
Genetic Kidney Diseases (GKDs) Modeling Using Genome Editing Technologies

 , , , and
, , , and
Abstract
:1. Introduction
1.1. Genetic Kidney Diseases
1.1.1. Autosomal Dominant Polycystic Kidney Disease (ADPKD)
1.1.2. Autosomal Recessive Polycystic Kidney Disease (ARPKD)
1.1.3. Alport Syndrome (AS)
1.1.4. Autosomal Dominant Tubulointerstitial Kidney Disease (ADTKD)
1.1.5. Gitelman (GS) and Bartter (BS) Syndromes
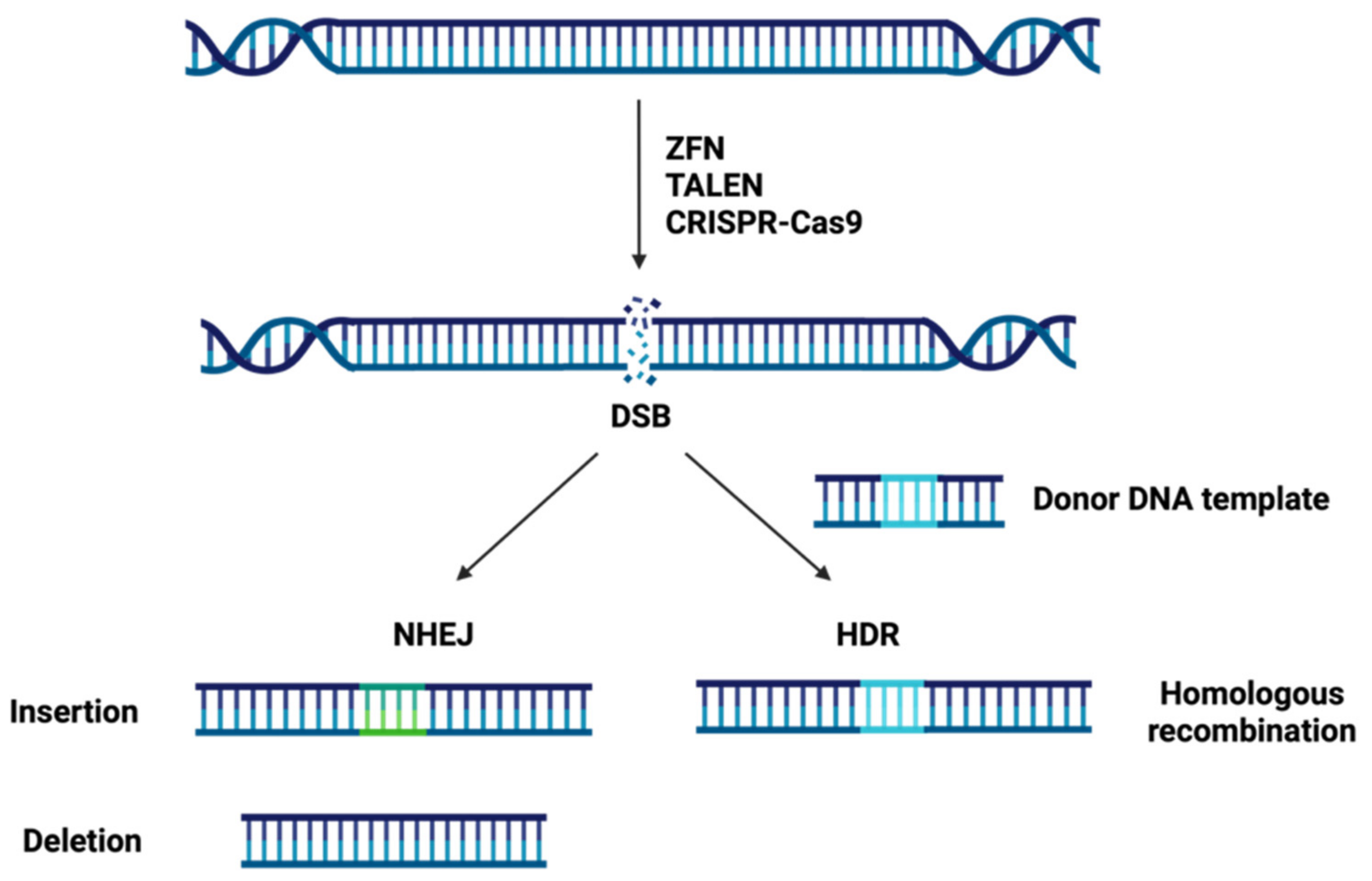
1.2. Site-Specific Nuclease Systems
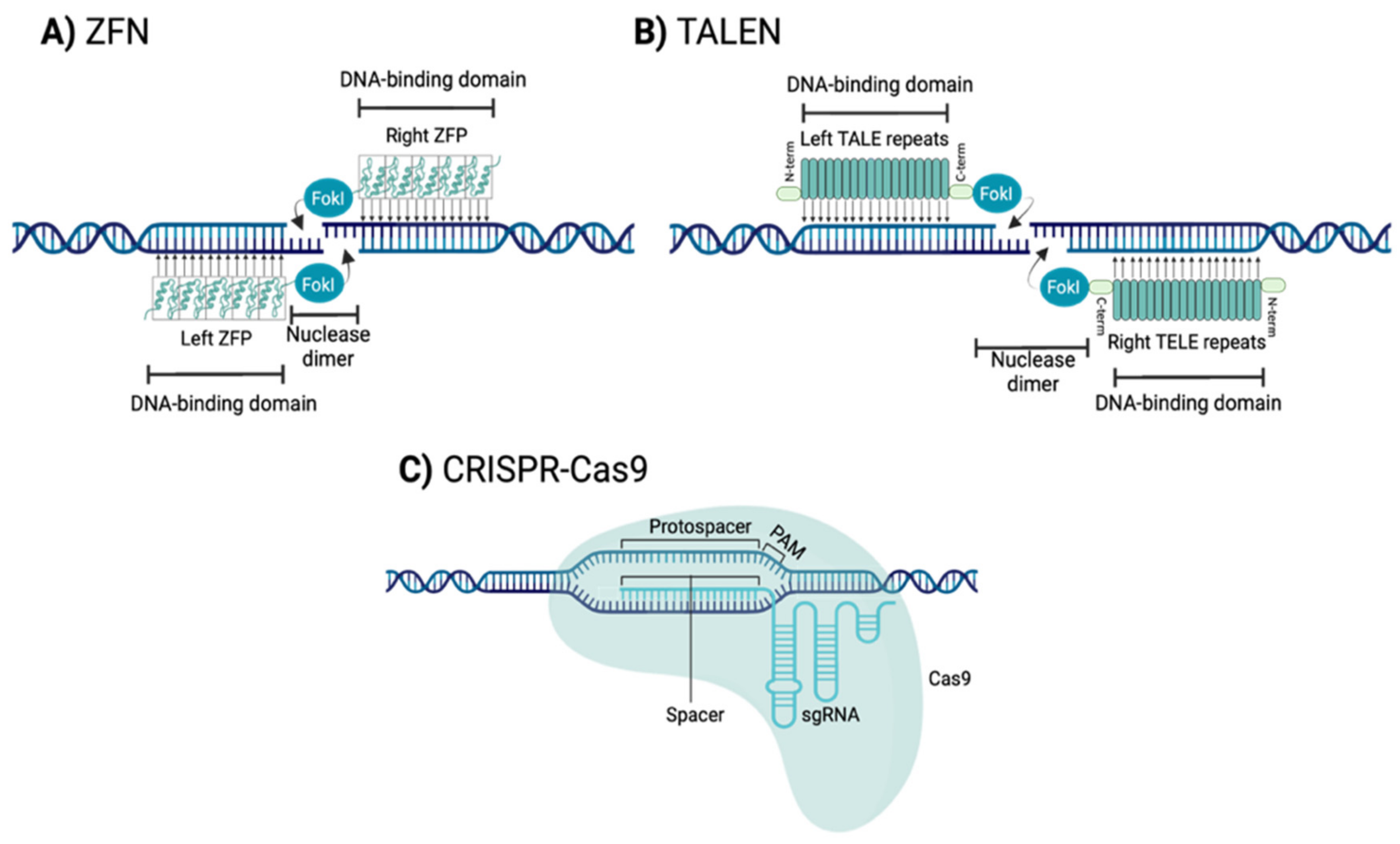
1.2.1. Zinc Finger Nucleases (ZFNs)
1.2.2. Transcription Activator-Like Effector Nucleases (TALENs)
1.2.3. Clustered Regularly Interspaced Short Palindromic Repeats-CRISPR Associated Protein 9 Nuclease (CRISPR-Cas9)
1.2.4. Comparison of the Three Types of Genome Editing Technologies
Sequence Selection and Assembly Evaluation
Delivery Strategies
Specificity and Efficiency
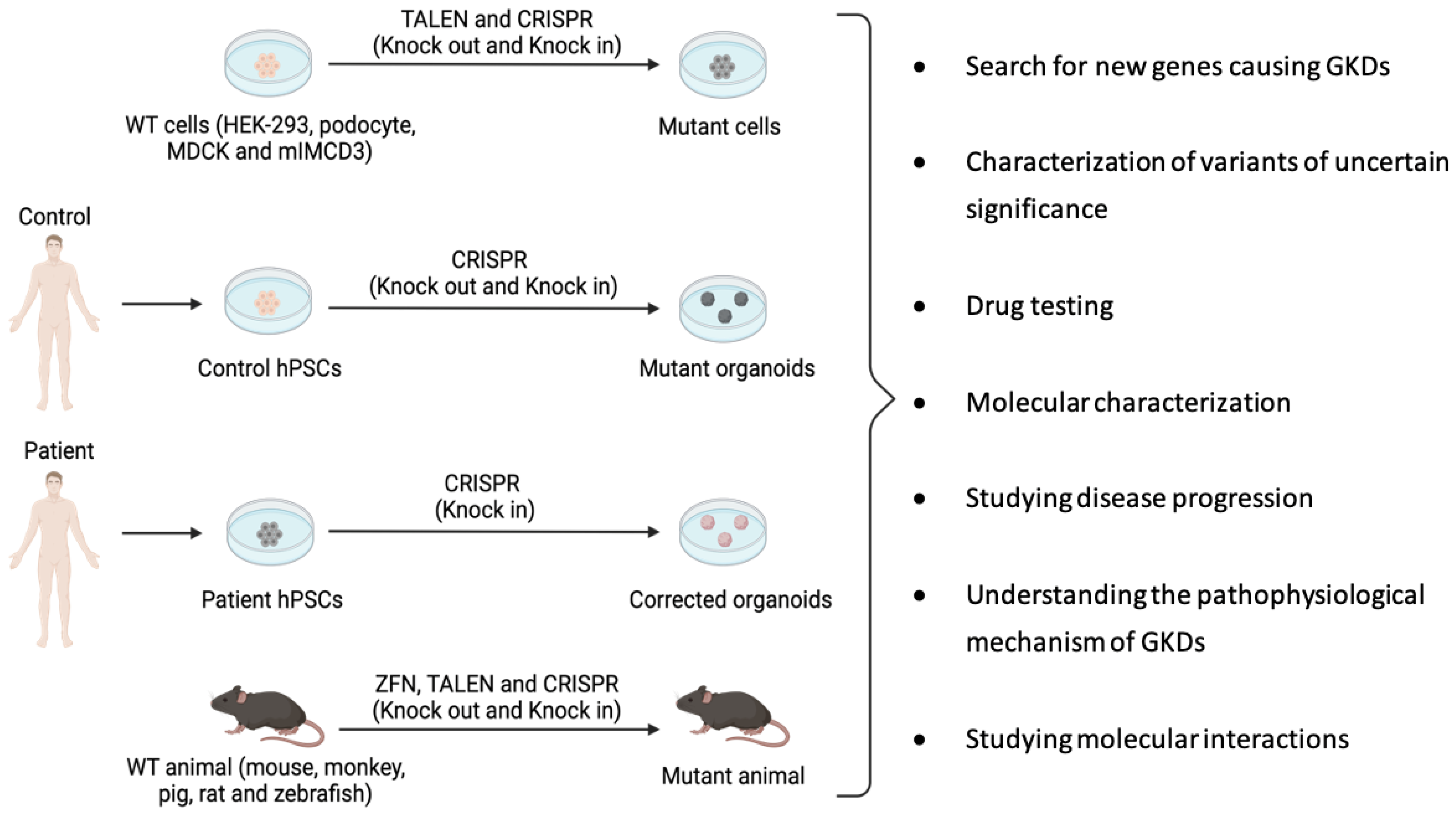
2. Site-Specific Nuclease Systems to Study Genetic Kidney Diseases
2.1. ADPKD Models Generated Using Genome Editing
2.1.1. ADPKD Models Generated Using ZFNs
2.1.2. ADPKD Models Generated Using TALENs
2.1.3. ADPKD Models Generated Using CRISPR-Cas9
2.2. ARPKD Models Generated Using Genome Editing
ARPKD Models Generated Using CRISPR-Cas9
2.3. AS Models Generated Using Genome Editing
AS Models Generated Using CRISPR-Cas9
2.4. ADTKD Models Generated Using Genome Editing
2.4.1. ADTKD Models Generated Using ZFNs
2.4.2. ADTKD Models Generated Using CRISPR-Cas9
2.5. GS and BS Models Generated Using Genome Editing
2.5.1. BS Models Generated Using ZFNs
2.5.2. BS Models Generated Using TALENs
3. Main Challenges to Overcome
3.1. Difficulty of Gene Delivery to the Kidney
3.2. Immune System against Gene Editing
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Devuyst, O.; Knoers, N.V.A.M.; Remuzzi, G.; Schaefer, F. Rare inherited kidney diseases: Challenges, opportunities, and perspectives. Lancet 2014, 383, 1844–1859. [Google Scholar] [CrossRef] [Green Version]
- Aymé, S.; Bockenhauer, D.; Day, S.; Devuyst, O.; Guay-Woodford, L.M.; Ingelfinger, J.R.; Klein, J.B.; Knoers, N.V.; Perrone, R.D.; Roberts, J.; et al. Common Elements in Rare Kidney Diseases: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2017, 92, 796–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivante, A.; Hildebrandt, F. Exploring the genetic basis of early-onset chronic kidney disease. Nat. Rev. Nephrol. 2016, 12, 133–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torra, R.; Furlano, M.; Ortiz, A.; Ars, E. Genetic kidney diseases as an underrecognized cause of chronic kidney disease: The key role of international registry reports. Clin. Kidney J. 2021, 14, 1879–1885. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.M.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Groopman, E.E.; Marasa, M.; Cameron-Christie, S.; Petrovski, S.; Aggarwal, V.S.; Rasouly, H.M.; Li, Y.; Zhang, J.; Nestor, J.; Krithivasan, P.; et al. Diagnostic Utility of Exome Sequencing for Kidney Disease. N. Engl. J. Med. 2019, 380, 142–151. [Google Scholar] [CrossRef]
- Connaughton, D.M.; Kennedy, C.; Shril, S.; Mann, N.; Murray, S.L.; Williams, P.A.; Conlon, E.; Nakayama, M.; van der Ven, A.T.; Ityel, H.; et al. Monogenic causes of chronic kidney disease in adults. Kidney Int. 2019, 95, 914–928. [Google Scholar] [CrossRef]
- Hofmeister, A.F.; Kömhoff, M.; Weber, S.; Grgic, I. Disease modeling in genetic kidney diseases: Mice. Cell Tissue Res. 2017, 369, 159–170. [Google Scholar] [CrossRef]
- Köttgen, A.; Gall, E.C.-L.; Halbritter, J.; Kiryluk, K.; Mallett, A.J.; Parekh, R.S.; Rasouly, H.M.; Sampson, M.G.; Tin, A.; Antignac, C.; et al. Genetics in chronic kidney disease: Conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2022. [Google Scholar] [CrossRef]
- van der Wijst, J.; Belge, H.; Bindels, R.J.M.; Devuyst, O. Learning Physiology From Inherited Kidney Disorders. Physiol. Rev. 2019, 99, 1575–1653. [Google Scholar] [CrossRef]
- UniProt. Available online: https://www.uniprot.org/ (accessed on 22 March 2022).
- Garcia-Gonzalez, M.A.; Jones, J.G.; Allen, S.K.; Palatucci, C.M.; Batish, S.D.; Seltzer, W.K.; Lan, Z.; Allen, E.; Qian, F.; Lens, X.M.; et al. Evaluating the clinical utility of a molecular genetic test for polycystic kidney disease. Mol. Genet. Metab. 2007, 92, 160–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordido, A.; Besada-Cerecedo, L.; García-González, M.A. The Genetic and Cellular Basis of Autosomal Dominant Polycystic Kidney Disease-A Primer for Clinicians. Front. Pediatr. 2017, 5, 279. [Google Scholar] [CrossRef] [PubMed]
- Porath, B.; Gainullin, V.G.; Gall, E.C.-L.; Dillinger, E.K.; Heyer, C.M.; Hopp, K.; Edwards, M.E.; Madsen, C.D.; Mauritz, S.R.; Banks, C.J.; et al. Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am. J. Hum. Genet. 2016, 98, 1193–1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gall, E.C.-L.; Olson, R.J.; Besse, W.; Heyer, C.M.; Gainullin, V.G.; Smith, J.M.; Audrézet, M.P.; Hopp, K.; Porath, B.; Shi, B.; et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am. J. Hum. Genet. 2018, 102, 832–844. [Google Scholar] [CrossRef] [Green Version]
- Senum, S.R.; Li, Y.S.M.; Benson, K.A.; Joli, G.; Olinger, E.; Lavu, S.; Madsen, C.D.; Gregory, A.V.; Neatu, R.; Kline, T.L.; et al. Monoallelic IFT140 pathogenic variants are an important cause of the autosomal dominant polycystic kidney-spectrum phenotype. Am. J. Hum. Genet. 2022, 109, 136–156. [Google Scholar] [CrossRef]
- Hildebrandt, F.; Benzing, T.; Katsanis, N. Ciliopathies. N. Engl. J. Med. 2011, 364, 1533–1543. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Walz, G. Sensitive cilia set up the kidney. Nat. Med. 2007, 13, 1409–1411. [Google Scholar] [CrossRef]
- Harris, P.C.; Torres, V.E. Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease. J. Clin. Investig. 2014, 124, 2315–2324. [Google Scholar] [CrossRef] [Green Version]
- Reiterová, J.; Štekrová, J.; Merta, M.; Kotlas, J.; Elišáková, V.; Lněnička, P.; Korabečná, M.; Kohoutová, M.; Tesař, V. Autosomal dominant polycystic kidney disease in a family with mosaicism and hypomorphic allele. BMC Nephrol. 2013, 14, 59. [Google Scholar] [CrossRef] [Green Version]
- Mehta, L.; Jim, B. Hereditary Renal Diseases. Semin. Nephrol. 2017, 37, 354–361. [Google Scholar] [CrossRef]
- Chebib, F.T.; Torres, V.E. Autosomal Dominant Polycystic Kidney Disease: Core Curriculum 2016. Am. J. Kidney Dis. 2016, 67, 792–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Gonzalez, M.A.; Outeda, P.; Zhou, Q.; Zhou, F.; Menezes, L.F.; Qian, F.; Huso, D.L.; Germino, G.G.; Piontek, K.B.; Watnick, T. Pkd1 and Pkd2 are required for normal placental development. PLoS ONE 2010, 5, e12821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, C.; Guay-Woodford, L.M.; Harris, P.C.; Horie, S.; Peters, D.J.M.; Torres, V.E. Polycystic kidney disease. Nat. Rev. Dis. Prim. 2018, 4. [Google Scholar] [CrossRef]
- Parfrey, P.S.; Bear, J.C.; Morgan, J.; Cramer, B.C.; McManamon, P.J.; Gault, M.H.; Churchill, D.N.; Singh, M.; Hewitt, R.; Somlo, S.; et al. The diagnosis and prognosis of autosomal dominant polycystic kidney disease. N. Engl. J. Med. 1990, 323, 1085–1090. [Google Scholar] [CrossRef] [PubMed]
- Onuchic, L.F.; Furu, L.; Nagasawa, Y.; Hou, X.; Eggermann, T.; Ren, Z.; Bergmann, C.; Senderek, J.; Esquivel, E.; Zeltner, R.; et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am. J. Hum. Genet. 2002, 70, 1305–1317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordido, A.; Vizoso-gonzalez, M.; Garcia-Gonzalez, M.A. Molecular Pathophysiology of Autosomal Recessive Polycystic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 6523. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Galeano, M.C.R.; Ott, E.; Kaeslin, G.; Kausalya, P.J.; Kramer, C.; Ortiz-Brüchle, N.; Hilger, N.; Metzis, V.; Hiersche, M.; et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet. 2017, 49, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Fu, Y.; Hui, K.; Moeckel, G.; Mai, W.; Li, C.; Liang, D.; Zhao, P.; Ma, J.; Chen, X.Z.; et al. Fibrocystin/polyductin modulates renal tubular formation by regulating polycystin-2 expression and function. J. Am. Soc. Nephrol. 2008, 19, 455–468. [Google Scholar] [CrossRef] [Green Version]
- Guay-Woodford, L.M.; Muecher, G.; Hopkins, S.D.; Avner, E.D.; Germino, G.G.; Guillot, A.P.; Herrin, J.; Holleman, R.; Irons, D.A.; Primack, W.; et al. The Severe Perinatal Form of Autosomal Recessive Polycystic Kidney Disease Maps to Chromosome 6p21.1-p12: Implications for Genetic Counseling. Am. J. Hum. Genet. 1995, 56, 1101. [Google Scholar]
- Zerres, K.; Rudnik-Schöneborn, S.; Steinkamm, C.; Becker, J.; Mücher, G. Autosomal recessive polycystic kidney disease. J. Mol. Med. 1998, 76, 303–309. [Google Scholar] [CrossRef]
- Capisonda, R.; Phan, V.; Traubuci, J.; Daneman, A.; Balfe, J.W.; Guay-Woodford, L.M. Autosomal recessive polycystic kidney disease: Outcomes from a single-center experience. Pediatr. Nephrol. 2003, 18, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Guay-Woodford, L.M.; Desmond, R.A. Autosomal recessive polycystic kidney disease: The clinical experience in North America. Pediatrics 2003, 111, 1072–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fonck, C.; Chauveau, D.; Gagnadoux, M.F.; Pirson, Y.; Grünfeld, J.P. Autosomal recessive polycystic kidney disease in adulthood. Nephrol. Dial. Transplant. 2001, 16, 1648–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stokman, M.F.; Renkema, K.Y.; Giles, R.H.; Schaefer, F.; Knoers, N.V.A.M.; Van Eerde, A.M. The expanding phenotypic spectra of kidney diseases: Insights from genetic studies. Nat. Rev. Nephrol. 2016, 12, 472–483. [Google Scholar] [CrossRef] [PubMed]
- Pinto e Vairo, F.; Prochnow, C.; Kemppainen, J.L.; Lisi, E.C.; Steyermark, J.M.; Kruisselbrink, T.M.; Pichurin, P.N.; Dhamija, R.; Hager, M.M.; Albadri, S.; et al. Genomics Integration Into Nephrology Practice. Kidney Med. 2021, 3, 785–798. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.; Fieldhouse, R.; Chan, M.M.Y.; Sadeghi-Alavijeh, O.; Burnett, L.; Izzi, V.; Persikov, A.V.; Gale, D.P.; Storey, H.; Savige, J. Prevalence Estimates of Predicted Pathogenic COL4A3-COL4A5 Variants in a Population Sequencing Database and Their Implications for Alport Syndrome. J. Am. Soc. Nephrol. 2021, 32, 2273–2290. [Google Scholar] [CrossRef]
- gnomAD. Available online: https://gnomad.broadinstitute.org/ (accessed on 28 April 2022).
- Martínez-Pulleiro, R.; García-Murias, M.; Fidalgo-Díaz, M.; García-González, M.Á. Molecular Basis, Diagnostic Challenges and Therapeutic Approaches of Alport Syndrome: A Primer for Clinicians. Int. J. Mol. Sci. 2021, 22, 110663. [Google Scholar] [CrossRef]
- Fallerini, C.; Dosa, L.; Tita, R.; Del Prete, D.; Feriozzi, S.; Gai, G.; Clementi, M.; La Manna, A.; Miglietti, N.; Mancini, R.; et al. Unbiased next generation sequencing analysis confirms the existence of autosomal dominant Alport syndrome in a relevant fraction of cases. Clin. Genet. 2014, 86, 252–257. [Google Scholar] [CrossRef]
- Morinière, V.; Dahan, K.; Hilbert, P.; Lison, M.; Lebbah, S.; Topa, A.; Bole-Feysot, C.; Pruvost, S.; Nitschke, P.; Plaisier, E.; et al. Improving mutation screening in familial hematuric nephropathies through next generation sequencing. J. Am. Soc. Nephrol. 2014, 25, 2740–2751. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, T.; Nozu, K.; Minamikawa, S.; Horinouchi, T.; Sakakibara, N.; Nagano, C.; Aoto, Y.; Ishiko, S.; Nakanishi, K.; Shima, Y.; et al. Comparison between conventional and comprehensive sequencing approaches for genetic diagnosis of Alport syndrome. Mol. Genet. Genom. Med. 2019, 7, e883. [Google Scholar] [CrossRef] [Green Version]
- Jais, J.P.; Knebelmann, B.; Giatras, I.; De Marchi, M.; Rizzoni, G.; Renieri, A.; Weber, M.; Gross, O.; Netzer, K.O.; Flinter, F.; et al. X-linked Alport syndrome: Natural history in 195 families and genotype- phenotype correlations in males. J. Am. Soc. Nephrol. 2000, 11, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Savige, J.; Colville, D.; Rheault, M.; Gear, S.; Lennon, R.; Lagas, S.; Finlay, M.; Flinter, F. Alport Syndrome in Women and Girls. Clin. J. Am. Soc. Nephrol. 2016, 11, 1713–1720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storey, H.; Savige, J.; Sivakumar, V.; Abbs, S.; Flinter, F.A. COL4A3/COL4A4 Mutations and Features in Individuals with Autosomal Recessive Alport Syndrome. J. Am. Soc. Nephrol. 2013, 24, 1945–1954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pescucci, C.; Mari, F.; Longo, I.; Vogiatzi, P.; Caselli, R.; Scala, E.; Abaterusso, C.; Gusmano, R.; Seri, M.; Miglietti, N.; et al. Autosomal-dominant Alport syndrome: Natural history of a disease due to COL4A3 or COL4A4 gene. Kidney Int. 2004, 65, 1598–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fallerini, C.; Baldassarri, M.; Trevisson, E.; Morbidoni, V.; La Manna, A.; Lazzarin, R.; Pasini, A.; Barbano, G.; Pinciaroli, A.R.; Garosi, G.; et al. Alport syndrome: Impact of digenic inheritance in patients management. Clin. Genet. 2017, 92, 34–44. [Google Scholar] [CrossRef]
- Savige, J. Should We Diagnose Autosomal Dominant Alport Syndrome When There Is a Pathogenic Heterozygous COL4A3 or COL4A4 Variant? Kidney Int. Rep. 2018, 3, 1239–1241. [Google Scholar] [CrossRef] [Green Version]
- Devuyst, O.; Olinger, E.; Weber, S.; Eckardt, K.U.; Kmoch, S.; Rampoldi, L.; Bleyer, A.J. Autosomal dominant tubulointerstitial kidney disease. Nat. Rev. Dis. Prim. 2019, 5, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Eckardt, K.U.; Alper, S.L.; Antignac, C.; Bleyer, A.J.; Chauveau, D.; Dahan, K.; Deltas, C.; Hosking, A.; Kmoch, S.; Rampoldi, L.; et al. Autosomal dominant tubulointerstitial kidney disease: Diagnosis, classification, and management—A KDIGO consensus report. Kidney Int. 2015, 88, 676–683. [Google Scholar] [CrossRef] [Green Version]
- Cormican, S.; Connaughton, D.M.; Kennedy, C.; Murray, S.; Živná, M.; Kmoch, S.; Fennelly, N.K.; O’Kelly, P.; Benson, K.A.; Conlon, E.T.; et al. Autosomal dominant tubulointerstitial kidney disease (ADTKD) in Ireland. Ren. Fail. 2019, 41, 832–841. [Google Scholar] [CrossRef] [Green Version]
- Bohle, A.; Mackensen-Haen, S.; Van Gise, H. Significance of tubulointerstitial changes in the renal cortex for the excretory function and concentration ability of the kidney: A morphometric contribution. Am. J. Nephrol. 1987, 7, 421–433. [Google Scholar] [CrossRef]
- Ayasreh, N.; Bullich, G.; Miquel, R.; Furlano, M.; Ruiz, P.; Lorente, L.; Valero, O.; García-González, M.A.; Arhda, N.; Garin, I.; et al. Autosomal Dominant Tubulointerstitial Kidney Disease: Clinical Presentation of Patients With ADTKD-UMOD and ADTKD-MUC1. Am. J. Kidney Dis. 2018, 72, 411–418. [Google Scholar] [CrossRef]
- Heidet, L.; Decramer, S.; Pawtowski, A.; Morinière, V.; Bandin, F.; Knebelmann, B.; Lebre, A.S.; Faguer, S.; Guigonis, V.; Antignac, C.; et al. Spectrum of HNF1B mutations in a large cohort of patients who harbor renal diseases. Clin. J. Am. Soc. Nephrol. 2010, 5, 1079–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolar, N.A.; Golzio, C.; Živná, M.; Hayot, G.; Van Hemelrijk, C.; Schepers, D.; Vandeweyer, G.; Hoischen, A.; Huyghe, J.R.; Raes, A.; et al. Heterozygous Loss-of-Function SEC61A1 Mutations Cause Autosomal-Dominant Tubulo-Interstitial and Glomerulocystic Kidney Disease with Anemia. Am. J. Hum. Genet. 2016, 99, 174–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downie, M.L.; Lopez Garcia, S.C.; Kleta, R.; Bockenhauer, D. Inherited Tubulopathies of the Kidney: Insights from Genetics. Clin. J. Am. Soc. Nephrol. 2021, 16, 620–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, D.B.; Nelson-Williams, C.; Bia, M.J.; Ellison, D.; Karet, F.E.; Molina, A.M.; Vaara, I.; Iwata, F.; Cushner, H.M.; Koolen, M.; et al. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat. Genet. 1996, 12, 24–30. [Google Scholar] [CrossRef]
- Simon, D.B.; Karet, F.E.; Hamdan, J.M.; Di Pietro, A.; Sanjad, S.A.; Lifton, R.P. Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused by mutations in the Na-K-2Cl cotransporter NKCC2. Nat. Genet. 1996, 13, 183–188. [Google Scholar] [CrossRef]
- Simon, D.B.; Karet, F.E.; Rodriguez-Soriano, J.; Hamdan, J.H.; DiPietro, A.; Trachtman, H.; Sanjad, S.A.; Lifton, R.P. Genetic heterogeneity of Bartter’s syndrome revealed by mutations in the K+ channel, ROMK. Nat. Genet. 1996, 14, 152–156. [Google Scholar] [CrossRef]
- Simon, D.B.; Bindra, R.S.; Mansfield, T.A.; Nelson-Williams, C.; Mendonca, E.; Stone, R.; Schurman, S.; Nayir, A.; Alpay, H.; Bakkaloglu, A.; et al. Mutations in the chloride channel gene, CLCNKB, cause Bartter’s syndrome type III. Nat. Genet. 1997, 17, 171–178. [Google Scholar] [CrossRef]
- Birkenhäger, R.; Otto, E.; Schürmann, M.J.; Vollmer, M.; Ruf, E.M.; Maier-Lutz, I.; Beekmann, F.; Fekete, A.; Omran, H.; Feldmann, D.; et al. Mutation of BSND causes Bartter syndrome with sensorineural deafness and kidney failure. Nat. Genet. 2001, 29, 310–314. [Google Scholar] [CrossRef]
- Laghmani, K.; Beck, B.B.; Yang, S.-S.; Seaayfan, E.; Wenzel, A.; Reusch, B.; Vitzthum, H.; Priem, D.; Demaretz, S.; Bergmann, K.; et al. Polyhydramnios, Transient Antenatal Bartter’s Syndrome, and MAGED2 Mutations. N. Engl. J. Med. 2016, 374, 1853–1863. [Google Scholar] [CrossRef]
- Schlingmann, K.P.; Konrad, M.; Jeck, N.; Waldegger, P.; Reinalter, S.C.; Holder, M.; Seyberth, H.W.; Waldegger, S. Salt wasting and deafness resulting from mutations in two chloride channels. N. Engl. J. Med. 2004, 350, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Bichet, D.G.; Fujiwara, T.M. Reabsorption of sodium chloride—Lessons from the chloride channels. N. Engl. J. Med. 2004, 350, 1281–1283. [Google Scholar] [CrossRef] [PubMed]
- Nuñez-Gonzalez, L.; Carrera, N.; Garcia-Gonzalez, M.A. Molecular Basis, Diagnostic Challenges and Therapeutic Approaches of Bartter and Gitelman Syndromes: A Primer for Clinicians. Int. J. Mol. Sci. 2021, 22, 11414. [Google Scholar] [CrossRef]
- Bao, M.; Cai, J.; Yang, X.; Ma, W. Genetic screening for Bartter syndrome and Gitelman syndrome pathogenic genes among individuals with hypertension and hypokalemia. Clin. Exp. Hypertens. 2019, 41, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Orphanet: Bartter Syndrome. Available online: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?Lng=GB&Expert=112 (accessed on 14 March 2022).
- Besouw, M.T.P.; Kleta, R.; Bockenhauer, D. Bartter and Gitelman syndromes: Questions of class. Pediatr. Nephrol. 2020, 35, 1815–1824. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zhang, J.; Lang, Z.; Botella, J.R.; Zhu, J.K. Genome Editing—Principles and Applications for Functional Genomics Research and Crop Improvement. Crit. Rev. Plant Sci. 2017, 36, 291–309. [Google Scholar] [CrossRef]
- Puchta, H. The repair of double-strand breaks in plants: Mechanisms and consequences for genome evolution. J. Exp. Bot. 2005, 56, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.M.; Musunuru, K. Expanding the genetic editing tool kit: ZFNs, TALENs, and CRISPR-Cas9. J. Clin. Investig. 2014, 124, 4154–4161. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.G.; Cha, J.; Chandrasegaran, S. Hybrid restriction enzymes: Zinc finger fusions to Fok I cleavage domain. Proc. Natl. Acad. Sci. USA 1996, 93, 1156–1160. [Google Scholar] [CrossRef] [Green Version]
- Beerli, R.R.; Barbas, C.F. Engineering polydactyl zinc-finger transcription factors. Nat. Biotechnol. 2002, 20, 135–141. [Google Scholar] [CrossRef]
- Carroll, D.; Morton, J.J.; Beumer, K.J.; Segal, D.J. Design, construction and in vitro testing of zinc finger nucleases. Nat. Protoc. 2006, 1, 1329–1341. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Clark, K.J.; Campbell, J.M.; Panetta, M.R.; Guo, Y.; Ekker, S.C. Making designer mutants in model organisms. Development 2014, 141, 4042–4054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE nuclease architecture for efficient genome editing. Nat. Biotechnol. 2011, 29, 143–150. [Google Scholar] [CrossRef]
- Bogdanove, A.J.; Schornack, S.; Lahaye, T. TAL effectors: Finding plant genes for disease and defense. Curr. Opin. Plant Biol. 2010, 13, 394–401. [Google Scholar] [CrossRef]
- Guilinger, J.P.; Pattanayak, V.; Reyon, D.; Tsai, S.Q.; Sander, J.D.; Joung, J.K.; Liu, D.R. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat. Methods 2014, 11, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [Green Version]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Szczelkun, M.D.; Tikhomirova, M.S.; Sinkunas, T.; Gasiunas, G.; Karvelis, T.; Pschera, P.; Siksnys, V.; Seidel, R. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl. Acad. Sci. USA 2014, 111, 9798–9803. [Google Scholar] [CrossRef] [Green Version]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Jinek, M.; Jiang, F.; Taylor, D.W.; Sternberg, S.H.; Kaya, E.; Ma, E.; Anders, C.; Hauer, M.; Zhou, K.; Lin, S.; et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 2014, 343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anzalone, A.V.; Koblan, L.W.; Liu, D.R. Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors. Nat. Biotechnol. 2020, 38, 824–844. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, C.L.; Foley, J.E.; Wright, D.A.; Müller-Lerch, F.; Rahman, S.H.; Cornu, T.I.; Winfrey, R.J.; Sander, J.D.; Fu, F.; Townsend, J.A.; et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat. Methods 2008, 5, 374–375. [Google Scholar] [CrossRef] [PubMed]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, R.; Peng, J.; Yan, Y.; Cao, P.; Wang, J.; Qiu, C.; Tang, L.; Liu, D.; Tang, L.; Jin, J.; et al. Efficient gene editing in adult mouse livers via adenoviral delivery of CRISPR/Cas9. FEBS Lett. 2014, 588, 3954–3958. [Google Scholar] [CrossRef]
- Li, H.; Yang, Y.; Hong, W.; Huang, M.; Wu, M.; Zhao, X. Applications of genome editing technology in the targeted therapy of human diseases: Mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef]
- Chen, L.; Tang, L.; Xiang, H.; Jin, L.; Li, Q.; Dong, Y.; Wang, W.; Zhang, G. Advances in genome editing technology and its promising application in evolutionary and ecological studies. Gigascience 2014, 3, 2047-217X. [Google Scholar] [CrossRef] [Green Version]
- Miyagi, A.; Lu, A.; Humphreys, B.D. Gene Editing: Powerful New Tools for Nephrology Research and Therapy. J. Am. Soc. Nephrol. 2016, 27, 2940–2947. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, B.; Schwimmer, L.J.; Fuller, R.P.; Ye, Y.; Asawapornmongkol, L.; Barbas, C.F. Modular system for the construction of zinc-finger libraries and proteins. Nat. Protoc. 2010, 5, 791–810. [Google Scholar] [CrossRef] [Green Version]
- Valton, J.; Dupuy, A.; Daboussi, F.; Thomas, S.; Maréchal, A.; Macmaster, R.; Melliand, K.; Juillerat, A.; Duchateau, P. Overcoming transcription activator-like effector (TALE) DNA binding domain sensitivity to cytosine methylation. J. Biol. Chem. 2012, 287, 38427–38432. [Google Scholar] [CrossRef] [Green Version]
- Ates, I.; Rathbone, T.; Stuart, C.; Bridges, P.H.; Cottle, R.N. Delivery approaches for therapeutic genome editing and challenges. Genes 2020, 11, 1113. [Google Scholar] [CrossRef] [PubMed]
- Yip, B.H. Recent advances in CRISPR/Cas9 delivery strategies. Biomolecules 2020, 10, 839. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Wilson, D.R.; Green, J.J. Non-Viral Delivery To Enable Genome Editing. Trends Biotechnol. 2019, 37, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, F.; Dang, L.; Liang, C.; Wang, C.; He, B.; Liu, J.; Li, D.; Wu, X.; Xu, X.; et al. In Vivo delivery systems for therapeutic genome editing. Int. J. Mol. Sci. 2016, 17, 626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faneca, H. Non-Viral Gene Delivery Systems. Pharmaceutics 2021, 13, 446. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Que, H.; Luo, M.; Zhao, X.; Wei, X. Genome editing via non-viral delivery platforms: Current progress in personalized cancer therapy. Mol. Cancer 2022, 21, 1–15. [Google Scholar] [CrossRef]
- Wang, M.; Glass, Z.A.; Xu, Q. Non-viral delivery of genome-editing nucleases for gene therapy. Gene Ther. 2017, 24, 144–150. [Google Scholar] [CrossRef]
- Sakellari, G.I.; Zafeiri, I.; Batchelor, H.; Spyropoulos, F. Formulation design, production and characterisation of solid lipid nanoparticles (SLN) and nanostructured lipid carriers (NLC) for the encapsulation of a model hydrophobic active. Food Hydrocoll. Health 2021, 1, 100024. [Google Scholar] [CrossRef]
- Pensado, A.; Seijo, B.; Sanchez, A. Current strategies for DNA therapy based on lipid nanocarriers. Expert Opin. Drug Deliv. 2014, 11, 1721–1731. [Google Scholar] [CrossRef]
- Lim, S.; Koo, J.H.; Choi, J.M. Use of Cell-Penetrating Peptides in Dendritic Cell-Based Vaccination. Immune Netw. 2016, 16, 33–43. [Google Scholar] [CrossRef] [Green Version]
- Hirakawa, M.P.; Krishnakumar, R.; Timlin, J.A.; Carney, J.P.; Butler, K.S. Gene editing and CRISPR in the clinic: Current and future perspectives. Biosci. Rep. 2020, 40, BSR20200127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Moscou, M.J.; Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef]
- Zhang, X.H.; Tee, L.Y.; Wang, X.G.; Huang, Q.S.; Yang, S.H. Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol. Ther. Nucleic Acids 2015, 4, e264. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Volz, S.E.; Zhang, F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015, 33, 139–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, S.B.; Kim, D.Y.; Ko, J.H.; Kim, J.S.; Kim, Y.S. Improving CRISPR Genome Editing by Engineering Guide RNAs. Trends Biotechnol. 2019, 37, 870–881. [Google Scholar] [CrossRef]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [Green Version]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef]
- Korablev, A.; Lukyanchikova, V.; Serova, I.; Battulin, N. On-Target CRISPR/Cas9 Activity Can Cause Undesigned Large Deletion in Mouse Zygotes. Int. J. Mol. Sci. 2020, 21, 3604. [Google Scholar] [CrossRef]
- Cruz, N.M.; Freedman, B.S. CRISPR Gene Editing in the Kidney. Am. J. Kidney Dis. 2018, 71, 874–883. [Google Scholar] [CrossRef]
- Koslowski, S.; Latapy, C.; Auvray, P.; Blondel, M.; Meijer, L. An Overview of In Vivo and In Vitro Models for Autosomal Dominant Polycystic Kidney Disease: A Journey from 3D-Cysts to Mini-Pigs. Int. J. Mol. Sci. 2020, 21, 4537. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Sanchez-Niño, M.D.; Izquierdo, M.C.; Martin-Cleary, C.; Garcia-Bermejo, L.; Moreno, J.A.; Ruiz-Ortega, M.; Draibe, J.; Cruzado, J.M.; Garcia-Gonzalez, M.A.; et al. Translational value of animal models of kidney failure. Eur. J. Pharmacol. 2015, 759, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Pandey, U.B.; Nichols, C.D. Human disease models in Drosophila melanogaster and the role of the fly in therapeutic drug discovery. Pharmacol. Rev. 2011, 63, 411–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strange, K. Drug Discovery in Fish, Flies, and Worms. ILAR J. 2016, 57, 133–143. [Google Scholar] [CrossRef]
- WareJoncas, Z.; Campbell, J.M.; Martínez-Gálvez, G.; Gendron, W.A.C.; Barry, M.A.; Harris, P.C.; Sussman, C.R.; Ekker, S.C. Precision Gene Editing and Applications in Nephrology. Nat. Rev. Nephrol. 2018, 14, 663. [Google Scholar] [CrossRef]
- He, J.; Li, Q.; Fang, S.; Guo, Y.; Liu, T.; Ye, J.; Yu, Z.; Zhang, R.; Zhao, Y.; Hu, X.; et al. PKD1 mono-allelic knockout is sufficient to trigger renal cystogenesis in a mini-pig model. Int. J. Biol. Sci. 2015, 11, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Hofherr, A.; Busch, T.; Huber, N.; Nold, A.; Bohn, A.; Viau, A.; Bienaimé, F.; Kuehn, E.W.; Arnold, S.J.; Köttgen, M. Efficient genome editing of differentiated renal epithelial cells. Pflugers Arch. 2017, 469, 303–311. [Google Scholar] [CrossRef] [Green Version]
- Freedman, B.S.; Brooks, C.R.; Lam, A.Q.; Fu, H.; Morizane, R.; Agrawal, V.; Saad, A.F.; Li, M.K.; Hughes, M.R.; Werff, R.V.; et al. Modelling kidney disease with CRISPR-mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat. Commun. 2015, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Cruz, N.M.; Song, X.; Czerniecki, S.M.; Gulieva, R.E.; Churchill, A.J.; Kim, Y.K.; Winston, K.; Tran, L.M.; Diaz, M.A.; Fu, H.; et al. Organoid cystogenesis reveals a critical role of microenvironment in human polycystic kidney disease. Nat. Mater. 2017, 16, 1112–1119. [Google Scholar] [CrossRef] [Green Version]
- Kuraoka, S.; Tanigawa, S.; Taguchi, A.; Hotta, A.; Nakazato, H.; Osafune, K.; Kobayashi, A.; Nishinakamura, R. PKD1-Dependent Renal Cystogenesis in Human Induced Pluripotent Stem Cell-Derived Ureteric Bud/Collecting Duct Organoids. J. Am. Soc. Nephrol. 2020, 31, 2355–2371. [Google Scholar] [CrossRef]
- Shamshirgaran, Y.; Jonebring, A.; Svensson, A.; Leefa, I.; Bohlooly, Y.M.; Firth, M.; Woollard, K.J.; Hofherr, A.; Rogers, I.M.; Hicks, R. Rapid target validation in a Cas9-inducible hiPSC derived kidney model. Sci. Rep. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Tsukiyama, T.; Kobayashi, K.; Nakaya, M.; Iwatani, C.; Seita, Y.; Tsuchiya, H.; Matsushita, J.; Kitajima, K.; Kawamoto, I.; Nakagawa, T.; et al. Monkeys mutant for PKD1 recapitulate human autosomal dominant polycystic kidney disease. Nat. Commun. 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, M.; Umeyama, K.; Nakano, K.; Matsunari, H.; Fukuda, T.; Matsumoto, K.; Tajiri, S.; Yamanaka, S.; Hasegawa, K.; Okamoto, K.; et al. Generation of heterozygous PKD1 mutant pigs exhibiting early-onset renal cyst formation. Lab. Investig. 2022, 102, 560–569. [Google Scholar] [CrossRef] [PubMed]
- Chumley, P.; Zhou, J.; Mrug, S.; Chacko, B.; Parant, J.M.; Challa, A.K.; Wilson, L.S.; Berryhill, T.F.; Barnes, S.; Kesterson, R.A.; et al. Truncating PKHD1 and PKD2 mutations alter energy metabolism. Am. J. Physiol. Renal Physiol. 2019, 316, F414–F425. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Wang, X.; Sussman, C.R.; Hopp, K.; Irazabal, M.V.; Bakeberg, J.L.; LaRiviere, W.B.; Manganiello, V.C.; Vorhees, C.V.; Zhao, H.; et al. Modulation of Polycystic Kidney Disease Severity by Phosphodiesterase 1 and 3 Subfamilies. J. Am. Soc. Nephrol. 2016, 27, 1312–1320. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Yamada, S.; Lariviere, W.B.; Ye, H.; Bakeberg, J.L.; Irazabal, M.V.; Chebib, F.T.; Van Deursen, J.; Harris, P.C.; Sussman, C.R.; et al. Generation and phenotypic characterization of Pde1a mutant mice. PLoS ONE 2017, 12, e0181087. [Google Scholar] [CrossRef]
- Low, J.H.; Li, P.; Chew, E.G.Y.; Zhou, B.; Suzuki, K.; Zhang, T.; Lian, M.M.; Liu, M.; Aizawa, E.; Esteban, C.R.; et al. Generation of Human PSC-Derived Kidney Organoids with Patterned Nephron Segments and a De Novo Vascular Network. Cell Stem Cell 2019, 25, 373–387.e9. [Google Scholar] [CrossRef]
- Shan, D.; Rezonzew, G.; Mullen, S.; Roye, R.; Zhou, J.; Chumley, P.; Revell, D.Z.; Challa, A.; Kim, H.; Lockhart, M.E.; et al. Heterozygous Pkhd1 C642* mice develop cystic liver disease and proximal tubule ectasia that mimics radiographic signs of medullary sponge kidney. Am. J. Physiol. Renal Physiol. 2019, 316, F463–F472. [Google Scholar] [CrossRef]
- Arkhipov, S.N.; Potter, D.L.; Geurts, A.M.; Pavlov, T.S. Knockout of P2rx7 purinergic receptor attenuates cyst growth in a rat model of ARPKD. Am. J. Physiol. Renal Physiol. 2019, 317, F1649–F1655. [Google Scholar] [CrossRef]
- Zhang, H.D.; Huang, J.N.; Liu, Y.Z.; Ren, H.; Xie, J.Y.; Chen, N. Endoplasmic reticulum stress and proteasome pathway involvement in human podocyte injury with a truncated COL4A3 mutation. Chin. Med. J. 2019, 132, 1823–1832. [Google Scholar] [CrossRef]
- Daga, S.; Donati, F.; Capitani, K.; Croci, S.; Tita, R.; Giliberti, A.; Valentino, F.; Benetti, E.; Fallerini, C.; Niccheri, F.; et al. New frontiers to cure Alport syndrome: COL4A3 and COL4A5 gene editing in podocyte-lineage cells. Eur. J. Hum. Genet. 2020, 28, 480–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuang, X.; Sun, L.; Wu, Y.; Huang, W. A novel missense mutation of COL4A5 gene alter collagen IV α5 chain to cause X-linked Alport syndrome in a Chinese family. Transl. Pediatr. 2020, 9, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Hashikami, K.; Asahina, M.; Nozu, K.; Iijima, K.; Nagata, M.; Takeyama, M. Establishment of X-linked Alport syndrome model mice with a Col4a5 R471X mutation. Biochem. Biophys. Rep. 2018, 17, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Funk, S.D.; Bayer, R.H.; Malone, A.F.; McKee, K.K.; Yurchenco, P.D.; Miner, J.H. Pathogenicity of a human laminin β2 mutation revealed in models of alport syndrome. J. Am. Soc. Nephrol. 2018, 29, 949–960. [Google Scholar] [CrossRef] [Green Version]
- Moreno, C.; Hoffman, M.; Stodola, T.J.; Didier, D.N.; Lazar, J.; Geurts, A.M.; North, P.E.; Jacob, H.J.; Greene, A.S. Creation and characterization of a renin knockout rat. Hypertension 2011, 57, 614–619. [Google Scholar] [CrossRef] [Green Version]
- Przepiorski, A.; Sander, V.; Tran, T.; Hollywood, J.A.; Sorrenson, B.; Shih, J.H.; Wolvetang, E.J.; McMahon, A.P.; Holm, T.M.; Davidson, A.J. A Simple Bioreactor-Based Method to Generate Kidney Organoids from Pluripotent Stem Cells. Stem Cell Rep. 2018, 11, 470–484. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.G.; Dang, L.T.; Marsh, G.; Roach, A.M.; Levine, Z.G.; Monti, A.; Reyon, D.; Feigenbaum, L.; Duffield, J.S. Uromodulin p.Cys147Trp mutation drives kidney disease by activating ER stress and apoptosis. J. Clin. Investig. 2017, 127, 3954–3969. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, Z.; Shin, M.K.; Horwitz, S.B.; Levorse, J.M.; Zhu, L.; Sharif-Rodriguez, W.; Streltsov, D.Y.; Dajee, M.; Hernandez, M.; et al. Heterozygous disruption of renal outer medullary potassium channel in rats is associated with reduced blood pressure. Hypertension 2013, 62, 288–294. [Google Scholar] [CrossRef] [Green Version]
- Grill, A.; Schießl, I.M.; Gess, B.; Fremter, K.; Hammer, A.; Castrop, H. Salt-losing nephropathy in mice with a null mutation of the Clcnk2 gene. Acta Physiol. 2016, 218, 198–211. [Google Scholar] [CrossRef]
- Hopp, K.; Ward, C.J.; Hommerding, C.J.; Nasr, S.H.; Tuan, H.F.; Gainullin, V.G.; Rossetti, S.; Torres, V.E.; Harris, P.C. Functional polycystin-1 dosage governs autosomal dominant polycystic kidney disease severity. J. Clin. Investig. 2012, 122, 4257–4273. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Peissel, B.; Babakhanlou, H.; Pavlova, A.; Geng, L.; Fan, X.; Larson, C.; Brent, G.; Zhou, J. Perinatal lethality with kidney and pancreas defects in mice with a targetted Pkd1 mutation. Nat. Genet. 1997, 17, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Lian, X.; Wu, X.; Li, Z.; Zhang, Y.; Song, K.; Cai, G.; Li, Q.; Lin, S.; Chen, X.; Bai, X.Y. The combination of metformin and 2-deoxyglucose significantly inhibits cyst formation in miniature pigs with polycystic kidney disease. Br. J. Pharmacol. 2019, 176, 711–724. [Google Scholar] [CrossRef] [Green Version]
- Torres, V.E.; Harris, P.C. Strategies targeting cAMP signaling in the treatment of polycystic kidney disease. J. Am. Soc. Nephrol. 2014, 25, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Chebib, F.T.; Sussman, C.R.; Wang, X.; Harris, P.C.; Torres, V.E. Vasopressin and disruption of calcium signalling in polycystic kidney disease. Nat. Rev. Nephrol. 2015, 11, 451–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gattone, V.H.; Wang, X.; Harris, P.C.; Torres, V.E. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat. Med. 2003, 9, 1323–1326. [Google Scholar] [CrossRef]
- Czerniecki, S.M.; Cruz, N.M.; Harder, J.L.; Menon, R.; Annis, J.; Otto, E.A.; Gulieva, R.E.; Islas, L.V.; Kim, Y.K.; Tran, L.M.; et al. High-Throughput Screening Enhances Kidney Organoid Differentiation from Human Pluripotent Stem Cells and Enables Automated Multidimensional Phenotyping. Cell Stem Cell 2018, 22, 929–940.e4. [Google Scholar] [CrossRef] [Green Version]
- Romero-Guevara, R.; Ioannides, A.; Xinaris, C. Kidney Organoids as Disease Models: Strengths, Weaknesses and Perspectives. Front. Physiol. 2020, 11, 1384. [Google Scholar] [CrossRef]
- Wu, H.; Uchimura, K.; Donnelly, E.L.; Kirita, Y.; Morris, S.A.; Humphreys, B.D. Comparative Analysis and Refinement of Human PSC-Derived Kidney Organoid Differentiation with Single-Cell Transcriptomics. Cell Stem Cell 2018, 23, 869–881.e8. [Google Scholar] [CrossRef] [Green Version]
- Cordido, A.; Nunez-Gonzalez, L.; Martinez-Moreno, J.M.; Lamas-Gonzalez, O.; Rodriguez-Osorio, L.; Perez-Gomez, M.V.; Martin-Sanchez, D.; Outeda, P.; Chiaravalli, M.; Watnick, T.; et al. TWEAK Signaling Pathway Blockade Slows Cyst Growth and Disease Progression in Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2021, 32, 1913–1932. [Google Scholar] [CrossRef]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Grantham, J.J.; Higashihara, E.; Perrone, R.D.; Krasa, H.B.; Ouyang, J.; Czerwiec, F.S. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2012, 367, 2407–2418. [Google Scholar] [CrossRef] [Green Version]
- Menezes, L.F.; Zhou, F.; Patterson, A.D.; Piontek, K.B.; Krausz, K.W.; Gonzalez, F.J.; Germino, G.G. Network analysis of a Pkd1-mouse model of autosomal dominant polycystic kidney disease identifies HNF4α as a disease modifier. PLoS Genet. 2012, 8, e1003053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palygin, O.; Ilatovskaya, D.V.; Levchenko, V.; Klemens, C.A.; Dissanayake, L.; Williams, A.M.; Pavlov, T.S.; Staruschenko, A. Characterization of purinergic receptor expression in ARPKD cystic epithelia. Purinergic Signal. 2018, 14, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Kruegel, J.; Rubel, D.; Gross, O. Alport syndrome—Insights from basic and clinical research. Nat. Rev. Nephrol. 2013, 9, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Hertz, J.M.; Thomassen, M.; Storey, H.; Flinter, F. Clinical utility gene card for: Alport syndrome—Update 2014. Eur. J. Hum. Genet. 2015, 23, 713. [Google Scholar] [CrossRef] [Green Version]
- Massa, F.; Garbay, S.; Bouvier, R.; Sugitani, Y.; Noda, T.; Gubler, M.C.; Heidet, L.; Pontoglio, M.; Fischer, E. Hepatocyte nuclear factor 1β controls nephron tubular development. Development 2013, 140, 886–896. [Google Scholar] [CrossRef] [Green Version]
- Bates, J.M.; Raffi, H.M.; Prasadan, K.; Mascarenhas, R.; Laszik, Z.; Maeda, N.; Hultgren, S.J.; Kumar, S. Tamm-Horsfall protein knockout mice are more prone to urinary tract infection: Rapid communication. Kidney Int. 2004, 65, 791–797. [Google Scholar] [CrossRef] [Green Version]
- Renigunta, A.; Renigunta, V.; Saritas, T.; Decher, N.; Mutig, K.; Waldegger, S. Tamm-Horsfall glycoprotein interacts with renal outer medullary potassium channel ROMK2 and regulates its function. J. Biol. Chem. 2011, 286, 2224–2235. [Google Scholar] [CrossRef] [Green Version]
- Mo, L.; Liaw, L.; Evan, A.P.; Sommer, A.J.; Lieske, J.C.; Wu, X.R. Renal calcinosis and stone formation in mice lacking osteopontin, Tamm-Horsfall protein, or both. Am. J. Physiol. Renal Physiol. 2007, 293, F1935–F1943. [Google Scholar] [CrossRef]
- Welling, P.A.; Ho, K. A comprehensive guide to the ROMK potassium channel: Form and function in health and disease. Am. J. Physiol. Renal Physiol. 2009, 297, F849–F863. [Google Scholar] [CrossRef] [Green Version]
- Bartter, F.C.; Pronove, P.; Gill, J.R.; Maccardle, R.C. Hyperplasia of the juxtaglomerular complex with hyperaldosteronism and hypokalemic alkalosis. A new syndrome. Am. J. Med. 1962, 33, 811–828. [Google Scholar] [CrossRef]
- Greger, R. Ion transport mechanisms in thick ascending limb of Henle’s loop of mammalian nephron. Physiol. Rev. 1985, 65, 760–797. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.; Zhang, C.; Tian, X.; Coman, D.; Hyder, F.; Ma, M.; Somlo, S. Renal plasticity revealed through reversal of polycystic kidney disease in mice. Nat. Genet. 2021, 53, 1649–1663. [Google Scholar] [CrossRef] [PubMed]
- Rubin, J.D.; Nguyen, T.V.; Allen, K.L.; Ayasoufi, K.; Barry, M.A. Comparison of Gene Delivery to the Kidney by Adenovirus, Adeno-Associated Virus, and Lentiviral Vectors After Intravenous and Direct Kidney Injections. Hum. Gene Ther. 2019, 30, 1559–1571. [Google Scholar] [CrossRef] [PubMed]
- Kida, Y. Peritubular Capillary Rarefaction: An Underappreciated Regulator of CKD Progression. Int. J. Mol. Sci. 2020, 21, 8255. [Google Scholar] [CrossRef] [PubMed]
- Rubin, J.D.; Barry, M.A. Improving Molecular Therapy in the Kidney. Mol. Diagn. Ther. 2020, 24, 375–396. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, H.; Higuchi, N.; Nishikawa, Y.; Hirahara, H.; Iino, N.; Kameda, S.; Kawachi, H.; Yaoita, E.; Gejyo, F.; Miyazaki, J.I. Kidney-targeted naked DNA transfer by retrograde renal vein injection in rats. Hum. Gene Ther. 2002, 13, 455–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kameda, S.; Maruyama, H.; Higuchi, N.; Iino, N.; Nakamura, G.; Miyazaki, J.; Gejyo, F. Kidney-targeted naked DNA transfer by retrograde injection into the renal vein in mice. Biochem. Biophys. Res. Commun. 2004, 314, 390–395. [Google Scholar] [CrossRef]
- Liu, G.W.; Pippin, J.W.; Eng, D.G.; Lv, S.; Shankland, S.J.; Pun, S.H. Nanoparticles exhibit greater accumulation in kidney glomeruli during experimental glomerular kidney disease. Physiol. Rep. 2020, 8, e14545. [Google Scholar] [CrossRef]
- Williams, R.M.; Shah, J.; Ng, B.D.; Minton, D.R.; Gudas, L.J.; Park, C.Y.; Heller, D.A. Mesoscale nanoparticles selectively target the renal proximal tubule epithelium. Nano Lett. 2015, 15, 2358–2364. [Google Scholar] [CrossRef] [Green Version]
- Naumenko, V.; Nikitin, A.; Kapitanova, K.; Melnikov, P.; Vodopyanov, S.; Garanina, A.; Valikhov, M.; Ilyasov, A.; Vishnevskiy, D.; Markov, A.; et al. Intravital microscopy reveals a novel mechanism of nanoparticles excretion in kidney. J. Control. Release 2019, 307, 368–378. [Google Scholar] [CrossRef]
- Rocca, C.J.; Ur, S.N.; Harrison, F.; Cherqui, S. rAAV9 combined with renal vein injection is optimal for kidney-targeted gene delivery: Conclusion of a comparative study. Gene Ther. 2014, 21, 618–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Xu, Y.; Bai, Z.; Ma, D.; Niu, Q.; Meng, J.; Fan, S.; Zhang, L.; Hao, Z.; Zhang, X.; et al. Transparenchymal Renal Pelvis Injection of Recombinant Adeno-Associated Virus Serotype 9 Vectors Is a Practical Approach for Gene Delivery in the Kidney. Hum. Gene Ther. Methods 2018, 29, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Lieber, A.; He, C.Y.; Meuse, L.; Schowalter, D.; Kirillova, I.; Winther, B.; Kay, M.A. The role of Kupffer cell activation and viral gene expression in early liver toxicity after infusion of recombinant adenovirus vectors. J. Virol. 1997, 71, 8798–8807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worgall, S.; Wolff, G.; Falck-Pedersen, E.; Crystal, R.G. Innate immune mechanisms dominate elimination of adenoviral vectors following in vivo administration. Hum. Gene Ther. 1997, 8, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Johnson, J.E.; Richardson, J.A.; Hiesberger, T.; Igarashi, P. A minimal Ksp-cadherin promoter linked to a green fluorescent protein reporter gene exhibits tissue-specific expression in the developing kidney and genitourinary tract. J. Am. Soc. Nephrol. 2002, 13, 1824–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watnick, T.J.; Torres, V.E.; Gandolph, M.A.; Qian, F.; Onuchic, L.F.; Klinger, K.W.; Landes, G.; Germino, G.G. Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease. Mol. Cell 1998, 2, 247–251. [Google Scholar] [CrossRef]
- Doronin, K.; Shashkova, E.V.; May, S.M.; Hofherr, S.E.; Barry, M.A. Chemical modification with high molecular weight polyethylene glycol reduces transduction of hepatocytes and increases efficacy of intravenously delivered oncolytic adenovirus. Hum. Gene Ther. 2009, 20, 975–988. [Google Scholar] [CrossRef]
- Barry, M.A.; Weaver, E.A.; Chen, C.Y. Mining the adenovirus “virome” for systemic oncolytics. Curr. Pharm. Biotechnol. 2012, 13, 1804–1808. [Google Scholar] [CrossRef]
- Krasnykh, V.; Dmitriev, I.; Mikheeva, G.; Miller, C.R.; Belousova, N.; Curiel, D.T. Characterization of an adenovirus vector containing a heterologous peptide epitope in the HI loop of the fiber knob. J. Virol. 1998, 72, 1844–1852. [Google Scholar] [CrossRef] [Green Version]
- Dmitriev, I.; Krasnykh, V.; Miller, C.R.; Wang, M.; Kashentseva, E.; Mikheeva, G.; Belousova, N.; Curiel, D.T. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. J. Virol. 1998, 72, 9706–9713. [Google Scholar] [CrossRef] [Green Version]
- Wickham, T.J.; Tzeng, E.; Shears, L.L.; Roelvink, P.W.; Li, Y.; Lee, G.M.; Brough, D.E.; Lizonova, A.; Kovesdi, I. Increased in vitro and in vivo gene transfer by adenovirus vectors containing chimeric fiber proteins. J. Virol. 1997, 71, 8221–8229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, S.; Ohno, S.I.; Harada, Y.; Oikawa, K.; Fujita, K.; Mineo, S.; Gondo, A.; Kanno, Y.; Kuroda, M. rAAV6-mediated miR-29b delivery suppresses renal fibrosis. Clin. Exp. Nephrol. 2019, 23, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Chew, W.L. Immunity to CRISPR Cas9 and Cas12a therapeutics. Wiley Interdiscip. Rev. Syst. Biol. Med. 2018, 10, e1408. [Google Scholar] [CrossRef] [PubMed]
- Barbalat, R.; Ewald, S.E.; Mouchess, M.L.; Barton, G.M. Nucleic acid recognition by the innate immune system. Annu. Rev. Immunol. 2011, 29, 185–214. [Google Scholar] [CrossRef] [PubMed]
- McCullough, K.T.; Boye, S.L.; Fajardo, D.; Calabro, K.; Peterson, J.J.; Strang, C.E.; Chakraborty, D.; Gloskowski, S.; Haskett, S.; Samuelsson, S.; et al. Somatic Gene Editing of GUCY2D by AAV-CRISPR/Cas9 Alters Retinal Structure and Function in Mouse and Macaque. Hum. Gene Ther. 2019, 30, 571–589. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Wu, Y.; Gemberling, M.P.; Oliver, M.L.; Waller, M.A.; Bohning, J.D.; Robinson-Hamm, J.N.; Bulaklak, K.; Rivera, R.M.C.; Collier, J.H.; et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019, 25, 427–432. [Google Scholar] [CrossRef]
- Chew, W.L.; Tabebordbar, M.; Cheng, J.K.W.; Mali, P.; Wu, E.Y.; Ng, A.H.M.; Zhu, K.; Wagers, A.J.; Church, G.M. A multifunctional AAV-CRISPR-Cas9 and its host response. Nat. Methods 2016, 13, 868–874. [Google Scholar] [CrossRef] [Green Version]
- Rosenblum, D.; Gutkin, A.; Kedmi, R.; Ramishetti, S.; Veiga, N.; Jacobi, A.M.; Schubert, M.S.; Friedmann-Morvinski, D.; Cohen, Z.R.; Behlke, M.A.; et al. CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy. Sci. Adv. 2020, 6, eabc9450. [Google Scholar] [CrossRef]
- Xue, H.; Guo, P.; Wen, W.-C.; Wong, H. Lipid-Based Nanocarriers for RNA Delivery. Curr. Pharm. Des. 2015, 21, 3140–3147. [Google Scholar] [CrossRef]
- Dammes, N.; Goldsmith, M.; Ramishetti, S.; Dearling, J.L.J.; Veiga, N.; Packard, A.B.; Peer, D. Conformation-sensitive targeting of lipid nanoparticles for RNA therapeutics. Nat. Nanotechnol. 2021, 16, 1030–1038. [Google Scholar] [CrossRef]
- Ma, Y.; Cai, F.; Li, Y.; Chen, J.; Han, F.; Lin, W. A review of the application of nanoparticles in the diagnosis and treatment of chronic kidney disease. Bioact. Mater. 2020, 5, 732–743. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, J.; Jiang, K.; Chung, E.J. Improving kidney targeting: The influence of nanoparticle physicochemical properties on kidney interactions. J. Control. Release 2021, 334, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.B.; Song, G.L.; Liu, D.; Li, S.J.; Wu, J.H.; Kang, X.Q.; Qi, J.; Jin, F.Y.; Wang, X.J.; Xu, X.L.; et al. Sialic acid-modified solid lipid nanoparticles as vascular endothelium-targeting carriers for ischemia-reperfusion-induced acute renal injury. Drug Deliv. 2017, 24, 1856–1867. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Zhang, H.; Yin, N.; Zhang, Y.; Gou, J.; Yin, T.; He, H.; Ding, H.; Zhang, Y.; Tang, X. Sialic acid-modified dexamethasone lipid calcium phosphate gel core nanoparticles for target treatment of kidney injury. Biomater. Sci. 2020, 8, 3871–3884. [Google Scholar] [CrossRef] [PubMed]
- Van Alem, C.M.A.; Boonstra, M.; Prins, J.; Bezhaeva, T.; Van Essen, M.F.; Ruben, J.M.; Vahrmeijer, A.L.; Van Der Veer, E.P.; De Fijter, J.W.; Reinders, M.E.; et al. Local delivery of liposomal prednisolone leads to an anti-inflammatory profile in renal ischaemia-reperfusion injury in the rat. Nephrol. Dial. Transplant 2018, 33, 44–53. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Tang, H.; You, X.; Huang, K.; Dhinakar, A.; Kang, Y.; Yu, Q.; Wu, J. BAPTA-AM Nanoparticle for the Curing of Acute Kidney Injury Induced by Ischemia/Reperfusion. J. Biomed. Nanotechnol. 2018, 14, 868–883. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
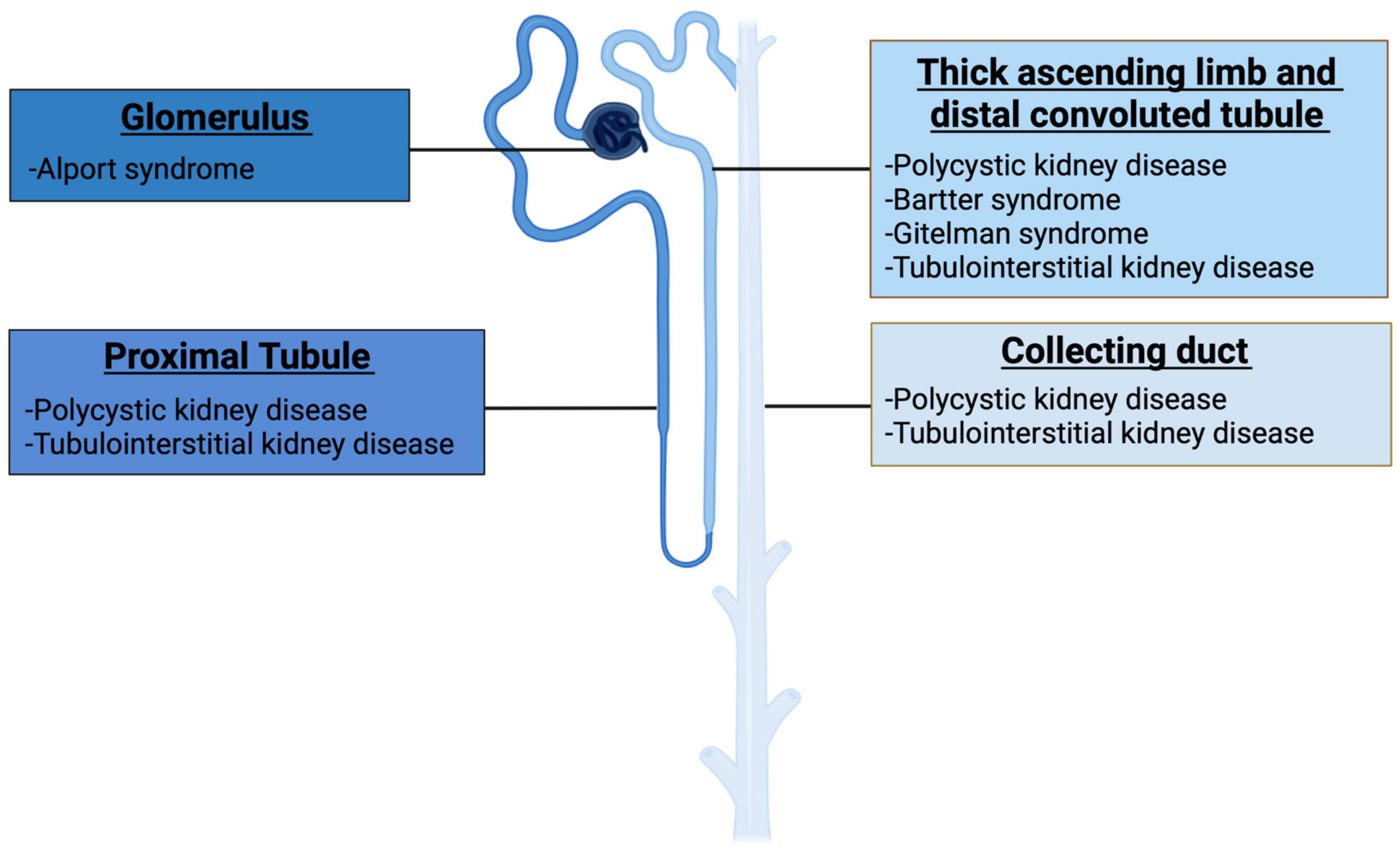
| Disease | Estimated Incidende (per 100,000 Population) | Gene | Protein | Function |
|---|---|---|---|---|
| Autosomal dominant polycystic kidney disease (CL) | ~100 individuals | PKD1 PKD2 GANAβ DNAJB11 IFT140 | Polycystin-1 Polycystin-2 Glucosidase II subunit α DnaJ homolog subfamily B member 11 Intraflagellar transport protein 140 homolog | Calcium ion transmembrane transport Calcium ion transmembrane transport Alpha-glucosidase activity Misfolded protein binding Cilium assembly |
| Autosomal recessive polycystic kidney disease (CL) | ~5 individuals | PKHD1 DZIP1L | Polyductin Cilium assembly protein DZIP1L | Cell-cell adhesion Metal ion binding |
| Alport syndrome (GL) | ~50 individuals | COL4A3 COL4A4 COL4A5 | Collagen alpha-3 (IV) chain Collagen alpha-4 (IV) chain Collagen alpha-5 (IV) chain | Extracellular matrix organization |
| Autosomal dominant tubulointerstitial kidney disease (TL) | N/A | UMOD MUC1 REN HNF1B SEC61A1 | Uromodulin Mucin-1 Renin Hepatocyte nuclear factor 1β α1 subunit of the SEC61 | Cellular defense response DNA damage response Cell maturation Transcription factor Endoplasmatic reticulum organization |
| Gitelman syndrome (TL) | ~2 individuals | SLC12A3 | Solute carrier family 12 member 3 | Ion transport |
| Bartter syndrome (TL) | <1 individual | SLC12A1 KCNJ1 CLCNKA CLCNKB BSND MAGED2 | Solute carrier family 12 member 1 ATP-sensitive inward rectifier potassium channel 1 Chloride channel protein ClC-Ka Chloride channel protein ClC-Kb Barttin Melanoma-associated antigen D2 | Ion transport Potassium ion transport Chloride transport Chloride transport Chloride transport Sodium ion absorption |
| ZFNs | TALENs | CRISPR-Cas9 | References | |
|---|---|---|---|---|
| Recognition site | Zinc finger motif | RVD region of tandem TALE repeat | Single-strand guide RNA | [72,77,80] |
| Endonuclease | FokI nuclease | FokI nuclease | Cas9 nuclease | [72,77,80] |
| Target sequence size | 9–18 bp per ZFP monomer | 14–20 bp per TALE monomer | 20 bp guide sequence | [71,79,83] |
| Targeting limitations | Difficult to target non-G-rich sites | 5ʹ targeted base must be a T for each TALEN monomer | Targeted site must precede a PAM sequence | [69,86,87] |
| Delivery (in vivo) | AAVs, LVs, AdVs | LVs, AdVs | AAVs, LVs | [69,88] |
| Specificity | Tolerating a small number of positional mismatches | Tolerating a small number of positional mismatches | Tolerating multiple consecutive mismatches | [89] |
| Efficiency | ~12% | ~76% | ~81% | [69,90] |
| Cost (Single experiment) | $5000 | $500 | $30 | [91] |
| Overall evaluation | Good | Very good | Excellent |
| Gene | Tool | Model | Key Finding | Refs. |
|---|---|---|---|---|
| Autosomal dominant polycystic kidney disease | ||||
| PKD1 | ZFN | Mini pig | Heterozygous PKD1 mini pigs develop cysts. | [119] |
| TALEN and CRISPR | MDCK and mIMCD3 | Protocol for creating knockout cell lines | [120] | |
| CRISPR | Kidney organoids | Knockout of PKD1 causes cysts | [121] | |
| CRISPR | Kidney organoids | Growing kidney organoids in suspension culture enhances cystogenesis | [122] | |
| CRISPR | UB organoids | cAMP signaling is involved in cystogenesis | [123] | |
| CRISPR | Kidney organoids | By using knockout pools it is possible to generate cystogenesis | [124] | |
| CRISPR | Monkey | Heterozygous PKD1 monkeys show cystogenesis perinatally | [125] | |
| CRISPR | Pig | Heterozygous PKD1 pigs develop many pathological conditions similar to ADPKD patients | [126] | |
| PKD2 | TALEN | MDCK and mIMCD3 | Protocol to creation knockout cell lines | [120] |
| CRISPR | Kidney organoids | Knockout of PKD2 causes cysts | [121] | |
| CRISPR | Kidney organoids | Growing organoids in suspension culture enhances cystogenesis | [122] | |
| CRISPR | Kidney organoids | By using knockout pools it is possible to generate cystogenesis | [124] | |
| CRISPR | HEK-293 | Knockout of PKD2 does not alter energy metabolism | [127] | |
| Pde1a | TALEN | Mouse | Knockout of Pde1a aggravates cystogenesis | [128] |
| TALEN | Mouse | Knockout of Pde1a aggravates cystogenesis | [129] | |
| GANAβ | CRISPR | RCTE | Knockout of GANAβ causes PC1 and PC2 maturation and localization defects | [14] |
| Autosomal recessive polycystic kidney disease | ||||
| PKHD1 | CRISPR | HEK-293 | Knockout of PKHD1 alters energy metabolism | [127] |
| CRISPR | Kidney organoids | CRISPR-knockin as a method to correct pathogenic variants. | [130] | |
| CRISPR | Mouse | Heterozygous Pkhd1 develop proximal tubule ectasia | [131] | |
| dzip1l | CRISPR | Zebrafish | DZIP1L is involved in the formation of primary cilia | [28] |
| P2rx7 | CRISPR | Mouse | P2X7 contributes to cyst growth by increasing pannexin-1-dependent ATP release into the lumen | [132] |
| Alport Syndrome | ||||
| COL4A3 | CRISPR | Mouse podocytes | Knockout of Col4a causes endoplasmic reticulum stress and apoptosis | [133] |
| CRISPR | Human podocytes | Innovative protocol for COL4A3 correction by HDR | [134] | |
| COL4A5 | CRISPR | Human podocytes | Innovative protocol for COL4A5 correction by HDR | [134] |
| CRISPR | Human podocytes | CRISPR-knockin as a method for confirming the pathogenicity of missense variants | [135] | |
| CRISPR | Mouse | Heterozygous Col4a5 male mice develop many pathological conditions similar to AS patients | [136] | |
| Lamb2 | CRISPR | Mouse | Heterozygous mutations in a gene encoding GBM components aggravate AS phenotype | [137] |
| Autosomal dominant tubulointerstitial kidney disease | ||||
| Ren | ZFN | SS rat | Knockout of Ren causes poor renal function | [138] |
| HNF1B | CRISPR | Kidney organoids | Knockout of HNF1B prevents proper formation of certain components of the nephron | [139] |
| sec61al2 | CRISPR | Zebrafish | Mutations in sec61al2 causes pronephric tubules defects | [55] |
| Umod | CRISPR | Mouse | Heterozygous Umod mice develop many pathological conditions similar to ADTKD-UMOD patient | [140] |
| Gitelman and Bartter syndromes | ||||
| Kcnj1 | ZFN | SS rat | Knockout of Kcnj1 protects against salt-induced hypertension and renal injury | [141] |
| Clcnk2 | TALEN | Mouse | ClC-K2-deficient mice develop many pathological conditions similar to BS patient | [142] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-García, F.; Martínez-Pulleiro, R.; Carrera, N.; Allegue, C.; Garcia-Gonzalez, M.A. Genetic Kidney Diseases (GKDs) Modeling Using Genome Editing Technologies. Cells 2022, 11, 1571. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11091571
Gómez-García F, Martínez-Pulleiro R, Carrera N, Allegue C, Garcia-Gonzalez MA. Genetic Kidney Diseases (GKDs) Modeling Using Genome Editing Technologies. Cells. 2022; 11(9):1571. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11091571
Chicago/Turabian StyleGómez-García, Fernando, Raquel Martínez-Pulleiro, Noa Carrera, Catarina Allegue, and Miguel A. Garcia-Gonzalez. 2022. "Genetic Kidney Diseases (GKDs) Modeling Using Genome Editing Technologies" Cells 11, no. 9: 1571. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11091571