developing effective anti-diabetic and anti-obesity treatments [125]. The role of PTP1B in breast cancer can be contentious; it has been shown to be a suppressor as well as an oncogene. While there are a few reports suggesting the prognostic action of PTP1B in cancers [126,127], there are several reports that counteract and suggest otherwise [128,129,130]. The gene encoding for PTP1B, PTPN1, is located on chromosome number 20q13, which is often augmented in breast cancers [128], and it is overexpressed in at least 70% of all breast cancers [131,132]. PTP1B expression has been shown to promote tumorigenesis in breast cancers, and several in vivo studies have shown that PTP1B knockout results in delayed tumour formation [129,133,134]. Inhibition of PTP1B results in decreased cell adhesion by downregulation of claudin-1, FAK, and E-cadherin and induces anoikis [120,135,136]. Recently, many anticancer agents have targeted for breast cancer in vitro and in vivo and have shown to act via inhibition of PTP1B activity. Some recent reports are presented in Table 2. Although, none of these studies report the direct involvement of PTP1B inhibition against cancer stem cells, there are a few reports in the literature suggesting the role of PTP1B in breast cancer stem cell specific pathways. The inhibition of PTP1B in D492 and HMLE cells induces loss of extracellular matrix attachment and apoptosis. The same report also suggested greater sensitivity of breast mesenchymal cells to PTP1B inhibitors, thereby signifying the role of PTP1B inhibitors for targeting cancer stem cell population in breast cancers [120]. Another possible mechanism of action of PTP1B inhibition against BCSCs involve the role of chemokine CCL5. CCL5 is secreted by the mesenchymal stem cells in breast cancer cells, which then enhances their motility, invasion, and migration [137]. PTP1B upregulates the expression of CCL5 in breast cancer cells; therefore, PTP1B inhibition can be a promising approach for the treatment of breast cancer [132].
5.3. PTP1B Inhibition of AMPK Activation: Can Vanadium Compounds Activate AMPK?
There is increasing evidence suggesting the link between PTP inhibition and AMPK activation. Several biochemical consequences of PTP inhibition are believed to be executed via AMPK activation [149]. PTP1B is known to negatively modulate AMPK in peripheral tissues [33,150,151], and PTP1B deficiency is known to cause AMPK activation and autophagy [152]. Many studies suggesting the development of novel PTP1B inhibitors for the management and treatment of diabetes also report enhancement of AMPK activity along with PTP1B inhibition [124,153,154]. Hence, there appears to be a clear link between PTP1B inhibition and the simultaneous AMPK activation.
This leads to an interesting hypothesis—can insulin mimetic vanadium compounds with PTP1B inhibitory activity also activate AMPK? This can have important implications in developing effective anticancer treatments, since vanadium compounds are also known for their anticancer action; however, their efficacy on cancer stem cells has not yet been explored in detail.
5.4. Vanadium-Mediated AMPK Activation—Reduced Adipogenesis
Obesity is commonly associated with diabetes, and many anti-diabetic drugs are known to inhibit adipocyte differentiation. The role of vanadium in AMPK activation has mainly been focussed on vanadium-mediated inhibition of adipogenesis. This arises as a direct consequence of the fact that adipocyte differentiation depends on several growth factors including IGF and insulin. Adipogenesis is the process by which mesenchymal stem cells differentiate and mature into adipocytes and is often considered a hallmark for cancer stem cells. It is a complex process involving the interplay of several proteins, such as PPARγ, and transforming growth factors, such as KLF and IGF. Vanadium compounds are known to inhibit adipogenesis and, thus, many insulin mimetic agents can also be used to treat obesity. The most generally accepted mechanism of vanadium-induced inhibition of adipocyte differentiation involves AMPK activation along with a few other pathways. Adipose tissues form the bulk of the breast cancer microenvironment. It is involved in the synthesis of various adipokines and signalling molecules (such as IL6, leptin, adipsin, and tumour necrosis factor-α) that play crucial roles in breast tumour formation and progression. Leptin signalling plays an important role in tumour progression and maintenance of the cancer stem cell population by upregulating the cancer stem cell signalling pathways, Notch and Wnt [155]. Similarly, IL6 also has implications in inducing epithelial–mesenchymal transitions and increasing the breast cancer stem cell population. Therefore, within the breast cancer microenvironment, adipocytes play a pivotal role in inducing and maintaining the cancer stem cell population, and inhibition of adipogenesis can prove to be a successful pathway for the treatment of breast cancer stem cells [156,157].
AMPK can undergo activation in the adipose tissue in response to stress such as malnutrition, exercising, and extreme cold. It can also be activated by endogenous molecules, such as high-density lipoproteins and IL6, in the adipose tissue including other activation pathways discussed above. Along with LKB1, AMPK in the adipose tissue can be phosphorylated at Thr172 by adipokines such as leptin and adiponectin. AMPK activation directly affects lipogenesis and fat accumulation. Activated AMPK phosphorylates several substrates resulting in the inhibition of the synthesis of fatty acids, triglycerides and cholesterol, and it promotes fatty acid oxidation.
The role and application of vanadium compounds in AMPK-mediated inhibition of adipogenesis are currently not very clearly understood. However, there are a few recent reports suggesting AMPK activation in insulin mimetic action as well as decreased adipogenesis by vanadium compounds [158,159,160]. Some recently reported vanadium compounds with significant adipogenesis inhibition activity are collected in Figure 7 and Table 3. Insulin mimetic vanadium compounds, such as VOSO4 and BMOV, can activate the GLUT translocator in adipose tissues [113]. Recently, a study reported the upregulation of hepatic AMPK levels and Akt cascade by a synergistic combination of metformin and bis(α-furancarboxylato)oxovanadium(IV) (BFOV) for the treatment of fatty liver disease [161]. Such reports of synergistic action of metformin and vanadium salts are not uncommon [162]. The most extensively studied vanadium compound in this context is vanadium(IV)chlorodipicolinate. It is known to inhibit adipogenesis in 3T3-L1 preadipocytes via LKB1-mediated activation of AMPK and the subsequent downregulation of ACC, FAS, FABP4, and PPARγ. It can also inhibit the storage of lipids and triglycerides in cells and upregulate microtube-associated light-chain proteins, inducing autophagy in hepatocytes by activation of the LKB1/AMPK pathway [163,164]. Vanadium-mediated insulin mimetic and anti-obesity action following AMPK action has also been reported using groundwater enriched with vanadium [165]. Similar results were also reported using vanadium extracted from Brassica napus cultured in vanadium-rich Jeju water. The vanadium-containing extract was reported to exhibit anti-diabetic effects as well as reduced triglyceride accumulation and adipogenesis in in vivo models. The suggested mechanism of action involved AMPK-mediated upregulation of glycogen synthesis by enhanced triacylglycerol lipase activity resulting in increased glucose uptake in adipocytes [166].
Vanadium catalysed AMPK activation and reduced adipocyte differentiation might also involve simultaneous activation of the peroxisome proliferator-activated receptor (PPARγ) [167,168,169]. PPARγ is a controller of metabolic pathways, and its activation is associated with the consequent activation of AMPK [160]. The anti-diabetic thiazolidinediones also exert their therapeutic effects by activating PPARγ. There are few reports suggesting the vanadium-mediated activation of PPARγ and AMPK [160].
Although the exact role of PPARγ in tumorigenesis is ambiguous, it can be modulated in a cell line specific manner to achieve anticancer properties [170]. PPARγ agonists have shown promising results in impeding cancer and reducing cancer stem cell population [171,172,173,174]. Cancer stem cells, particularly breast cancer stem cells, exhibit overexpression of inflammatory factors, such as NFκB, IL6 and IL8, that play a pivotal role in upregulating mammosphere regulatory genes (SLUG, Notch3 and Jagged1) and assist in the maintenance of the breast cancer stem cell population [175]. Upregulation of PPARγ negatively impacts on the cancer stem cell population by impeding the activity of these inflammatory growth factors. Thus, many PPARγ ligands and activators find potent anticancer applications, the most common being cisplatin.
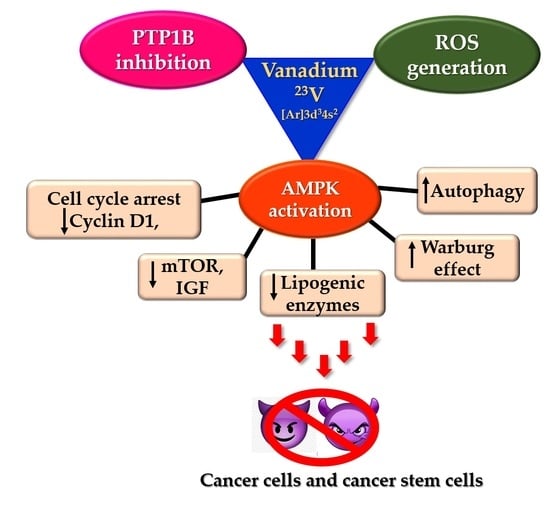
Thus, insulin mimetic vanadium compounds inhibit PTP1B activity and activate AMPK and PPARγ to suppress adipogenesis. Additionally, vanadium compounds are generally redox active under physiological conditions and can readily interconvert between VO(IV) and VO(V) using Fenton-like reactions within the cell. This redox interconversion results in the generation of ROS. These ROS can, in turn, further cause PTP inhibition as well as induce cytotoxicity [114,176]. Consequently, the anticancer properties of oxidovanadium complexes are well documented in the literature [111,176,177,178]. Vanadium (IV) compounds have been shown to inhibit cell adhesion to the extracellular matrix and cell migration in osteosarcoma cells [179]. Notably, there are also a few reports suggesting the role of vanadium-mediated inhibition of TGFβ-induced EMT. Dysregulated EMT in cancer cells is attributed as one of the main causes of cancer metastasis and the generation of the mesenchymal cancer stem cell phenotype. The insulin mimetic activity of sodium vanadate results in downregulation of protein tyrosine phosphatase and the activated insulin receptor substrate -1 in A549 cells. The activated IRS-1 subsequently suppresses TGFβ-mediated EMT [180]. Similar results were also reported using vanadium(V)–peroxido–betaine complex on A549 cells in vitro and ex vivo. The study reported vanadium-mediated inhibition of TGFβ-induced EMT and disruption of mitochondrial membrane potential in tumour cells. Furthermore, the vanadium (V) drug was found to act synergistically with the clinically used drug carboplatin with reduced expression of cancer stem cell markers [181]. However, the role of vanadium in targeting breast cancer stem cells has yet not been explored, but there are many insulin mimetic vanadium compounds in clinical trials [109,182,183] that could have the potential of successfully treating breast cancer and cancer stem cells.
6. Conclusions
Non-oncology drugs have gained increasing attention recently for their possible anticancer action. These drugs with well-defined pharmacokinetic and pharmacodynamic profiles fast track into clinical trials. The most successful non-oncology drug for cancer so far is metformin. Metformin has shown potent activity against many cancers, particularly breast cancer and breast cancer stem cells. The drug exerts its anti-diabetic and anticancer action through AMPK activation. AMPK is an energy-sensing molecule playing pivotal roles in metabolic processes. The phosphorylation and activation of AMPK have several implications that result in reduced cell proliferation and apoptosis in cancer cells. Activated AMPK phosphorylates several downstream substrates resulting in downregulation of cyclins, cell cycle arrest, suppression of fatty acid, cholesterol, triglyceride syntheses, inhibition of the mTOR pathway and induction of autophagy and Warburg effect antagonism. All these factors are involved in breast cancer as well as breast cancer stem cells and, thus, there are ample reports suggesting the inhibition of one or more of these pathways causing anticancer activity. AMPK activation also has implications in impeding adipogenesis, and many vanadium-based insulin mimetic agents can inhibit adipocyte differentiation following AMPK activation. Vanadium-based insulin mimetic compounds are known PTP1B inhibitors, and few were/are in clinical trials for the treatment of type 2 diabetes and obesity. There is clear evidence of the link between the interplay of PTP1B inhibition and AMPK activation in the prognosis and treatment of breast cancer. Although the anticancer applications of vanadium compounds are well documented, not much has been reported regarding the action of vanadium on cancer stem cells. Vanadium compounds possess all the required abilities to target and inhibit cancer and cancer stem cells. The recent development of vanadium-catalysed suppression of adipogenesis and the vast history of vanadium-mediated PTP1B inhibition and ROS generation can be carefully manipulated to target cancer stem cells, particularly breast cancer stem cells. This review presents the possibility of employing vanadium-based PTP1B inhibitors for activation of AMPK and treatment of breast cancer by targeting their stem cell population.
Author Contributions
Conceptualisation, B.U.; writing—original draft preparation, B.U.; writing—review and editing, B.U. and H.A.; supervision, H.A.; funding acquisition, H.A. All authors have read and agreed to the published version of the manuscript.
Funding
This work is based on the research supported by the South African Research Chairs Initiative of the Department of Science and Technology and National Research Foundation of South Africa (Grant No: 98337) as well as grants received from the University of Johannesburg (URC), the National Research Foundation (NRF), and the CSIR (Council for Scientific and Industrial Research)—NLC (National Laser Centre) Laser Rental Pool Programme.
Acknowledgments
B.U. is thankful to the Laser Research Centre and H. Abrahamse for awarding NRF SARChI Post Doctoral Fellowship.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 22 November 2021).
- Dittmer, J. Breast cancer stem cells: Features, key drivers and treatment options. Semin. Cancer Biol. 2018, 53, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butti, R.; Gunasekaran, V.P.; Kumar, T.V.S.; Banerjee, P.; Kundu, G.C. Breast cancer stem cells: Biology and therapeutic implications. Int. J. Biochem. Cell Biol. 2019, 107, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Chen, Q.; Zou, Y.; Chen, H.; Qi, L.; Chen, Y. Stem cells and cellular origins of breast cancer: Updates in the rationale, controversies, and therapeutic implications. Front. Oncol. 2019, 9, 820. [Google Scholar] [CrossRef] [PubMed]
- Ponti, D.; Zaffaroni, N.; Capelli, C.; Daidone, M.G. Breast cancer stem cells: An overview. Eur. J. Cancer 2006, 42, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Patel, M.R.; Prescher, J.A.; Patsialou, A.; Qian, D.; Lin, J.; Wen, S.; Chang, Y.F.; Bachmann, M.H.; Shimono, Y.; et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc. Natl. Acad. Sci. USA 2010, 107, 18115–18120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, S.; Schuetz, J.D.; Bunting, K.D.; Colapietro, A.M.; Sampath, J.; Morris, J.J.; Lagutina, I.; Grosveld, G.C.; Osawa, M.; Nakauchi, H.; et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 2001, 7, 1028–1034. [Google Scholar] [CrossRef]
- Zeng, X.; Liu, C.; Yao, J.; Wan, H.; Wan, G.; Li, Y.; Chen, N. Breast cancer stem cells, heterogeneity, targeting therapies and therapeutic implications. Pharmacol. Res. 2021, 163, 105320. [Google Scholar] [CrossRef] [PubMed]
- Palomeras, S.; Ruiz-Martínez, S.; Puig, T. Targeting breast cancer stem cells to overcome treatment resistance. Molecules 2018, 23, 2193. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target Ther. 2020, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Wicha, M.S. Targeting breast cancer stem cells. J. Clin. Oncol. 2010, 28, 4006–4012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Castillo, B.; Lopez-Bonet, E.; Cuyàs, E.; Viñas, G.; Pernas, S.; Dorca, J.; Menendez, J.A. Cancer stem cell-driven efficacy of trastuzumab (Herceptin): Towards a reclassification of clinically HER2-positive breast carcinomas. Oncotarget 2015, 6, 32317–32338. [Google Scholar] [CrossRef]
- Korkaya, H.; Paulson, A.; Iovino, F.; Wicha, M.S. HER2 regulates the mammary stem/progenitor cell population driving tumorigenesis and invasion. Oncogene 2008, 27, 6120–6130. [Google Scholar] [CrossRef] [Green Version]
- Fu, L.; Jin, W.; Zhang, J.; Zhu, L.; Lu, J.; Zhen, Y.; Zhang, L.; Ouyang, L.; Liu, B.; Yu, H. Repurposing non-oncology small-molecule drugs to improve cancer therapy: Current situation and future directions. Acta Pharm. Sin. B 2021. [Google Scholar] [CrossRef]
- Waissengrin, B.; Wolf, I.; Zahavi, T.; Salmon-Divon, M.; Sonnenblick, A. The effect of non-oncology drugs on clinical and genomic risk in early luminal breast cancer. J. Clin. Oncol. 2021, 39, e12505. [Google Scholar] [CrossRef]
- Zhang, Z.; Ji, J.; Liu, H. Drug repurposing in oncology: Current evidence and future direction. Curr. Med. Chem. 2021, 28, 2175–2194. [Google Scholar] [CrossRef] [PubMed]
- Corsello, S.M.; Nagari, R.T.; Spangler, R.D.; Rossen, J.; Kocak, M.; Bryan, J.G.; Humeidi, R.; Peck, D.; Wu, X.; Tang, A.A.; et al. Discovering the anticancer potential of non-oncology drugs by systematic viability profiling. Nat. Cancer 2020, 1, 235–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beijersbergen, R.L. Old drugs with new tricks. Nat. Cancer 2020, 1, 153–155. [Google Scholar] [CrossRef] [PubMed]
- Gandini, S.; Puntoni, M.; Heckman-Stoddard, B.M.; Dunn, B.K.; Ford, L.; DeCensi, A.; Szabo, E. Metformin and cancer risk and mortality: A systematic review and meta-analysis taking into account biases and confounders. Cancer Prev. Res. 2014, 7, 867–885. [Google Scholar] [CrossRef] [Green Version]
- Evans, J.M.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. Br. Med. J. 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dowling, R.J.O.; Goodwin, P.J.; Stambolic, V. Understanding the benefit of metformin use in cancer treatment. BMC Med. 2011, 9, 33. [Google Scholar] [CrossRef] [Green Version]
- Kasznicki, J.; Sliwinska, A.; Drzewoski, J. Metformin in cancer prevention and therapy. Ann. Transl. Med. 2014, 2, 57. [Google Scholar] [CrossRef]
- Gillies, R.J.; Pilot, C.; Marunaka, Y.; Fais, S. Targeting acidity in cancer and diabetes. Biochim. Biophys. Acta Rev. Cancer 2019, 1871, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Sahra, I.B.; Regazzetti, C.; Robert, G.; Laurent, K.; le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef] [Green Version]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The beneficial effects of metformin on cancer prevention and therapy: A comprehensive review of recent advances. Cancer Manag. Res. 2019, 11, 3295–3313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jalving, M.; Gietema, J.A.; Lefrandt, J.D.; de Jong, S.; Reyners, A.K.L.; Gans, R.O.B.; Vries, E.G.E.D. Metformin: Taking away the candy for cancer? Eur. J. Cancer 2010, 46, 2369–2380. [Google Scholar] [CrossRef] [PubMed]
- Saini, N.; Yang, X. Metformin as an anti-cancer agent: Actions and mechanisms targeting cancer stem cells. Acta Biochim. Biophys. Sin. 2018, 50, 133–143. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Wu, W.; Yang, F.; Liu, L.; Yang, Z.; Liu, L.; Tang, W.; Sun, F.; Lin, H. Marine sponge-derived smenospongine preferentially eliminates breast cancer stem-like cells via p38/AMPKα pathways. Cancer Med. 2018, 7, 3965–3976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, H.; Orhan, Y.C.; Zha, X.; Esencan, E.; Chatterton, R.T.; Bulun, S.E. AMP-activated protein kinase and energy balance in breast cancer. Am. J. Transl. Res. 2017, 9, 197–213. [Google Scholar]
- Song, C.W.; Lee, H.; Dings, R.P.M.; Williams, B.; Powers, J.; dos Santos, T.; Choi, B.H.; Park, H.J. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci. Rep. 2012, 2, 362. [Google Scholar] [CrossRef] [Green Version]
- Xue, B.; Pulinilkunnil, T.; Murano, I.; Bence, K.K.; He, H.; Minokoshi, Y.; Asakura, K.; Lee, A.; Haj, F.; Furukawa, N.; et al. Neuronal protein tyrosine phosphatase 1B deficiency results in inhibition of hypothalamic AMPK and isoform-specific activation of AMPK in peripheral tissues. Mol. Cell. Biol. 2009, 29, 4563–4573. [Google Scholar] [CrossRef] [Green Version]
- Pinter, K.; Jefferson, A.; Czibik, G.; Watkins, H.; Redwood, C. Subunit composition of AMPK trimers present in the cytokinetic apparatus: Implications for drug target identification. Cell Cycle 2012, 11, 917–921. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Yang, G.; Kim, Y.; Kim, J.; Ha, J. AMPK activators: Mechanisms of action and physiological activities. Exp. Mol. Med. 2016, 48, e224. [Google Scholar] [CrossRef] [Green Version]
- Baron, S.J.; Li, J.; Russell, R.R.; Neumann, D.; Miller, E.J.; Tuerk, R.; Wallimann, T.; Hurley, R.L.; Witters, L.A.; Young, L.H. Dual mechanisms regulating AMPK kinase action in the ischemic heart. Circ. Res. 2005, 96, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Yang, W.; Wu, F.; Wang, C.; Yu, L.; Tang, L.; Qiu, B.; Li, Y.; Guo, L.; Wu, M.; et al. Prognostic significance of AMPK activation and therapeutic effects of metformin in hepatocellular carcinoma. Clin. Cancer Res. 2013, 19, 5372–5380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.Y.; He, Y.Y. Targeting the AMP-activated protein kinase for cancer prevention and therapy. Front. Oncol. 2013, 3, 175. [Google Scholar] [CrossRef] [Green Version]
- Chuang, H.-C.; Chou, C.-C.; Kulp, S.K.; Chen, C.-S. AMPK as a potential anticancer target—Friend or foe? Curr. Pharm. Des. 2014, 20, 2607–2618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harhaji-Trajkovic, L.; Vilimanovich, U.; Kravic-Stevovic, T.; Bumbasirevic, V.; Trajkovic, V. AMPK-mediated autophagy inhibits apoptosis in cisplatin-treated tumour cells. J. Cell. Mol. Med. 2009, 13, 3644–3654. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, N.; Liu, P.; Xie, X. AMPK and cancer. In AMP-activated Protein Kinase; Cordero, M.D., Viollet, B., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 203–226. ISBN 978-3-319-43589-3. [Google Scholar]
- Vara-Ciruelos, D.; Russell, F.M.; Hardie, G. The strange case of AMPK and cancer: Dr Jekyll or Mr Hyde? Open Biol. 2019, 9, 190099. [Google Scholar] [CrossRef] [Green Version]
- Yi, Y.; Chen, D.; Ao, J.; Zhang, W.; Yi, J.; Ren, X.; Fei, J.; Li, F.; Niu, M.; Chen, H.; et al. Transcriptional suppression of AMPKα1 promotes breast cancer metastasis upon oncogene activation. Proc. Natl. Acad. Sci. USA 2020, 117, 8013–8021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, C.; Zou, H.; Xiao, T.; Liu, X.; Wang, Q.; Cheng, J.; Fu, S.; Peng, J.; Xie, X.; Fu, J. TQFL12, a novel synthetic derivative of TQ, inhibits triple-negative breast cancer metastasis and invasion through activating AMPK/ACC pathway. J. Cell Mol. Med. 2021, 25, 10101–10110. [Google Scholar] [CrossRef] [PubMed]
- Penugurti, V.; Khumukcham, S.S.; Padala, C.; Dwivedi, A.; Kamireddy, K.R.; Mukta, S.; Bhopal, T.; Manavathi, B. HPIP protooncogene differentially regulates metabolic adaptation and cell fate in breast cancer cells under glucose stress via AMPK and RNF2 dependent pathways. Cancer Lett. 2021, 518, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Tran, Q.H.; Hoang, D.H.; Song, M.; Choe, W.; Kang, I.; Kim, S.S.; Ha, J. Melatonin and doxorubicin synergistically enhance apoptosis via autophagy-dependent reduction of AMPKα1 transcription in human breast cancer cells. Exp Mol Med. 2021, 53, 1413–1422. [Google Scholar] [CrossRef]
- Seto-Tetsuo, F.; Arioka, M.; Miura, K.; Inoue, T.; Igawa, K.; Tomooka, K.; Takahashi-Yanaga, F.; Sasaguri, T. DIF-1 inhibits growth and metastasis of triple-negative breast cancer through AMPK-mediated inhibition of the mTORC1-S6K signaling pathway. Oncogene 2021, 40, 5579–5589. [Google Scholar] [CrossRef]
- Kim, J.H.; Lee, J.O.; Kim, N.; Lee, H.J.; Lee, Y.W.; Kim, H.I.; Kim, S.J.; Park, S.H.; Kim, H.S. Paclitaxel suppresses the viability of breast tumor MCF7 cells through the regulation of EF1á and FOXO3a by AMPK signaling. Int. J. Oncol. 2015, 47, 1874–1880. [Google Scholar] [CrossRef] [Green Version]
- Cao, W.; Li, J.; Hao, Q.; Vadgama, J.V.; Wu, Y. AMP-activated protein kinase: A potential therapeutic target for triple-negative breast cancer 11 medical and health sciences 1112 oncology and carcinogenesis. Breast Cancer Res. 2019, 21, 29. [Google Scholar] [CrossRef] [Green Version]
- Zadra, G.; Batista, J.L.; Loda, M. Dissecting the dual role of AMPK in cancer: From experimental to human studies. Mol. Cancer Res. 2015, 13, 1059–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, C.C.; Lee, K.H.; Lai, I.L.; Wang, D.; Mo, X.; Kulp, S.K.; Shapiro, C.L.; Chen, C.S. AMPK reverses the mesenchymal phenotype of cancer cells by targeting the Akt-MDM2-Foxo3a signaling axis. Cancer Res. 2014, 74, 4783–4795. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Hsu, E.C.; Guh, J.H.; Yang, H.C.; Wang, D.; Kulp, S.K.; Shapiro, C.L.; Chen, C.S. Targeting energy metabolic and oncogenic signaling pathways in triple-negative breast cancer by a novel adenosine monophosphate-activated protein kinase (AMPK) activator. J. Biol. Chem. 2011, 286, 39247–39258. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Storr, S.J.; Johnson, K.; Green, A.R.; Rakha, E.A.; Ellis, I.O.; Morgan, D.A.L.; Martin, S.G. Involvement of metformin and AMPK in the radioresponse and prognosis of luminal versus basal-like breast cancer treated with radiotherapy. Oncotarget 2014, 5, 12936–12949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Sante, G.; Page, J.; Jiao, X.; Nawab, O.; Cristofanilli, M.; Skordalakes, E.; Pestell, R.G. Recent advances with cyclin-dependent kinase inhibitors: Therapeutic agents for breast cancer and their role in immuno- oncology. Expert Rev. Anticancer Ther. 2019, 19, 569–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, G.; Dai, M.; Zhang, C.; Poulet, S.; Moamer, A.; Wang, N.; Boudreault, J.; Ali, S.; Lebrun, J.J. TGFβ/cyclin D1/Smad-mediated inhibition of BMP4 promotes breast cancer stem cell self-renewal activity. Oncogenesis 2021, 10, 21. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.; Al-Odaini, A.A.; Fils-Aimé, N.; Villatoro, M.A.; Guo, J.; Arakelian, A.; Rabbani, S.A.; Ali, S.; Lebrun, J.J. Erratum to: Cyclin D1 cooperates with p21 to regulate TGFβ-mediated breast cancer cell migration and tumor local invasion. Breast Cancer Res. 2017, 19, 43. [Google Scholar] [CrossRef] [Green Version]
- Millar, E.K.A.; Dean, J.L.; McNeil, C.M.; O’Toole, S.A.; Henshall, S.M.; Tran, T.; Lin, J.; Quong, A.; Comstock, C.E.S.; Witkiewicz, A.; et al. Cyclin D1b protein expression in breast cancer is independent of cyclin D1a and associated with poor disease outcome. Oncogene 2009, 28, 1812–1820. [Google Scholar] [CrossRef] [Green Version]
- Jiao, J.; Huang, L.; Ye, F.; Shi, M.F.; Cheng, X.D.; Wang, X.Y.; Hu, D.X.; Xie, X.; Lu, W.G. Cyclin D1 affects epithelial-mesenchymal transition in epithelial ovarian cancer stem cell-like cells. OncoTargets Ther. 2013, 6, 667–677. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J.; Beach, D.; Shapiro, G.I. Targeting CDK4 and CDK6: From discovery to therapy. Cancer Discov. 2016, 6, 353–367. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.; Rychahou, P.; Sviripa, V.M.; Weiss, H.L.; Liu, C.; Watt, D.S.; Evers, B.M. Induction of AMPK activation by N,N′-diarylurea FND-4b decreases growth and increases apoptosis in triple negative and estrogen-receptor positive breast cancers. PLoS ONE 2019, 14, e0209392. [Google Scholar] [CrossRef] [Green Version]
- Zhuang, Y.; Keith, W.K. Cell cycle arrest in Metformin treated breast cancer cells involves activation of AMPK, downregulation of cyclin D1, and requires p27Kip1 or p21Cip1. J. Mol. Signal. 2008, 3, 18. [Google Scholar] [CrossRef] [Green Version]
- Zang, Y.; Yu, L.F.; Nan, F.J.; Feng, L.Y.; Li, J. AMP-activated protein kinase is involved in neural stem cell growth suppression and cell cycle arrest by 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside and glucose deprivation by down-regulating phospho-retinoblastoma protein and cyclin D. J. Biol. Chem. 2009, 284, 6175–6184. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Sheng, H.; Zhang, X.; Qi, Q.; Chan, C.B.; Li, L.; Shan, C.; Ye, K. Cellular energy stress induces AMPK-mediated regulation of glioblastoma cell proliferation by PIKE-A phosphorylation. Cell Death Dis. 2019, 10, 222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Shao, S.H.; Xu, Z.-X.; Hennessy, B.; Ding, Z.; Larrea, M.; Kondo, S.; Dumont, D.J.; Gutterman, J.U.; Walker, C.L.; et al. The energy sensing LKB1–AMPK pathway regulates p27kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007, 9, 218–224. [Google Scholar] [CrossRef]
- Casimiro, M.C.; di Sante, G.; di Rocco, A.; Loro, E.; Pupo, C.; Pestell, T.G.; Bisetto, S.; Velasco-Velázquez, M.A.; Jiao, X.; Li, Z.; et al. Cyclin D1 restrains oncogene-induced autophagy by regulating the AMPK–LKB1 signaling axis. Cancer Res. 2017, 77, 3391–3405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dohmen, M.; Krieg, S.; Agalaridis, G.; Zhu, X.; Shehata, S.N.; Pfeiffenberger, E.; Amelang, J.; Bütepage, M.; Buerova, E.; Pfaff, C.M.; et al. AMPK-dependent activation of the Cyclin Y/CDK16 complex controls autophagy. Nat. Commun. 2020, 11, 1032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aryal, P.; Kim, K.; Park, P.H.; Ham, S.; Cho, J.; Song, K. Baicalein induces autophagic cell death through AMPK/ULK1 activation and downregulation of mTORC1 complex components in human cancer cells. FEBS J. 2014, 281, 4644–4658. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Sun, D.; Gao, S.; Gao, Y.; Ye, J.; Liu, P. Cyclovirobuxine D induces autophagy-associated cell death via the Akt/mTOR pathway in MCF-7 human breast cancer cells. J. Pharmacol. Sci. 2014, 125, 74–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, X.; Hu, X.; Tan, X.; Cheng, W.; Wang, Q.; Chen, X.; Guan, Y.; Chen, C.; Jing, X. Metformin induced AMPK activation, G0/G1 phase cell cycle arrest and the inhibition of growth of Esophageal squamous cell carcinomas in vitro and in vivo. PLoS ONE 2015, 10, e0133349. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.T.; Ho, H.J.; Lin, J.T.; Shieh, J.J.; Wu, C.Y. Simvastatin-induced cell cycle arrest through inhibition of STAT3/SKP2 axis and activation of AMPK to promote p27 and p21 accumulation in hepatocellular carcinoma cells. Cell Death Dis. 2017, 8, e2626. [Google Scholar] [CrossRef] [Green Version]
- Queiroz, E.A.I.F.; Puukila, S.; Eichler, R.; Sampaio, S.C.; Forsyth, H.L.; Lees, S.J.; Barbosa, A.M.; Dekker, R.F.H.; Fortes, Z.B.; Khaper, N. Metformin induces apoptosis and cell cycle arrest mediated by oxidative stress, AMPK and FOXO3a in MCF-7 breast cancer cells. PLoS ONE 2014, 9, e98207. [Google Scholar] [CrossRef]
- Liu, Z.; Ren, L.; Liu, C.; Xia, T.; Zha, X.; Wang, S. Phenformin induces cell cycle change, apoptosis, and mesenchymal-epithelial transition and regulates the AMPK/mTOR/p70s6k and MAPK/ERK pathways in breast cancer cells. PLoS ONE 2015, 10, e0131207. [Google Scholar] [CrossRef]
- Cai, J.; Qiong, G.; Li, C.; Sun, L.; Luo, Y.; Yuan, S.; Gonzalez, F.J.; Xu, J. Manassantin B attenuates obesity by inhibiting adipogenesis and lipogenesis in an AMPK dependent manner. FASEB J. 2021, 35, e21496. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Lv, Y.; Zheng, M.; Yin, L.; Wang, X.; Fu, Y.; Yu, B.; Li, J. Polyphenols from blue honeysuckle (Lonicera caerulea var. edulis) berry inhibit lipid accumulation in adipocytes by suppressing lipogenesis. J. Ethnopharmacol. 2021, 279, 114403. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Chen, G.; Hu, T.; Mo, X.; Hou, X.; Cao, K.; Wang, L.; Pan, Z.; Wu, Q.; Li, X.; et al. Resveratrol ameliorates lipid accumulation and inflammation in human SZ95 sebocytes via the AMPK signaling pathways in vitro. J. Dermatol. Sci. 2021, 103, 156–166. [Google Scholar] [CrossRef] [PubMed]
- McFadden, J.W.; Corl, B.A. Activation of AMP-activated protein kinase (AMPK) inhibits fatty acid synthesis in bovine mammary epithelial cells. Biochem. Biophys. Res. Commun. 2009, 390, 388–393. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schul, A. Greasing the wheels of the cancer machine: The role of lipid metabolism in cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Guo, D.; Hildebrandt, I.J.; Prins, R.M.; Soto, H.; Mazzotta, M.M.; Dang, J.; Czernin, J.; Shyy, J.Y.J.; Watson, A.D.; Phelps, M.; et al. The AMPK agonist AICAR inhibits the growth of EGFRvIII-expressing glioblastomas by inhibiting lipogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 12932–12937. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.-H.; Tsai, S.-J.; Wang, Y.-J.; Pan, M.-H.; Kao, J.-Y.; Way, T.-D. EGCG inhibits protein synthesis, lipogenesis, andcell cycle progression through activation of AMPK inp53 positive and negative human hepatoma cells. Mol. Nutr. Food Res. 2009, 53, 1156–1165. [Google Scholar] [CrossRef]
- Zadra, G.; Priolo, C.; Patnaik, A.; Loda, M. New strategies in prostate cancer: Targeting lipogenic pathways and the energy sensor AMPK. Clin. Cancer Res. 2010, 16, 3322–3328. [Google Scholar] [CrossRef] [Green Version]
- Zadra, G.; Photopoulos, C.; Tyekucheva, S.; Heidari, P.; Weng, Q.P.; Fedele, G.; Liu, H.; Scaglia, N.; Priolo, C.; Sicinska, E.; et al. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol. Med. 2014, 6, 519–538. [Google Scholar] [CrossRef]
- Wang, Y.; Pan, H.; Chen, D.; Guo, D.; Wang, X. Targeting at cancer energy metabolism and lipid droplet formation as new treatment strategies for epigallocatechin-3-gallate (EGCG) in colorectal cancer cells. J. Funct. Foods 2021, 83, 104570. [Google Scholar] [CrossRef]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin inhibits hepatic mTORC1 signaling via dose-dependent mechanisms involving AMPK and the TSC Complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Pamu, S.; Cui, Q.; Chan, T.H.; Dou, Q.P. Novel epigallocatechin gallate (EGCG) analogs activate AMP-activated protein kinase pathway and target cancer stem cells. Bioorgan. Med. Chem. 2012, 20, 3031–3037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Floyd, S.; Favre, C.; Lasorsa, F.M.; Leahy, M.; Trigiante, G.; Stroebel, P.; Marx, A.; Loughran, G.; O’Callaghan, K.; Marobbio, C.M.T.; et al. The insulin-like growth factor-I–mTOR signaling pathway induces the mitochondrial pyrimidine nucleotide carrier to promote cell growth. Mol. Biol. Cell 2007, 18, 3545–3555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, P.; Xu, X.Y. PI3K/Akt/mTOR signaling pathway in cancer stem cells: From basic research to clinical application. Am. J. Cancer Res. 2015, 5, 1602–1609. [Google Scholar]
- Francipane, M.G.; Lagasse, E. Therapeutic potential of mTOR inhibitors for targeting cancer stem cells. Br. J. Clin. Pharmacol. 2016, 82, 1180–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Min, Y. Pre-clinical evidence that salinomycin is active against retinoblastoma via inducing mitochondrial dysfunction, oxidative damage and AMPK activation. J. Bioenerg. Biomembr. 2021, 53, 513–523. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, W.; Yan, Z.; Zhao, W.; Mi, J.; Li, J.; Yan, H. Metformin induces autophagy and G0/G1 phase cell cycle arrest in myeloma by targeting the AMPK/mTORC1 and mTORC2 pathways. J. Exp. Clin. Cancer Res. 2018, 37, 63. [Google Scholar] [CrossRef]
- Inoki, K.; Zhu, T.; Guan, K.-L. TSC2 mediates cellular energy response to control cell growth and survival ken. Cells 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Park, H.J.; Park, C.S.; Oh, E.T.; Choi, B.H.; Williams, B.; Lee, C.K.; Song, C.W. Response of breast cancer cells and cancer stem cells to metformin and hyperthermia alone or combined. PLoS ONE 2014, 9, e87979. [Google Scholar] [CrossRef]
- Kawakita, E.; Yang, F.; Kumagai, A.; Takagaki, Y.; Kitada, M.; Yoshitomi, Y.; Ikeda, T.; Nakamura, Y.; Ishigaki, Y.; Kanasaki, K.; et al. Metformin mitigates DPP-4 inhibitor-induced breast cancer metastasis via suppression of mTOR signaling. Mol. Cancer Res. 2020, 16, 61–73. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. Correction to: ‘The Warburg EFFECT: How does it benefit cancer cells?’. Trends Biochem. Sci. 2016, 41, 287. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. AMPK is a negative regulator of the Warburg EFFECT and suppresses tumor growth in vivo opposes tumor development, and its loss fosters tumor progression in part by regulating cellular metabolic pathways that support cell growth and proliferation. Cell Metab. Jan. 2013, 8, 113–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Wong, C.C.; Zhang, X.; Kang, W.; Nakatsu, G.; Zhao, Q.; Chen, H.; Go, M.Y.Y.; Chiu, P.W.Y.; Wang, X.; et al. CAB39L elicited an anti-Warburg effect via a LKB1-AMPK-PGC1α axis to inhibit gastric tumorigenesis. Oncogene 2018, 37, 6383–6398. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.L.; Zhu, Y.J.; Hu, C.H.; You, L.; Wu, J.; He, X.Y.; Huang, W.J.; Wu, Z.H. Ghrelin affects gastric cancer progression by activating AMPK signaling pathway. Biochem. Genet. 2021, 59, 652–667. [Google Scholar] [CrossRef]
- Ci, X.; Zhou, J.; Lv, H.; Yu, Q.; Peng, L.; Hua, S. Betulin exhibits anti-inflammatory activity in lps-stimulated macrophages and endotoxin-shocked mice through an ampk/akt/nrf2-dependent mechanism. Cell Death Dis. 2017, 8, e2798. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Choi, S.S.; Park, M.H.; Jang, H.; Lee, Y.H.; Khim, K.W.; Oh, S.R.; Park, J.; Ryu, H.W.; Choi, J.H. Broussonetia papyrifera root bark extract exhibits anti-inflammatory effects on adipose tissue and improves insulin sensitivity potentially via AMPK activation. Nutrients 2020, 12, 773. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Farzaneh, M. Signaling pathways governing breast cancer stem cells behavior. Stem Cell Res. Ther. 2021, 12, 245–255. [Google Scholar] [CrossRef]
- Li, Y.H.; Luo, J.; Mosley, Y.Y.C.; Hedrick, V.E.; Paul, L.N.; Chang, J.; Zhang, G.; Wang, Y.K.; Banko, M.R.; Brunet, A.; et al. AMP-Activated protein kinase directly phosphorylates and destabilizes Hedgehog pathway transcription factor GLI1 in medulloblastoma. Cell Rep. 2015, 12, 599–609. [Google Scholar] [CrossRef] [Green Version]
- Kwan, H.T.; Chan, D.W.; Cai, P.C.H.; Mak, C.S.L.; Yung, M.M.H.; Leung, T.H.Y.; Wong, O.G.W.; Cheung, A.N.Y.; Ngan, H.Y.S. AMPK activators suppress cervical cancer cell growth through Inhibition of DVL3 mediated Wnt/b-catenin signaling activity. PLoS ONE 2013, 8, e53597. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.F.; Xie, C.W.; Yang, S.X.; Xiong, J.P. AMPK activators suppress breast cancer cell growth by inhibiting DVL3-facilitated Wnt/β-catenin signaling pathway activity. Mol. Med. Rep. 2017, 15, 899–907. [Google Scholar] [CrossRef] [Green Version]
- Bao, B.; Azmi, A.S.; Ali, S.; Zaiem, F.; Sarkar, F.H. Metformin may function as anti-cancer agent via targeting cancer stem cells: The potential biological significance of tumor-associated miRNAs in breast and pancreatic cancers. Ann. Transl. Med. 2014, 2, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Dronamraju, V.; Ibrahim, B.A.; Briski, K.P.; Sylvester, P.W. γ-tocotrienol suppression of the warburg effect is mediated by AMPK activation in human breast cancer cells. Nutr. Cancer 2019, 71, 1214–1228. [Google Scholar] [CrossRef] [PubMed]
- Zha, Q.B.; Zhang, X.Y.; Lin, Q.R.; Xu, L.H.; Zhao, G.X.; Pan, H.; Zhou, D.; Ouyang, D.Y.; Liu, Z.H.; He, X.H. Cucurbitacin e induces autophagy via downregulating mTORC1 signaling and upregulating AMPK activity. PLoS ONE 2015, 10, e0124355. [Google Scholar] [CrossRef]
- Turtoi, M.; Anghelache, M.; Patrascu, A.A.; Maxim, C.; Manduteanu, I.; Calin, M.; Popescu, D.L. Synthesis, characterization, and in vitro insulin-mimetic activity evaluation of valine schiff base coordination compounds of oxidovanadium(v). Biomedicines 2021, 9, 562. [Google Scholar] [CrossRef] [PubMed]
- Szklarzewicz, J.; Jurowska, A.; Hodorowicz, M.; Kazek, G.; Mordyl, B.; Menaszek, E.; Sapa, J. Characterization and antidiabetic activity of salicylhydrazone Schiff base vanadium(IV) and (V) complexes. Transit. Met. Chem. 2021, 46, 201–217. [Google Scholar] [CrossRef]
- Suman, S.G.; Gretarsdottir, J.M. Chemical and clinical aspects of metal-containing antidotes for poisoning by cyanide. In Essential Metals in Medicine: Therapeutic Use and Toxicity of Metal Ions in the Clinic; Carver, P.L., Ed.; De Gruyter: Berlin, Germany, 2019; pp. 359–392. [Google Scholar]
- Thompson, K.H.; Orvig, C. Vanadium in diabetes: 100 years from phase 0 to phase I. J. Inorg. Biochem. 2006, 100, 1925–1935. [Google Scholar] [CrossRef]
- Crans, D.C. Antidiabetic, chemical, and physical properties of organic vanadates as presumed transition-state inhibitors for phosphatases. J. Org. Chem. 2015, 80, 11899–11915. [Google Scholar] [CrossRef] [Green Version]
- Pessoa, J.C.; Etcheverry, S.; Gambino, D. Vanadium compounds in medicine. Coord. Chem. Rev. 2015, 301–302, 24–48. [Google Scholar] [CrossRef]
- Sakurai, H.; Kojima, Y.; Yoshikawa, Y.; Kawabe, K.; Yasui, H. Antidiabetic vanadium(IV) and zinc(II) complexes. Coord. Chem. Rev. 2002, 226, 187–198. [Google Scholar] [CrossRef]
- Kawabe, K.; Yoshikawa, Y.; Adachi, Y.; Sakurai, H. Possible mode of action for insulinomimetic activity of vanadyl(IV) compounds in adipocytes. Life Sci. 2006, 78, 2860–2866. [Google Scholar] [CrossRef]
- Irving, E.; Stoker, A.W. Vanadium compounds as PTP inhibitors. Molecules 2017, 22, 2269. [Google Scholar] [CrossRef] [Green Version]
- Leblanc, C.; Vilter, H.; Fournier, J.B.; Delage, L.; Potin, P.; Rebuffet, E.; Michel, G.; Solari, P.L.; Feiters, M.C.; Czjzek, M. Vanadium haloperoxidases: From the discovery 30 years ago to X-ray crystallographic and V K-edge absorption spectroscopic studies. Coord. Chem. Rev. 2015, 301–302, 134–146. [Google Scholar] [CrossRef] [Green Version]
- Crans, D.C. Fifteen years of dancing with vanadium. Pure Appl. Chem. 2005, 77, 1497–1527. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.-Y. Protein-tyrosine phosphatases: Biological function, structural characteristics, and mechanism of catalysis. Crit. Rev. Biochem. Mol. Biol. 1998, 33, 1–52. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.-X.; Zhang, Z.-Y. Targeting PTPs with small molecule inhibitors in cancer treatment. Cancer Metastasis Rev. 2008, 27, 263–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treviño, S.; Díaz, A.; Sánchez-Lara, E.; Sanchez-Gaytan, B.L.; Perez-Aguilar, J.M.; González-Vergara, E. Vanadium in biological action: Chemical, pharmacological aspects, and metabolic implications in diabetes mellitus. Biol. Trace Elem. Res. 2019, 188, 68–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilmarsdottir, B.; Briem, E.; Halldorsson, S.; Kricker, J.; Ingthorsson, S.; Gustafsdottir, S.; Mælandsmo, G.M.; Magnusson, M.K.; Gudjonsson, T. Inhibition of PTP1B disrupts cell—Cell adhesion and induces anoikis in breast epithelial cells. Cell Death Dis. 2017, 8, e2769. [Google Scholar] [CrossRef] [Green Version]
- Vieira, M.N.N.; Lyra e Silva, N.M.; Ferreira, S.T.; de Felice, F.G. Protein tyrosine phosphatase 1B (PTP1B): A potential target for Alzheimer’s therapy? Front. Aging Neurosci. 2017, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Niu, X.; Xiao, R.; Wang, N.; Wang, Z.; Zhang, Y.; Xia, Q.; Yang, X. The molecular mechanisms and rational design of anti-diabetic vanadium compounds. Curr. Top. Med. Chem. 2016, 16, 811–822. [Google Scholar] [CrossRef]
- Tamrakar, A.K.; Maurya, C.K.; Rai, A.K. PTP1B inhibitors for type 2 diabetes treatment: A patent review (2011–2014). Expert Opin. Ther. Pat. 2014, 24, 1101–1115. [Google Scholar] [CrossRef]
- Yang, J.L.; Ha, T.K.Q.; Lee, B.W.; Kim, J.; Oh, W.K. PTP1B inhibitors from the seeds of Iris sanguinea and their insulin mimetic activities via AMPK and ACC phosphorylation. Bioorgan. Med. Chem. Lett. 2017, 27, 5076–5081. [Google Scholar] [CrossRef] [PubMed]
- Beyene, Z. The protein tyrosine phosphatase PTB1B role in the development of obesity, diabetes, and cancer and its potential inhibitors. Int. J. Pharm. Bio-Med. Sci. 2021, 1, 88–101. [Google Scholar] [CrossRef]
- Soysal, S.; Obermann, E.C.; Gao, F.; Oertli, D.; Gillanders, W.E.; Viehl, C.T.; Muenst, S. PTP1B expression is an independent positive prognostic factor in human breast cancer. Breast Cancer Res. Treat. 2013, 137, 637–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Pan, Y.; Zhao, L.; Qi, F.; Liu, J. Protein tyrosine phosphatase 1B(PTP1B) promotes melanoma cells progression through Src activation. Bioengineered 2021, 12, 8396–8406. [Google Scholar] [CrossRef] [PubMed]
- Tonks, N.K.; Muthuswamy, S.K. A brake becomes an accelerator: PTP1B-A new therapeutic target for breast cancer. Cancer Cell 2007, 11, 214–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentires-Alj, M.; Neel, B.G. Protein-tyrosine phosphatase 1B is required for HER2/Neu-induced breast cancer. Cancer Res. 2007, 67, 2420–2424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, N.; Koveal, D.; Miller, D.H.; Xue, B.; Akshinthala, S.D.; Kragelj, J.; Jensen, M.R.; Gauss, C.-M.; Page, R.; Blackledge, M.; et al. Targeting the disordered C terminus of PTP1B with an allosteric inhibitor. Nat. Chem. Biol. 2014, 10, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Wiener, J.R.; Kerns, B.-J.M.; Harvey, E.L.; Conaway, M.R.; Lglehart, J.D.; Berchuck, A.; Bast, R.C., Jr. Overexpression of the protien tyrosine phosphatase PTP1B in human breast cancer: Assocation with p185 c-erbB-2 protein expression. J. Natl. Cancer Inst. 1994, 86, 372–378. [Google Scholar] [CrossRef]
- Liao, S.C.; Li, J.X.; Yu, L.; Sun, S.R. Protein tyrosine phosphatase 1B expression contributes to the development of breast cancer. J. Zhejiang Univ. Sci. B 2017, 18, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zeng, Q.; Qiu, J.; Pang, T.; Xian, J.; Zhang, X. Long non-coding RNA UCA1 promotes breast cancer by upregulating PTP1B expression via inhibiting miR-206. Cancer Cell Int. 2019, 19, 275. [Google Scholar] [CrossRef]
- Przychodzen, P.; Kuban-Jankowska, A.; Wyszkowska, R.; Barone, G.; lo Bosco, G.; Celso, F.L.; Kamm, A.; Daca, A.; Kostrzewa, T.; Gorska-Ponikowska, M. PTP1B phosphatase as a novel target of oleuropein activity in MCF-7 breast cancer model. Toxicol. Vitr. 2019, 61, 104624. [Google Scholar] [CrossRef] [PubMed]
- Arregui, C.O.; González, Á.; Burdisso, J.E.; Wusener, A.E.G. Protein tyrosine phosphatase PTP1B in cell adhesion and migration. Cell Adhes. Migr. 2013, 7, 418–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, K.A.; Biggins, L.; Sharpe, H.J. Protein tyrosine phosphatases in cell adhesion. Biochem. J. 2021, 478, 1061–1083. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Kostrzewa, T.; Wołosewicz, K.; Jamrozik, M.; Drzeżdżon, J.; Siemińska, J.; Jacewicz, D.; Górska-Ponikowska, M.; Kołaczkowski, M.; Łaźny, R.; Kuban-Jankowska, A. Curcumin and its new derivatives: Correlation between cytotoxicity against breast cancer cell lines, degradation of PTP1B phosphatase and ROS generation. Int. J. Mol. Sci. 2021, 22, 10368. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, G.; Mu, Y.; Wu, W.; Cao, B.; Wang, Z.; Yu, H.; Guan, P.; Han, L.; Li, L.; et al. Discovery of anti-TNBC agents targeting PTP1B: Total synthesis, structure-activity relationship, in vitro and in vivo investigations of jamunones. J. Med. Chem. 2021, 64, 6008–6020. [Google Scholar] [CrossRef]
- Kuban-Jankowska, A.; Kostrzewa, T.; Musial, C.; Barone, G.; Lo-Bosco, G.; Lo-Celso, F.; Gorska-Ponikowska, M. Green tea catechins induce inhibition of ptp1b phosphatase in breast cancer cells with potent anti-cancer properties: In vitro assay, molecular docking, and dynamics studies. Antioxidants 2020, 9, 1208. [Google Scholar] [CrossRef]
- Kuban-Jankowska, A.; Gorska-Ponikowska, M.; Wozniak, M. Lipoic acid decreases the viability of breast cancer cells and activity of PTP1B and SHP2. Anticancer Res. 2017, 37, 2893–2898. [Google Scholar] [CrossRef] [Green Version]
- To, D.C.; Hoang, D.T.; Tran, M.H.; Pham, M.Q.; Huynh, N.T.; Nguyen, P.H. PTP1B inhibitory flavonoids from orthosiphon stamineus benth. and their growth inhibition on human breast cancer cells. Nat. Prod. Commun. 2020, 15, 1–9. [Google Scholar] [CrossRef]
- Kuban-Jankowska, A.; Gorska-Ponikowska, M.; Sahu, K.K. Docosahexaenoic acid inhibits PTP1B phosphatase. Nutrients 2019, 11, 2554. [Google Scholar] [CrossRef] [Green Version]
- Uddin, M.N.; Sharma, G.; Yang, J.-L.; Choi, H.S.; Lim, S.-I.; Kang, K.W.; Oh, W.K. Oleanane triterpenes as protein tyrosine phosphatase 1B (PTP1B) inhibitors from Camellia japonica. Phytochemistry 2014, 103, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Kostrzewa, T.; Przychodzen, P.; Gorska-Ponikowska, M.; Kuban-Jankowska, A. Curcumin and cinnamaldehyde as PTP1B inhibitors with antidiabetic and anticancer potential. Anticancer Res. 2019, 39, 745–749. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Sharma, G.; Dao, T.T.; Uddin, M.N.; Kang, K.W.; Ndinteh, D.T.; Mbafor, J.T.; Oh, W.K. New prenylated isoflavonoids as protein tyrosine phosphatase 1B (PTP1B) inhibitors from Erythrina addisoniae. Bioorgan. Med. Chem. 2012, 20, 6459–6464. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.H.; Le, T.V.T.; Thuong, P.T.; Dao, T.T.; Ndinteh, D.T.; Mbafor, J.T.; Kang, K.W.; Oh, W.K. Cytotoxic and PTP1B inhibitory activities from Erythrina abyssinica. Bioorgan. Med. Chem. Lett. 2009, 19, 6745–6749. [Google Scholar] [CrossRef]
- Kuban-Jankowska, A.; Sahu, K.K.; Gorska-Ponikowska, M.; Tuszynski, J.A.; Wozniak, M. Inhibitory activity of iron chelators ATA and DFO on MCF-7 breast cancer cells and phosphatases PTP1B and SHP2. Anticancer Res. 2017, 37, 4799–4806. [Google Scholar]
- Chandel, S.; Manikandan, A.; Mehta, N.; Nathan, A.A.; Tiwari, R.K.; Mohapatra, S.B.; Chandran, M.; Jaleel, A.; Manoj, N.; Dixit, M. Protein tyrosine phosphatase-PEST (PTP-PEST) mediates hypoxia-induced endothelial autophagy and angiogenesis through AMPK activation. J. Cell Sci. 2020, 134, jcs250274. [Google Scholar] [CrossRef]
- Xu, Q.; Wu, N.; Li, X.; Guo, C.; Li, C.; Jiang, B.; Wang, H.; Shi, D. Inhibition of PTP1B blocks pancreatic cancer progression by targeting the PKM2/AMPK/mTOC1 pathway. Cell Death Dis. 2019, 10, 874. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Lee, Y.J.; Jin, F.; Park, Y.N.; Deng, Y.; Kang, Y.; Yang, J.H.; Chang, J.H.; Kim, D.Y.; Kim, J.A.; et al. Sirt1 negatively regulates FcϵRI-mediated mast cell activation through AMPK- and PTP1B-dependent processes. Sci. Rep. 2017, 7, 6444. [Google Scholar] [CrossRef]
- Ito, Y.; Hsu, M.-F.; Bettaieb, A.; Koike, S.; Mello, A.; Calvo-Rubio, M.; Villalba, J.M.; Haj, F.G. Protein tyrosine phosphatase 1B deficiency in podocytes mitigates hyperglycemia-induced renal injury. Metabolism 2017, 76, 56–69. [Google Scholar] [CrossRef]
- Yang, Z.; Wu, F.; He, Y.; Zhang, Q.; Zhang, Y.; Zhou, G.; Yang, H.; Zhou, P. A novel PTP1B inhibitor extracted from: Ganoderma lucidum ameliorates insulin resistance by regulating IRS1-GLUT4 cascades in the insulin signaling pathway. Food Funct. 2018, 9, 397–406. [Google Scholar] [CrossRef]
- Luo, J.; Hou, Y.; Xie, M.; Ma, W.; Shi, D.; Jiang, B. CYC31, A natural bromophenol PTP1B inhibitor, activates insulin signaling and improves long chain-fatty acid oxidation in C2C12 myotubes jiao. Mar. Drugs 2020, 18, 267. [Google Scholar] [CrossRef]
- Wolfson, B. Adipocyte activation of cancer stem cell signaling in breast cancer. World J. Biol. Chem. 2015, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Yang, X.; Zhao, Q.; Li, Z.; Fu, F.; Zhang, H.; Zheng, M.; Zhang, S. Molecular mechanism of stem cell differentiation into adipocytes and adipocyte differentiation of malignant tumor. Stem Cells Int. 2020, 2020, 8892300. [Google Scholar] [CrossRef]
- Goto, H.; Shimono, Y.; Funakoshi, Y.; Imamura, Y.; Toyoda, M.; Kiyota, N.; Kono, S.; Takao, S.; Mukohara, T.; Minami, H. Adipose-derived stem cells enhance human breast cancer growth and cancer stem cell-like properties through adipsin. Oncogene 2019, 38, 767–779. [Google Scholar] [CrossRef] [PubMed]
- Gruzewska, K.; Michno, A.; Pawelczyk, T.; Bielarczyk, H. Essentiality and toxicity of vanadium supplements in health and pathology. J. Physiol. Pharmacol. 2014, 65, 603–611. [Google Scholar] [PubMed]
- Zhang, S.; Yan, L.; Kim, S.M. Vanadium-protein complex inhibits human adipocyte differentiation through the activation of β-catenin and LKB1/AMPK signaling pathway. PLoS ONE 2020, 15, e0239547. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Wu, Y.; Wang, N.; Wang, Z.; Zhao, P.; Yang, X. Is the hypoglycemic action of vanadium compounds related to the suppression of feeding? Biol. Trace Elem. Res. 2014, 157, 242–248. [Google Scholar] [CrossRef]
- Liu, Q.; Li, L.; Gao, L.; Li, C.; Huan, Y.; Lei, L.; Cao, H.; Li, L.; Gao, A.; Liu, S.; et al. Combination of bis (α-furancarboxylato) oxovanadium (IV) and metformin improves hepatic steatosis through down-regulating inflammatory pathways in high-fat diet-induced obese C57BL/6J mice. Basic Clin. Pharmacol. Toxicol. 2021, 128, 747–757. [Google Scholar] [CrossRef]
- Silva-Nolasco, A.M.; Camacho, L.; Saavedra-Díaz, R.O.; Hernández-Abreu, O.; León, I.E.; Sánchez-Lombardo, I. Kinetic studies of sodium and metforminium decavanadates decomposition and in vitro cytotoxicity and insulin-like activity. Inorganics 2020, 8, 67. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, F.; Zhang, F.; Liu, P.; Xu, T.; Ding, W. Vanadium(IV)-chlorodipicolinate alleviates hepatic lipid accumulation by inducing autophagy via the LKB1/AMPK signaling pathway in vitro and in vivo. J. Inorg. Biochem. 2018, 183, 66–76. [Google Scholar] [CrossRef]
- Zhang, L.; Huang, Y.; Liu, F.; Zhang, F.; Ding, W. Vanadium(IV)-chlorodipicolinate inhibits 3T3-L1 preadipocyte adipogenesis by activating LKB1/AMPK signaling pathway. J. Inorg. Biochem. 2016, 162, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.L.; Chang, H.W. Natural vanadium-containing Jeju ground water stimulates glucose uptake through the activation of AMP-activated protein kinase in L6 myotubes. Mol. Cell. Biochem. 2012, 360, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.H.; Park, S.H.; Choi, G.H.; Park, I.J.; Lee, J.H.; Lee, O.H.; Kim, J.H.; Seo, Y.H.; Cho, J.H. Antidiabetic effect of an extract of nutricultured Brassica napus containing vanadium from a Jeju water concentrate. Food Sci. Biotechnol. 2019, 28, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Huang, M.; Zhao, P.; Yang, X. Vanadyl acetylacetonate upregulates PPARγ and adiponectin expression in differentiated rat adipocytes. J. Biol. Inorg. Chem. 2013, 18, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.; Dong, Y.; Wang, J.; Yang, X. Vanadyl acetylacetonate attenuates Aβ pathogenesis in APP/PS1 transgenic mice depending on the intervention stage. New J. Chem. 2019, 43, 17588–17594. [Google Scholar] [CrossRef]
- Zhao, P.; Yang, X. Vanadium compounds modulate PPARγ activity primarily by increasing PPARγ protein levels in mouse insulinoma NIT-1 cells. Metallomics 2013, 5, 836–843. [Google Scholar] [CrossRef]
- Cheng, H.S.; Yip, Y.S.; Lim, E.K.Y.; Wahli, W.; Tan, N.S. PPARs and tumor microenvironment: The emerging roles of the metabolic master regulators in tumor stromal-epithelial crosstalk and carcinogenesis. Cancers 2021, 13, 2153. [Google Scholar] [CrossRef]
- Kaur, S.; Nag, A.; Gangenahalli, G.; Sharma, K. Peroxisome proliferator activated receptor gamma sensitizes non-small cell lung carcinoma to gamma irradiation induced apoptosis. Front. Genet. 2019, 10, 554. [Google Scholar] [CrossRef] [Green Version]
- Im, C.-N. Targeting glioblastoma stem cells (GSCs) with peroxisome proliferator-activated receptor gamma (PPARγ) ligands. IUBMB Life 2016, 68, 173–177. [Google Scholar] [CrossRef] [Green Version]
- Lanlan, L.; Yang, Z.; Xu, Y.; Li, J.; Xu, D.; Zhang, L.; Sun, J.; Xia, S.; Zou, F.; Liu, Y. Inhibition of oxidative stress-elicited AKT activation facilitates PPARcAgonist-mediated inhibition of stemcell character and tumor growth of liver cancer cells. PLoS ONE 2013, 8, e73038. [Google Scholar]
- Augimeri, G.; Giordano, C.; Gelsomino, L.; Plastina, P.; Barone, I.; Catalano, S.; Andò, S.; Bonofiglio, D. The role of PPARγ ligands in breast cancer: From basic research to clinical studies. Cancers 2020, 12, 2623. [Google Scholar] [CrossRef] [PubMed]
- Yousefnia, S.; Momenzadeh, S.; Seyed Forootan, F.; Ghaedi, K.; Nasr Esfahani, M.H. The influence of peroxisome proliferator-activated receptor γ (PPARγ) ligands on cancer cell tumorigenicity. Gene 2018, 649, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Kioseoglou, E.; Petanidis, S.; Gabriel, C.; Salifoglou, A. The chemistry and biology of vanadium compounds in cancer therapeutics. Coord. Chem. Rev. 2015, 301–302, 87–105. [Google Scholar] [CrossRef]
- Amante, C.; de Sousa-Coelho, A.L.; Aureliano, M. Vanadium and melanoma: A systematic review. Metals 2021, 11, 828. [Google Scholar] [CrossRef]
- Noriega, L.; Castro, M.E.; Perez-Aguilar, J.M.; Caballero, N.A.; Sanchez-Gaytan, B.L.; González-Vergara, E.; Melendez, F.J. Oxidovanadium(V) complexes as promising anticancer photosensitizers. J. Inorg. Biochem. 2020, 203, 110862. [Google Scholar] [CrossRef]
- Molinuevo, M.S.; Cortizo, A.M.; Etcheverry, S.B. Vanadium(IV) complexes inhibit adhesion, migration and colony formation of UMR106 osteosarcoma cells. Cancer Chemother. Pharmacol. 2008, 61, 767–773. [Google Scholar] [CrossRef]
- Shi, J.; Wang, D.M.; Wang, C.M.; Hu, Y.; Liu, A.H.; Zhang, Y.L.; Sun, B.; Song, J.G. Insulin receptor substrate-1 suppresses transforming growth factor-β1–mediated epithelial-mesenchymal transition. Cancer Res. 2009, 69, 7180–7187. [Google Scholar] [CrossRef] [Green Version]
- Petanidis, S.; Kioseoglou, E.; Domvri, K.; Zarogoulidis, P.; Carthy, J.M.; Anestakis, D.; Moustakas, A.; Salifoglou, A. In vitro and ex vivo vanadium antitumor activity in (TGF-β)-inducedEMT. Synergistic activity with carboplatin and correlation with tumormetastasis in cancer patients. Int. J. Biochem. Cell Biol. 2016, 74, 121–134. [Google Scholar] [CrossRef]
- Smith, D.M.; Pickering, R.M.; Lewith, G.T. A systematic review of vanadium oral supplements for glycaemic control in type 2 diabetes mellitus. QJM 2008, 101, 351–358. [Google Scholar] [CrossRef]
- Thompson, K.H.; Lichter, J.; LeBel, C.; Scaife, M.C.; McNeill, J.H.; Orvig, C. Vanadium treatment of type 2 diabetes: A view to the future. J. Inorg. Biochem. 2009, 103, 554–558. [Google Scholar] [CrossRef]
Figure 1.
Breast cancer stem cells, or cancer-initiating cells, display an innate ability to evade and survive all treatment strategies owing to their stem-like properties. They function to maintain the cancer cell population while regenerating themselves continuously.
Figure 1.
Breast cancer stem cells, or cancer-initiating cells, display an innate ability to evade and survive all treatment strategies owing to their stem-like properties. They function to maintain the cancer cell population while regenerating themselves continuously.

Figure 2.
Mechanism of AMPK activation. AMPK can be activated via the LKB1 or CaMKKβ pathway (physiological activation) following phosphorylation at threonine 172 residue in the α subunit. It can also be activated exogenously by drugs such as quercetin and metformin that act on the mitochondria and increase the AMP:ATP ratio, which subsequently activates the LKB1-mediated phosphorylation and activation of AMPK. Lastly, it can also be activated allosterically by the binding of AMP at the AMPKγ subunit.
Figure 2.
Mechanism of AMPK activation. AMPK can be activated via the LKB1 or CaMKKβ pathway (physiological activation) following phosphorylation at threonine 172 residue in the α subunit. It can also be activated exogenously by drugs such as quercetin and metformin that act on the mitochondria and increase the AMP:ATP ratio, which subsequently activates the LKB1-mediated phosphorylation and activation of AMPK. Lastly, it can also be activated allosterically by the binding of AMP at the AMPKγ subunit.

Figure 3.
Known mechanisms of activated AMPK-mediated downregulation of cyclin D1, cell cycle arrest, and autophagy. AMPK phosphorylation activates various cyclin-dependent kinases (CDKs) to result in downregulation of cyclin D1 and cell cycle arrest in the G1/S phase as well autophagy, all of which have been implicated in anticancer therapy.
Figure 3.
Known mechanisms of activated AMPK-mediated downregulation of cyclin D1, cell cycle arrest, and autophagy. AMPK phosphorylation activates various cyclin-dependent kinases (CDKs) to result in downregulation of cyclin D1 and cell cycle arrest in the G1/S phase as well autophagy, all of which have been implicated in anticancer therapy.

Figure 4.
Activated AMPK-mediated downregulation of the mTORC1 complex can follow two pathways: through the phosphorylation and activation of tuberous sclerosis complex 2 (TSC 2) or via direct phosphorylation of raptor at S722 and S792. The deactivation of mTOR is subsequently followed by the inhibition of S6K1 and 4E-BP1, both of which are responsible for the initiation of protein translation and maintenance of cell size and growth.
Figure 4.
Activated AMPK-mediated downregulation of the mTORC1 complex can follow two pathways: through the phosphorylation and activation of tuberous sclerosis complex 2 (TSC 2) or via direct phosphorylation of raptor at S722 and S792. The deactivation of mTOR is subsequently followed by the inhibition of S6K1 and 4E-BP1, both of which are responsible for the initiation of protein translation and maintenance of cell size and growth.

Figure 5.
Cellular pathways associated with AMPK activation in cancer and cancer stem cells. The figure depicts various important molecular pathways mediated by AMPK activation that play pivotal roles against cancer and cancer stem cells.
Figure 5.
Cellular pathways associated with AMPK activation in cancer and cancer stem cells. The figure depicts various important molecular pathways mediated by AMPK activation that play pivotal roles against cancer and cancer stem cells.

Figure 6.
Structural similarity between phosphate and vanadate at physiological pH and the redox conversion between V (IV) and V (V) oxidation states.
Figure 6.
Structural similarity between phosphate and vanadate at physiological pH and the redox conversion between V (IV) and V (V) oxidation states.

Figure 7.
Structures of some vanadium (IV) complexes with reported AMPK activation activity.


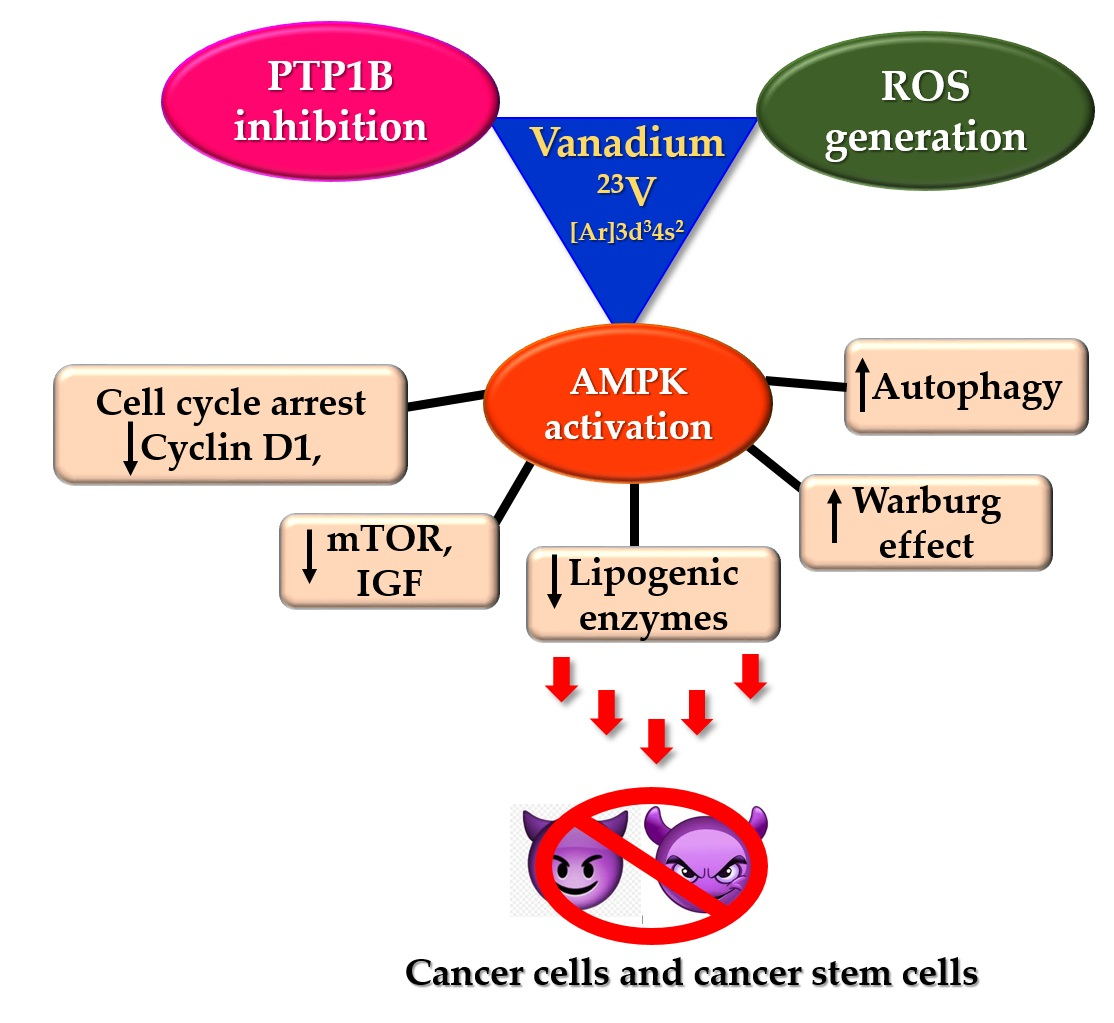{kind=link}
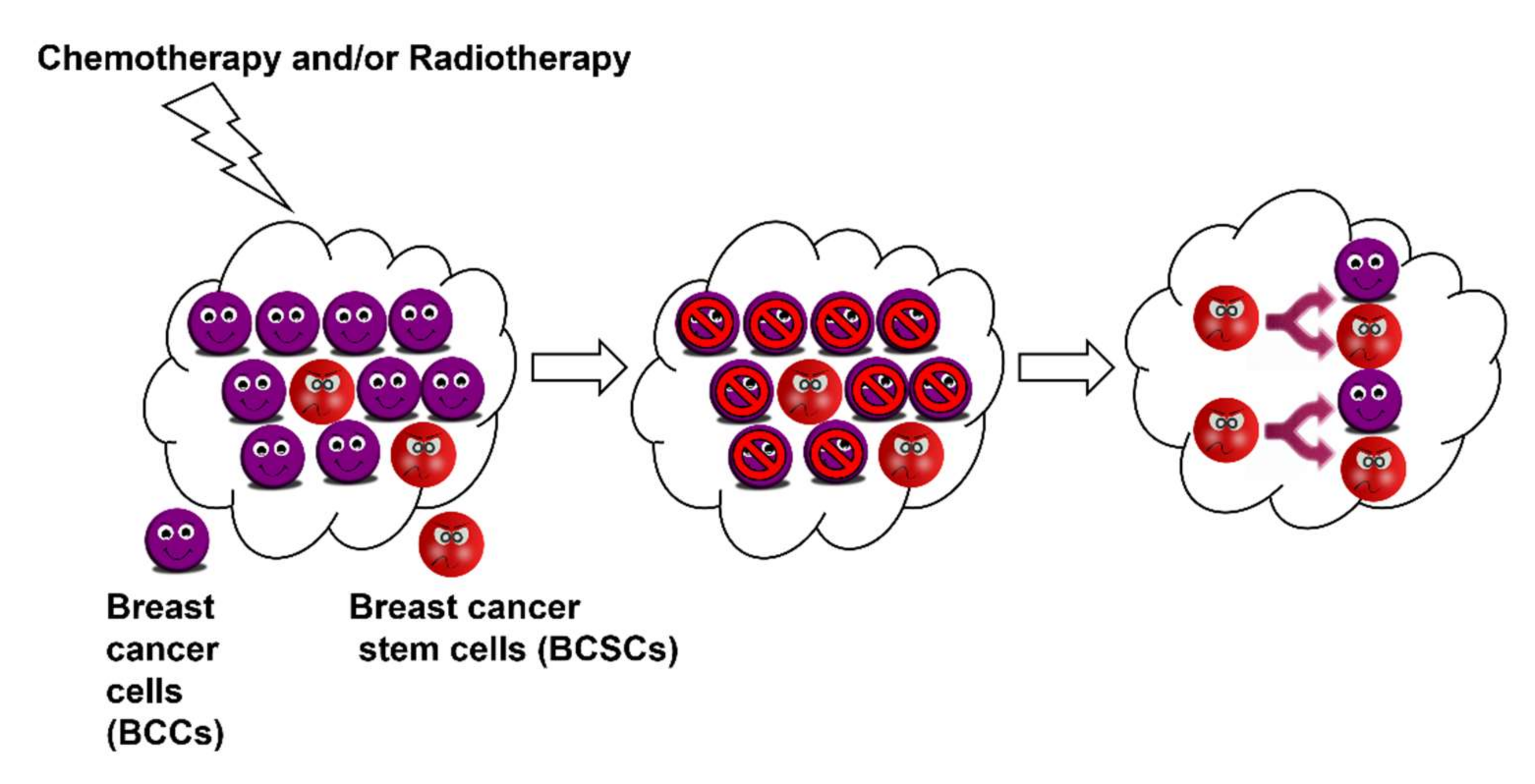{kind=link}
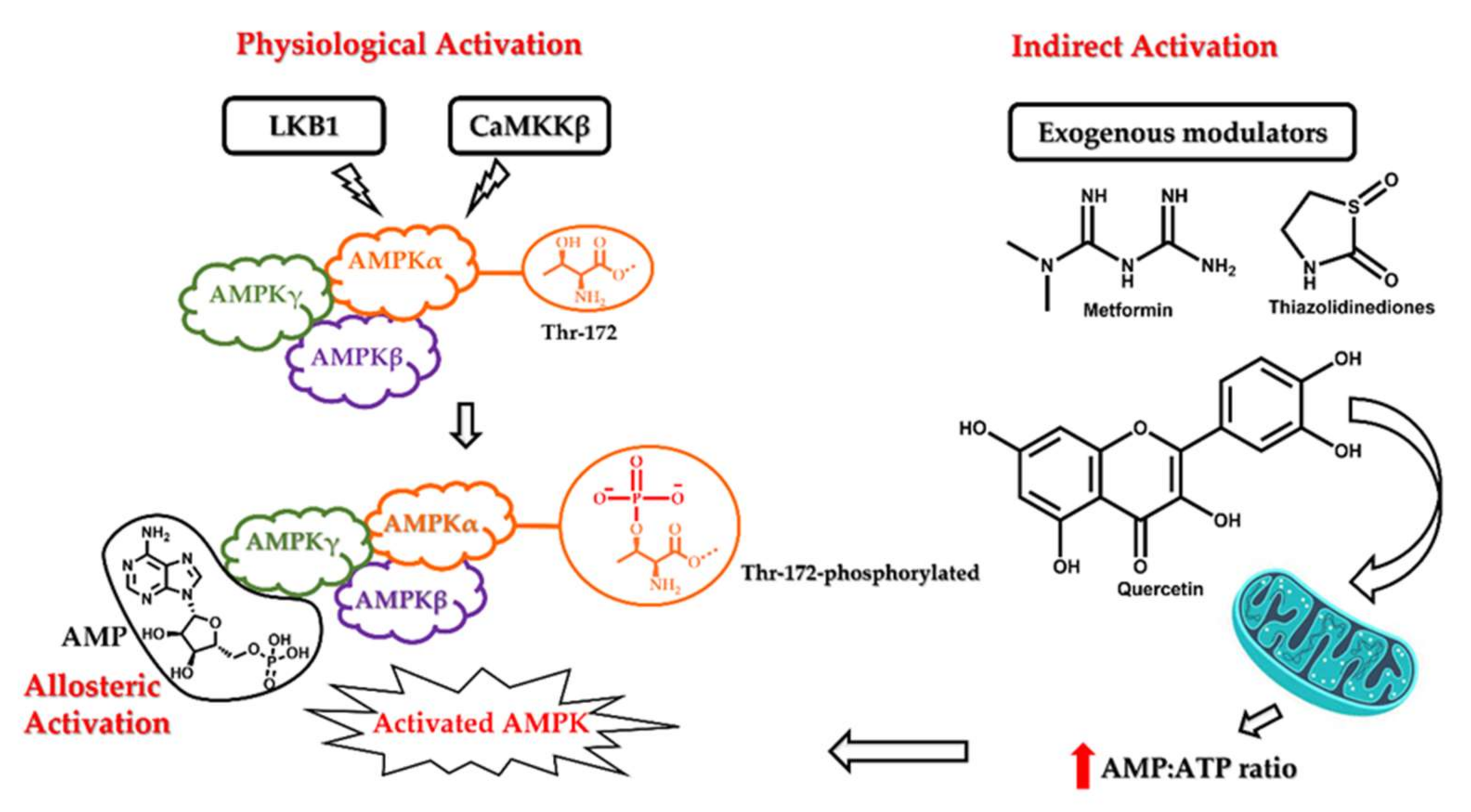{kind=link}
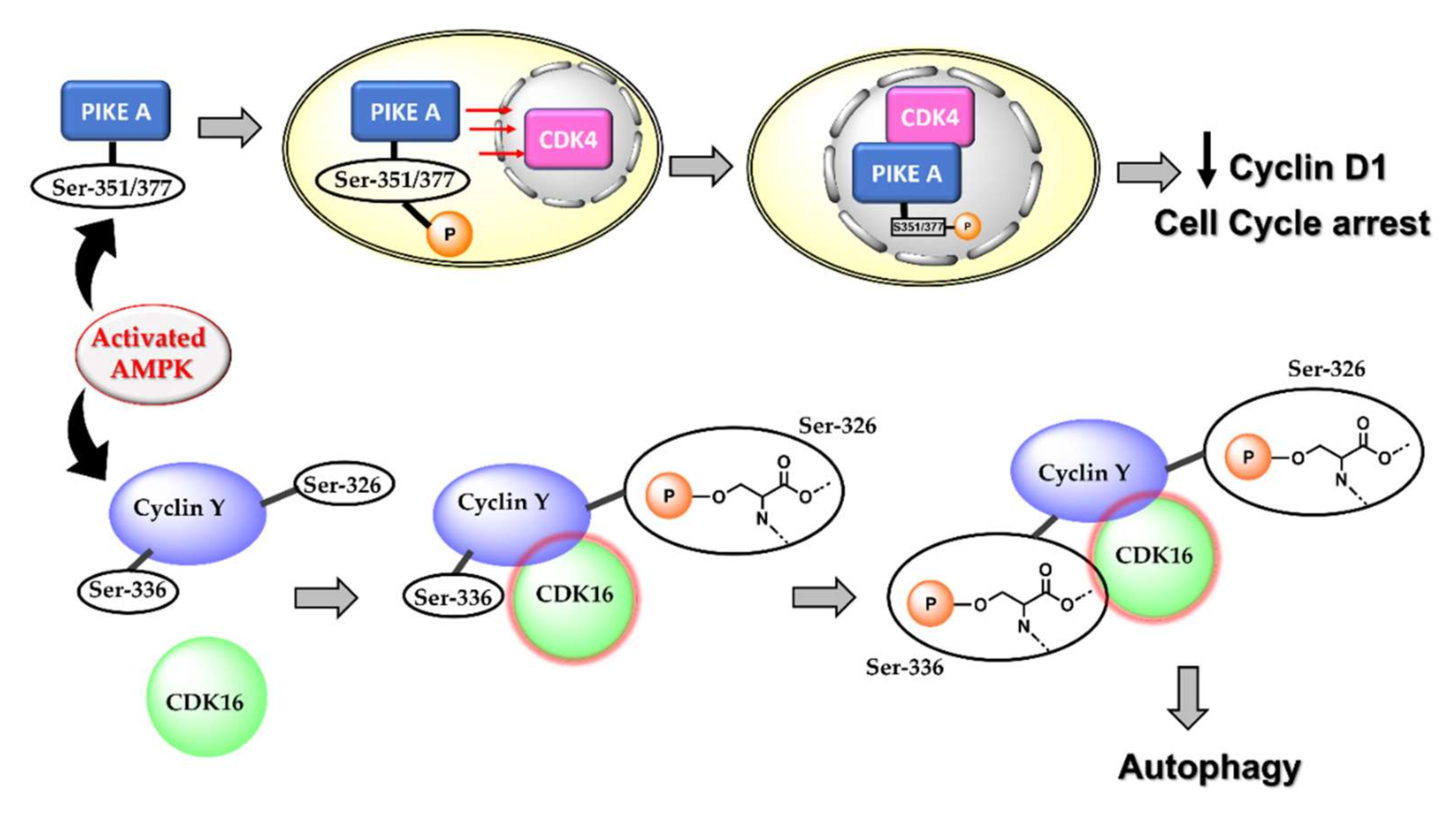{kind=link}
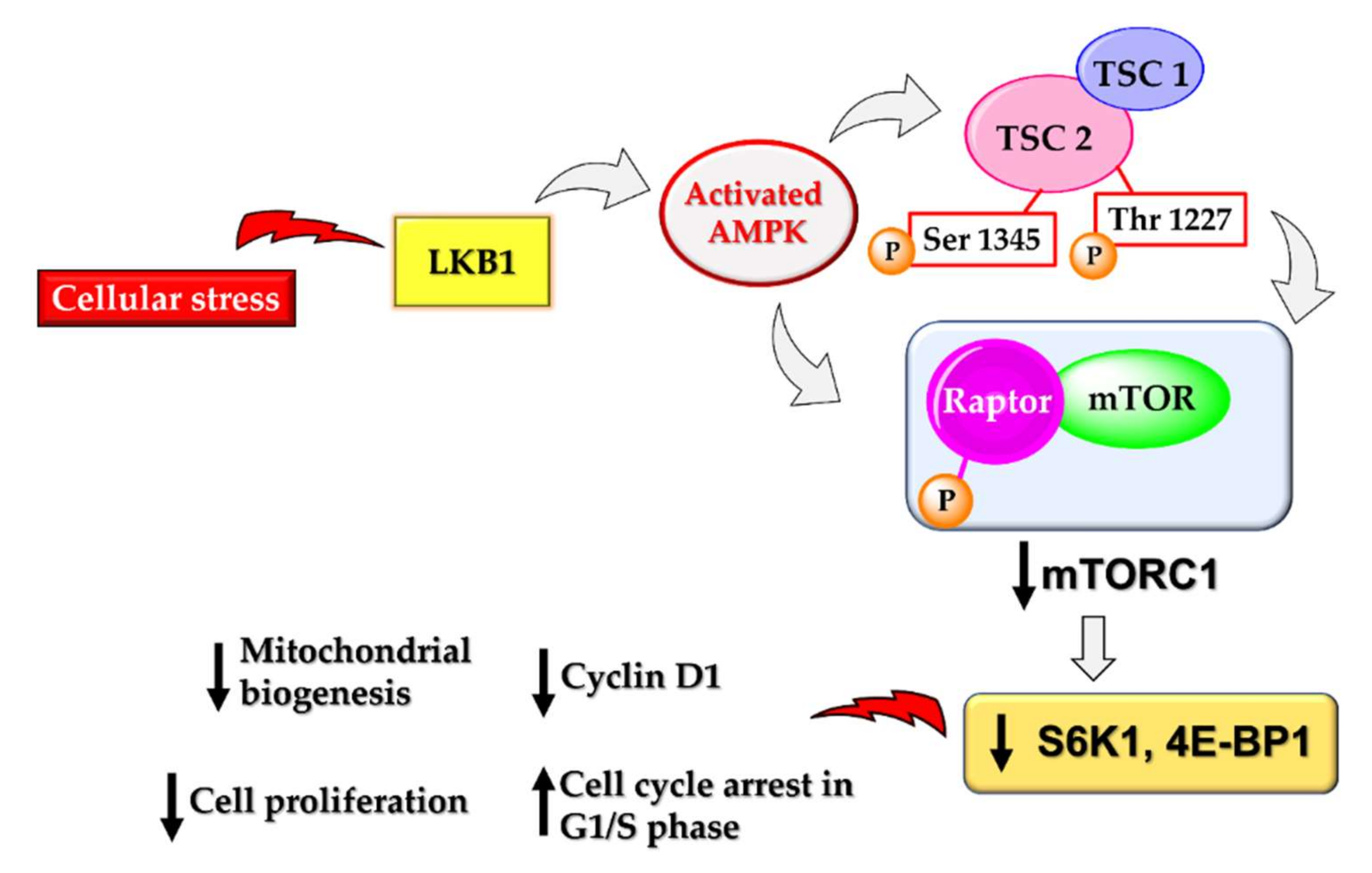{kind=link}
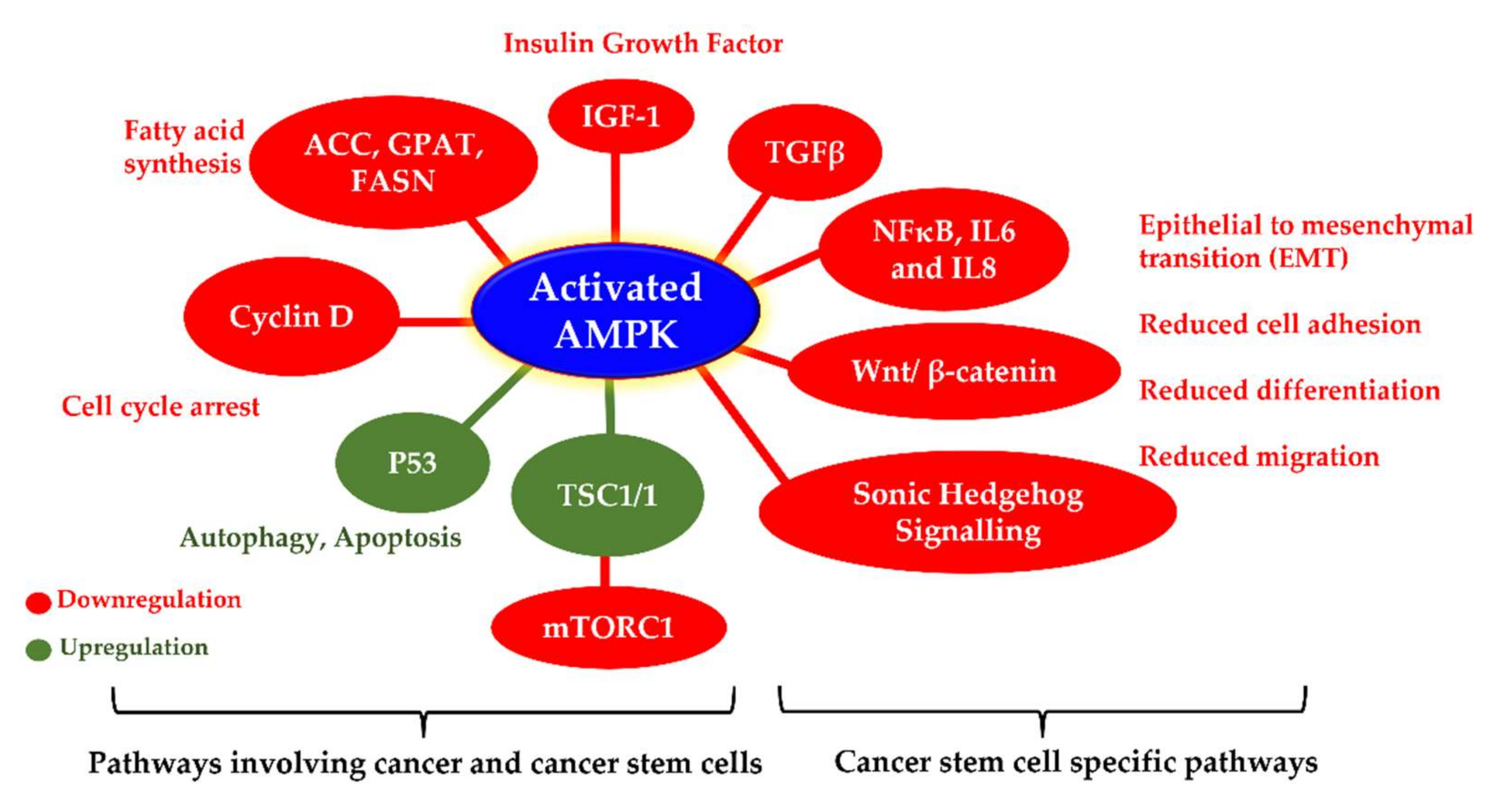{kind=link}
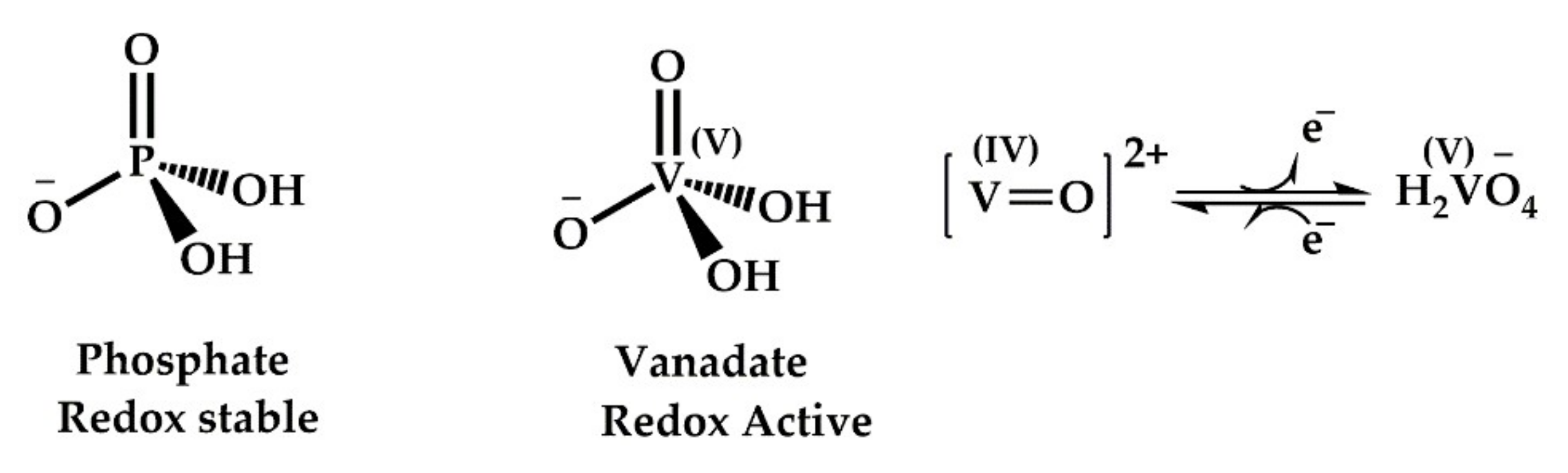{kind=link}
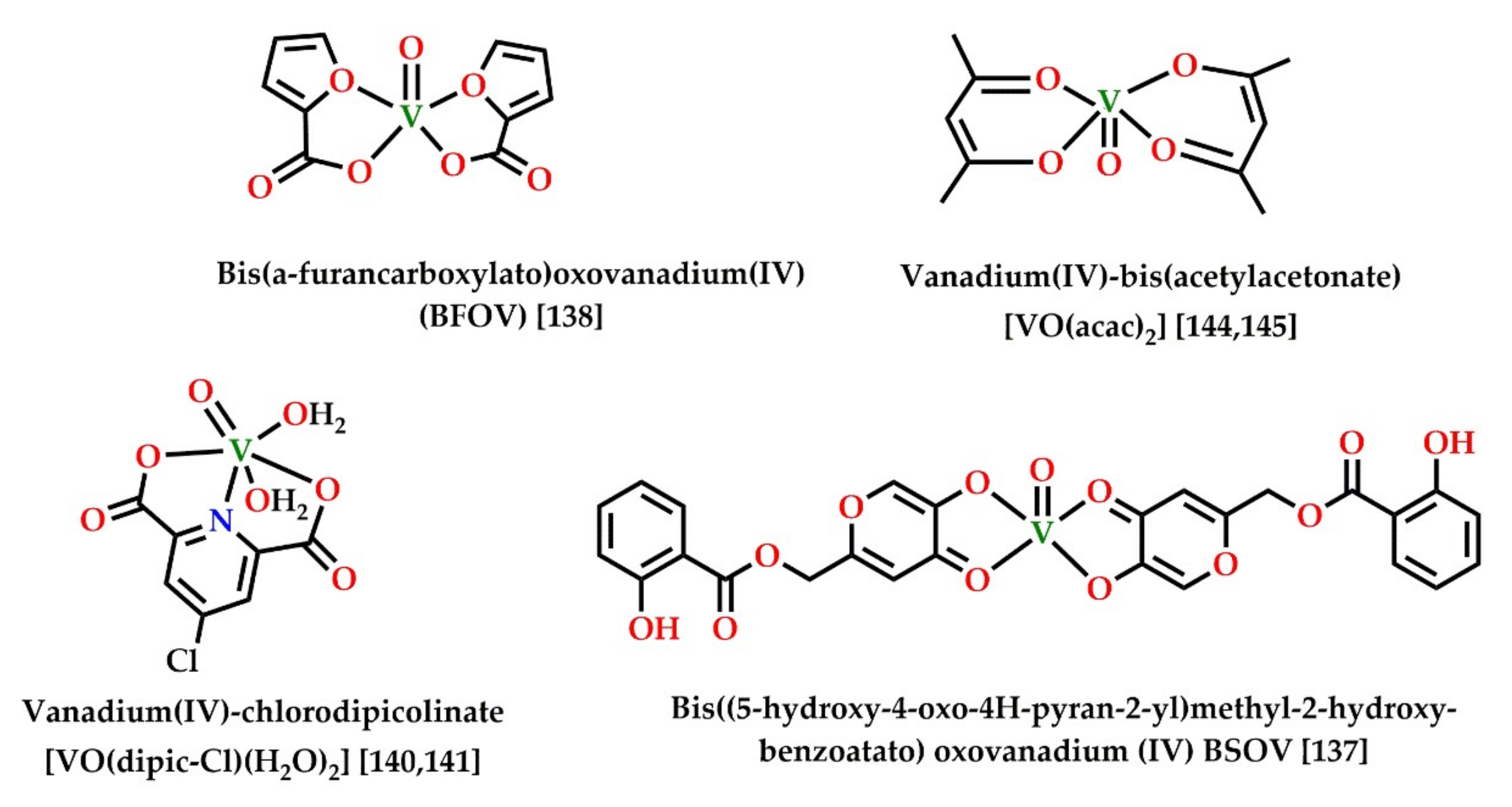{kind=link}
Table 1.
Some recently reported compounds with anticancer action following AMPK activation.
| Drug | Cell Lines | Activated AMPK-Mediated Action | Reference |
|---|---|---|---|
| FND-4b | MCF-7, T-47D, MDA-MB-231, HCC-1143, and HCC-1806 (CSCs) * | Downregulation of ACC, S6, and cyclin D1 activity | [56] |
| Metformin | EC109 and EC9706 | Cell cycle arrest in G0/G1 phase | [65] |
| Simvastatin | HepG2 and Hep3B | G0/G1 arrest by upregulating p21 | [66] |
| Marine sponge-derived smenospongine | MCF7, HBL100, and 16HBE (CSCs) * | Cell cycle arrest and downregulation of Nanog, Bmi1, and Sox2 | [30] |
| Phenformin | MCF7, ZR-75-1, MDA-MB-231, and SUM1315 | Downregulation of cyclin D1, cell cycle arrest at G1 phase, and downregulation of pERK in ER+ cells (MCF7 and ZR-75-1) only | [68] |
| Epigallocatechin gallate and analogues | MDA-MB-231 (CSCs) * Hep G2 and Hep 3B HCT116 and HT-29 | Cell cycle arrest and downregulation of mTOR Downregulation of mTOR and lipogenesis Inhibition of lipogenesis and energy metabolism | [75,78,84] |
| Metformin | MCF-7 and MDA-MB-231 cells MIA PaCa-2 (CSCs) * | Downregulation of cyclin D1, cell cycle arrest at G1 phase, and suppression of mTOR | [87] |
| Metformin | RPMI8226 and U266 | Induction of autophagy and G0/G1 cell cycle arrest and suppression of mTORC1 and mTORC2 | [85] |
| Salinomycin | RB 383, WERI-Rb-1 and RB116 (CSCs) * | Inhibition of mitochondrial respiration and mTOR | [88] |
| AICAR | Glioblastoma, in vivo | Inhibition of lipogenesis and mTOR | [74] |
| MT 63-78 | LNCaP, CL1, PC3, DU145, and HeLa | Inhibition of lipogenesis and mTOR | [77] |
| γ–Tocotrienol | MCF-7 and MDA-MB-231 | Warburg effect | [104] |
| Baicalein | PC-3, DU145, and MDA-MB-231 | Inhibition of mTOR and autophagy | [63] |
| Cyclovirobuxine D | MCF7 | AMPK autophagy | [64] |
| Cucurbitacin E | HeLa, MCF7, and DU145 | AMPK, autophagy, and reduced mTORC1 | [105] |
* CSCs: cancer stem cells.
Table 2.
Some recently reported PTP1B inhibitors with promising anticancer activity against breast cancer.
Table 2.
Some recently reported PTP1B inhibitors with promising anticancer activity against breast cancer.
| Compound | Cell Lines | Action | Reference |
|---|---|---|---|
| Oleuropein | MCF7 | Cytotoxicity | [134] |
| Curcumin and derivatives | MCF-7 and MDA-MB-231 | ROS generation, cytotoxicity | [138] |
| Jamunones | MCF-7 and MDA-MB-231, TNBC | Downregulation of (PI3K)/Akt pathway-mediated apoptosis; G0/G1 phase arrest | [139] |
| Green tea catechins (epigallocatechin and epigallocatechin gallate) | MCF-7 | Cytotoxicity | [140] |
| Alpha-lipoic acid (ALA) and its reduced form of dihydrolipoic acid (DHLA) | MCF7 | PTP and SHP2 inhibition and cytotoxicity | [141] |
| Flavonoids from Orthosiphon stamineus Benth. | MCF7, MCF7/TAMR, and MDA-MB-231 | Cytotoxicity | [142] |
| Docosahexaenoic acid | MCF7 | Cytotoxicity | [143] |
| Oleanane triterpenes from Camellia japonica | MCF7, MCF7/ADR, and MDA-MB-231 | Cytotoxicity | [144] |
| Curcumin and cinnamaldehyde | MCF 7 | Cytotoxicity | [145] |
| Isoflavonoids from Erythrina addisoniae | MCF7, MCF7/ADR, and MDA-MB-231 | Cytotoxicity | [146] |
| Pterocarpan derivatives from Erythrina abyssinica | MCF7, MCF7/TAMR, MCF7/ADR, and MDA-MB-231 | Cytotoxicity | [147] |
| Aurintricarboxylic acid (Fe chelator) | MCF7 | Inhibition of SHP2 phosphatases and cytotoxicity | [148] |
Table 3.
Vanadium compounds with reported AMPK phosphorylation and activation activity.
| Compounds | Action | Reference |
|---|---|---|
| BFOV (BFOV + metformin) | Activation of AMPK and reduced hepatic steatosis | [161] |
| (VO(acac)2) | Activation of AMPK, p38, and PPARγ, stimulation of adiponectin | [167,168] |
| Vanadium protein complex | Activation of AMPK and LKB1 and decreased adipogenesis | [159] |
| (VO(dipic-Cl)(H2O)2) | Activation of AMPK/LKB1, autophagy and reduced lipid accumulation and adipogenesis | [163,164] |
| BSOV | Activation of AMPK and PPARγ Insulin mimetic action | [160] |
| Vanadium-containing Jeju groundwater | Activation of AMPK and reduced adipogenesis | [165] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Uprety, B.; Abrahamse, H. Targeting Breast Cancer and Their Stem Cell Population through AMPK Activation: Novel Insights. Cells 2022, 11, 576. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030576
AMA Style
Uprety B, Abrahamse H. Targeting Breast Cancer and Their Stem Cell Population through AMPK Activation: Novel Insights. Cells. 2022; 11(3):576. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030576
Chicago/Turabian StyleUprety, Bhawna, and Heidi Abrahamse. 2022. "Targeting Breast Cancer and Their Stem Cell Population through AMPK Activation: Novel Insights" Cells 11, no. 3: 576. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11030576
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.