1. Introduction
Muscle atrophy is highly deleterious for cachexic patients as it alters both the quality of life and the efficiency of treatments [
1,
2]. The decrease in muscle mass is attributable to an alteration of proteostasis mainly due to a huge increase of protein degradation, which affects the size of muscle fibers rather than decreasing their number [
3,
4,
5]. In addition, skeletal muscle fibers are differentially affected by the atrophying program depending on their contractile and metablic properties, each fiber type being more sensitive to specific atrophying stimuli [
4,
5,
6]. For example, muscle disuse predominantly affects slow type I fibers while sepsis and cancer cachexia induces atrophy on fast-twitch type II fibers. Proteolysis activation is the main cause of muscle atrophy in numerous catabolic human diseases for the rapid degradation of contractile proteins, the ubiquitin-proteasome system (UPS) and autophagy being the main proteolytic systems involved [
3,
7].
The UPS is crucial as it controls the degradation of the bulk of cellular proteins and also represses protein synthesis [
3]. The UPS targets the proteins to be degraded by covalently linking a ubiquitin (Ub) chain to the substrates, which enables the recognition and the degradation of the targets by the 26S proteasome. The Ub chain is formed by an enzymatic cascade that activates Ub (E1 activating enzyme), recognizes the substrates (E3 ligases), and catalyzes an isopeptide linkage between the substrate and Ub or between 2 molecules of Ub (E2 ligases and some E3s). In atrophying skeletal muscles, several UPS genes (Ub, 26S Proteasome subunits, E2s, E3s, etc.) are up-regulated in most catabolic situations, which includes two muscle-specific E3 ligases, muscle atrophy F-box (MAFbx or atrogin1) and muscle ring finger-1 (MuRF1) [
8,
9,
10,
11].
MuRF1 is the only E3 ligase known to target the myofibrillar proteins but MuRF1 needs the presence of E2 enzymes for catalyzing the Ub chains [
10,
12,
13,
14,
15,
16]. Little is known about E2 enzymes in muscles (for a review see [
17]) but we found recently that at least 5 of them are able to bind MuRF1, including E2E1 [
15].
E2E1 belongs to the class III E2 enzymes, which means it possesses an N-terminal extension besides the UBC fold that characterizes E2 enzymes [
18]. E2E1 is a member of the UBE2E sub-family that comprises also E2E2 and E2E3. The three isoforms have a highly conserved UBC domain (92% identity) but variable N-terminal extension (26% identity) [
19,
20]. The variability of the latter suggests yet unidentified differential roles between the E2E enzymes that may be related to differential substrate interaction, location, or interacting proteins (E3 ligases, co-factors, etc.). For example, E2E1 and E2E2 can promote ISG15 modification on proteins while E2E3 cannot [
21]. Similarly, PTEN is targeted for degradation by E2E1 but not by E2E2 and E2E3 [
22], which means that each isoform possesses both common and distinct capacities/roles.
Two reports indicated that the N-terminal extension of E2E1 restricts ubiquitin transfer when Ub is covalently bound to the extension but this mechanism depends on the presence of some E3 ligases [
23,
24]. In these cases, monoubiquitylation predominates when using the full-length E2E1. However, other studies showed a fair activity of E2E1 for building polyUb chains using other E3 ligases and substrates [
23,
25,
26,
27,
28]. In addition, E2E1 preferentially catalyzes K48 (or K11) chains on chimeric proteins, i.e., the typical chains for targeting substrates for the subsequent degradation by the 26S proteasome [
29]. The discrepancies between the different studies may be linked either to the E3 ligases and/or to the substrates used in the assays.
E2E1 is so far described as mostly nuclear in different organs (human proteome Atlas (
https://www.proteinatlas.org)) and cells [
30]. The nuclear localization is driven by Importin-11 and is strictly dependent on the presence of Ub in the active site of E2E1 [
30]. This preferential location is in line with the mono-Ub of histone H2A by E2E1 in cooperation with the E3 ligase complex PRC1 [
31]. However, the location of E2E1 has never been addressed in skeletal muscle cells.
In a previous work, we found that among the E2 enzymes able to interact with MuRF1, E2E1 was peculiar in that only the presence of the substrate (telethonin) allowed for the clear interaction between MuRF1 and E2E1, which suggested a peculiar role during the atrophy process [
15]. However, the multiple locations of telethonin and MuRF1 within skeletal muscle cells did not allow us to determine the subcellular location of this interaction. In view of the paucity of data regarding E2E1, we first addressed the expression and location of E2E1 in the skeletal muscles of mice and then we determined its potential role in the muscle during catabolic conditions by using knockdown or overexpression approaches in both cultured cells and mice.
2. Materials and Methods
2.1. Constructs and Materials
Rat MuRF1 and murine UBE2E1 (E2E1) cDNAs were amplified by RT–PCR from either rat soleus muscles or murine C2C12 myotubes mRNA using Superscript II and Platinum Pfx DNA polymerase (Invitrogen, Carlsbad, CA, USA). cDNAs were cloned in the pcDNA3.1 expression vector (Invitrogen) for expression in mammalian cells. MuRF1 and E2E1 shRNA were produced using pLKO.1 clones purchased from Sigma (
Supplementary Table S1). Oligos used for miR production (
Supplementary Table S2) directed against E2E1 and the pcDNA™6.2-GW/EmGFP-miR vector were obtained from ThermoFisher Scientific. Combinations of at least 2 RNAi vectors were used for knocking down MuRF1 and E2E1.
Immunoblotting and immunohistochemistry (IHC) were performed using anti-flag (F1804), α-actin (HHF-35), PCNA (P8825), MHCI (M8421), and anti-laminin-α1 (L9393) from Sigma, St. Louis, MO, USA, E2E1 (TA309924), from Origene, Rockville, MD, USA, MHCI (clone BAF8), MHCIIa (clone N2), and MHCIIb (clone BFF3) from DSHB (University of Iowa, Iowa City, IA, USA) and anti-caspase 3 (9662) and cleaved caspase 3 (Asp175, 9661) from Cell Signaling Technology, Danvers, MA, USA.
2.2. Cell Culture and Knockdown Experiments
HEK293T cells and C2C12 myoblasts were cultured in Dulbecco’s Modified Eagle Medium and 10% (
v/
v) fetal bovine serum (FBS, Gibco, Invitrogen, Paislay, UK) and supplemented with L-glutamine, non-essential amino acids, and gentamycin (Gibco, growth medium, GM). HEK293T cells were plated in 6-well dishes and transfected by the calcium phosphate co-precipitation method [
32]. Cells were transfected or co-transfected with plasmid(s) encoding for green fluorescent protein (GFP, i.e., Mock), MuRF1 or E2E1, and were harvested after 48 h of transfection. Cells were lyzed and soluble proteins were obtained as previously described [
32]. Two independent experiments were performed.
For C2C12 myoblast differentiation into myotubes, cells were plated onto 24-well plates and grown to near confluence, and then shifted to DMEM containing 2% horse serum (differentiation medium, DM) for 5 days. A catabolic state was induced in myotubes by adding dexamethasone (Dex, 48 h) before harvesting the cells.
Lysate from MEF cells invalidated for the PERK kinase and treated for 24 h with 20 µM Resveratrol was used as a positive control of apoptosis (a gift from Dr. J. Averous). A total of 20 µg of soluble proteins (25 mM HEPES, pH 7.4, 400 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, NP40 1%, 1X Sigma inhibitor cocktail) were used for immunoblotting against pro-caspase 3 and cleaved caspase 3.
Knockdown experiments were carried out on HEK293T and C2C12 myotubes grown in multiwell cell culture plates and treated or not treated with Dex (C2C12 only). Myotubes were transfected or not at day 5 of differentiation with shRNAs targeting either MuRF1, UBE2E1 or no known protein sequence (scr-shRNA, negative control) using either a standards calcium phosphate approach for HEK293T cells as already described [
32] or a NEPA21 electroporator (SONIDEL) for C2C12 myotubes. Briefly, electroporation was performed as follows. pLKO.1 constructs were diluted at 0.46 µg/µL in OptiMEM (ThermoFisher Scientific, Waltham, MA, USA). At least 2 different shRNAs were mixed for optimal knockdown (>60%), which was tested by qRT-PCR. Another control group was systematically performed where only 1X PBS was present (mock transfection).
At day 5 of differentiation, C2C12 myotubes were rinsed twice with 1X PBS and then 300 µL of OptiMEM was added in the wells (24-wells plates). For each well, the myotubes were electroporated following the instructions of the manufacturer with the following parameters: 2 poring pulses, 225 V (5 ms, 10% decay, 50 msec interval); 5 transfer pulses, 30 V (50 ms, 40% decay, 50 ms interval). The DNA solution was removed and 500 µL of fresh growth medium deprived of antibiotic and serum was added in the well. C2C12 myotubes were incubated for 1.5 h at 37 °C under 5% CO
2 and the medium was replaced by the differentiation medium containing or not dexamethasone (50 µM). Briefly, myotubes were washed in 1X phosphate-buffered saline and 300 µL of lysis buffer (5 mM Tris, pH 7.5, 5 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 10 mM NEM, 1% Triton X-100/anti-proteases (Protease Inhibitor Mixture/Sigma)) was added. Cells were then scraped off the plate and sonicated for 30 s at maximum power using a UP50H sonicator (Hielscher, Teltow, Germany) as previously described [
33]. Cell lysates were then centrifuged at 10,000
g for 10 min at 4 °C and the supernatant (soluble fraction) was kept at −80 °C until use. The pellets enriched in myofibrillar proteins were resuspended in a homogenization buffer and sonicated to solubilize the proteins.
2.3. Animals and Knockdown Experiments
The experiments were conducted in accordance with the National Research Council Guide for the Care and Use of Laboratory Animals. National authorization to perform animal experiments for this project has been obtained (authorization #9204 for project 2017042115497506). All animals were maintained in a temperature-controlled room (22 ± 1 °C) with a 12:12 h light:dark cycle. Three-months old male C57BL/6 mice were housed for 1 week in a standard environment and then submitted or not to Dex treatment at 1 or 5 mg/kg/day for 5, 9, or 14 days. The length of the Dex-treatment was adapted to the dose for avoiding excessive weight loss of animals for complying with current legislation on animal care. Food intake was monitored in the first series of experiment and animals were pair-fed for avoiding discrepancies due to Dex treatment. At the end of the experiment, animals were killed by cervical dislocation and the skeletal muscles (EDL, gastrocnemius, soleus, and Tibialis anterior) were excised, weighed, frozen in liquid nitrogen and stored at −80 °C until use. Alternatively, skeletal muscles used for immunohistochemistry were frozen in liquid nitrogen-cooled isopentane. In vivo knockdown was performed as previously described by electroporation [
34]. Briefly, a target finder and design tool (Invitrogen) was used to identify target regions in the mouse MuRF1 and E2E1 genes amenable to siRNA. The sequences of siRNAs targeting MuRF1 and E2E1 were cloned into the BLOCK-iT Pol II miR RNAi Expression Vector Kit with EmGFP (Invitrogen number K4936-00). The oligos used are shown in
Supplementary Table S2. As a negative control, the pcDNA6.2-GW/EmGFP-miR-neg control was used according to manufacturer’s instructions.
2.4. Immunohistochemistry
The contractile type and the cross-sectional area of the T. anterior fibers were determined using serial cross-sections labeled with antibodies directed against the different myosin isoforms and co-labeled with anti-laminin-α1. For E2E1 immunohistochemistry, muscle cross-sections were either pre-incubated in 4% paraformaldehyde to preserve nuclear labeling or directly incubated with a rabbit polyclonal antibody directed against UBE2E1 (UBCH6; Origene, TA309924) to preferentially label muscle fibers. Negative controls were performed using either the secondary FITC-labeled antibody alone or no antibody, the sections being observed at an identical exposure time, i.e., 286 ms (
Supplementary Figure S1). Images were captured with a high-resolution cooled digital DP-72 camera coupled to a BX-51 microscope (Olympus, Tokyo, Japan) at a resolution of 0.64 μm/pixel. The CSA and contractile type of each fiber were obtained using the Visilog-6.9 software (Noesis) as previously described [
35].
2.5. Protein Extraction
Portions of the Tibialis anterior muscle (≈10 mg) were homogenized using a TissueRuptor
® (Kinematica, Littau-Luzern, Switzerland) in 200 µL of lysis buffer (PBS 1X pH 7.4, 5 mM EDTA pH 8.0, 1 mM PMSF, protease inhibitor cocktail (10 µL/mL of lysis buffer) (Sigma, St Louis, MO, USA), NEM 10 mM, Triton X100 1%). The soluble and the myofibrillar-enriched were separated as previously described [
16]. Muscle homogenates were centrifuged at 16,000
g (4 °C, 10 min) and the supernatants containing the cytoplasmic fraction were aliquoted and frozen at −80 °C until use. The pellets enriched in myofibrillar proteins were resuspended in 150 µL of lysis buffer containing 2% SDS and 5% glycerol using a sonicator (see above) and frozen at −80 °C until use.
The protein concentration was measured by absorption spectrophotometry (OD 562 nm) using the BCA kit (Pierce, Rockford, IL, USA) with BSA as a standard.
2.6. qRT-PCR
mRNA levels of muscle-specific E3 ligases (MuRF1, MAFbx) were determined by quantitative RT-PCR (see
Supplementary Table S3 for oligos used). The reverse transcription of total RNA into DNA was performed using the QuantiTect
® Reverse Transcription kit (Qiagen
®, Venlo, The Netherlands). qPCR was performed using the FastStart DNA Master SYBR Green I kit (Roche, Basel, Switzerland), according to the manufacturer’s instructions using a CFX96 thermocycler (BIORAD, Hercules, CA, USA). Calculations were made using the comparative ∆Ct method with YWHAZ, HPRT1, and 36B4 housekeeping genes. The oligos used are shown in
Supplementary Table S3.
2.7. Statistical Analysis
Statistical analyses were performed using the XLSTAT software (Addinsoft®, Paris, France). The tests were two-sided, with a Type I error set at α = 0.05. Variables are presented as means ± SE and a Student’s t-test or a 1-Way ANOVA (analysis of variance) were performed for cultured cell experiments. For in vivo transfection, the assumption of normality was assessed by the Shapiro–Wilk test and homoscedasticity verified by the Fisher-Snedecor test. Comparisons between groups were performed first using a 1-way ANOVA. When statistical significance was reached, it was followed by multiple post-hoc Tukey’s tests. The transfection of the scr-siRNA and the sh-E2E1 were performed in the left and right leg of the same animal respectively. Data analysis was therefore completed by a multivariate analysis in order to specifically distinguish the “UBE2E1 knockdown” effect from the “transfection” and the “leg” effects. We thus performed a 3-way ANOVA including the 3 factors of variability. The Least Square (LS) means of the surface in control and UBE2E1-knocked down legs were computed using this model and comparisons were made using multiple post-hoc Tukey’s tests. The conclusions of the 1-Way and the 3-Way ANOVA were similar.
3. Results and Discussion
We recently identified a MuRF1-E2 network that included E2E1 as a MuRF1 partner [
15]. Among the 5 E2 enzymes able to interact with MuRF1, E2E1 was peculiar as it interacted with MuRF1 only when telethonin was present, which was attributed to an allosteric mechanism [
15]. MuRF1 is a hallmark of the atrophying process in skeletal muscles and this E3 enzyme recognizes several sarcomeric proteins for their subsequent degradation by the 26S proteasome [
12,
13,
14,
15,
16]. Little is known about E2E1 and besides the expression levels, no data are available for skeletal muscles. For example, E2E1 is described as a nuclear enzyme in fibroblasts [
30] but its location in muscle cells in unknown. As MuRF1 and telethonin are located both in the cytosol and the nucleus [
36,
37,
38], our previous work did not give any clue about a putative role of E2E1 for targeting contractile proteins in the cytoplasm. Thus, in this work, the main objectives were (1) to address the E2E1 location in skeletal muscle cells; (2) to elucidate whether the MuRF1-E2E1 couple is able to target the main contractile proteins (α-actin and myosin heavy chain) for their subsequent degradation; and (3) to identify the putative role of E2E1 during skeletal muscle atrophy in vivo.
3.1. E2E1 Is Present in Both the Nuclei and the Cytoplasm of Mouse and Human Muscle Cells
The human proteome Atlas (
https://www.proteinatlas.org) is the widest source of information for the known location of E2E1 together with a single study that found an exclusive nuclear location of E2E1 in murine fibroblasts and in mouse embryo [
30]. According to the human proteome Atlas database, E2E1 is mostly nuclear in several organs (e.g., testis) but some cytoplasmic location was also confirmed in colon endothelial cells and in the kidney.
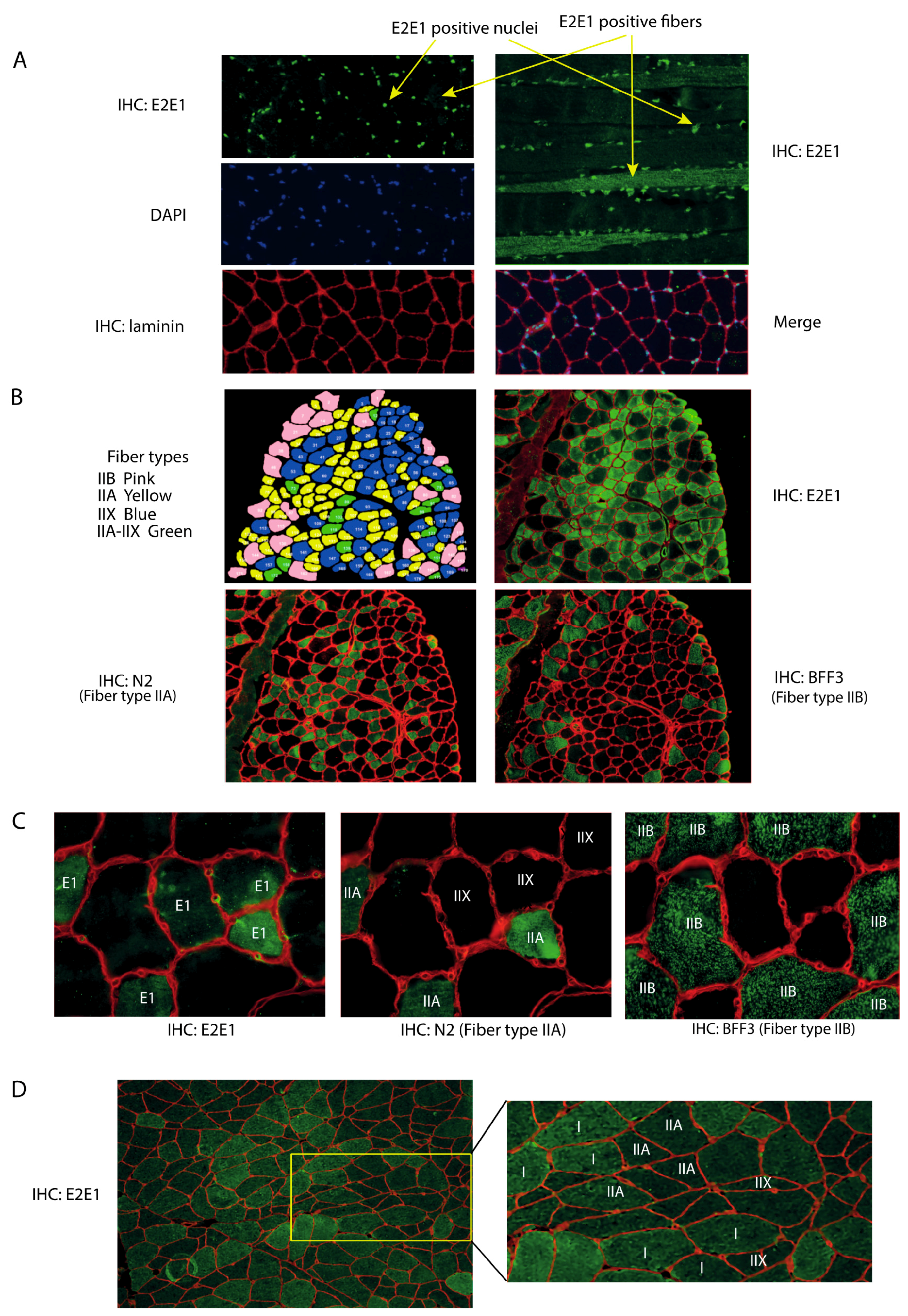
We attempted to locate E2E1 within skeletal muscles for appreciating its potential involvement in the degradation of the contractile apparatus. Indeed, an E2 enzyme exclusively located in the nucleus could not be directly involved in the degradation of contractile proteins in collaboration with MuRF1. Using an immunohistochemistry approach, we observed that E2E1 decorated the nuclei of the mice Tibialis anterior (T. anterior) muscle (
Figure 1A), like in other cell types [
30]. Interestingly, merging DAPI and E2E1 immunostaining indicated that most if not all the nuclei were positive, indicating that E2E1 was present in the nuclei of any type of fiber (
Figure 1A, left panel). Previous work has shown that E2E1 is localized in the nuclei of murine fibroblasts and HeLa cells through an active transport by importin-11 [
30]. Interestingly, the nuclear transport of E2E1 is tightly linked to the presence of Ub in the catalytic site of the E2 enzyme and the authors hypothesized that this may be a mechanism for either directly providing the delivery of the activated E2E1 to its nuclear targets or for protecting cytoplasmic targets of E2E1. Recent work confirmed that both hypotheses might be true as the tumor suppressor PTEN was degraded through E2E1 targeting (with the E3 ligase Nedd4) when trapped in the cytoplasm with E2E1, whereas the nuclear import of Ub-E2E1 protected PTEN from degradation [
22]. Interestingly, this mechanism was specific of E2E1 as the isoforms E2E2 and E2E3 were unable to ubiquitinylate PTEN. Another recent study found that nuclear E2E1 was part of the PRC1 ligase complex responsible for histone H2A mono-ubiquitylation, which, in turn, repressed the transcription of specific genes [
31]. The role of E2E1 in the nuclei of muscle cells is of obvious interest and should be addressed in future work but it was clearly beyond the goals of this study.
Interestingly, we found a faint although detectable cytoplasmic labeling of E2E1 in the T. anterior cross-sections (
Figure 1A, left upper panel). However, longitudinal sections clearly identified some fibers positive for E2E1 staining in the cytoplasm, including the myofibrillar region (
Figure 1A, right upper panel). By contrast, no signal was observed in the negative controls using either the secondary FITC-labeled antibody or no antibody (
Supplementary Figure S1). E2E1 was uniformly present in the positive fibers and hardly detectable in the negative ones but we cannot rule out that low levels of E2E1 may be present in these fibers. Indeed we did not use long exposure times (≥1 s) because this induces fiber auto-fluorescence due to the presence of myoglobin. As discussed above, the presence/absence of E2E1 in the cytoplasm might be controlled by differential nuclear import fluxes and/or by the modulation of E2E1 expression between fibers. The presence of E2E1 in the cytoplasm of some fibers may be an adaptive mechanism for targeting specific proteins for their degradation.
Within a skeletal muscle, cells do not harbor an identical proteome but rather exhibit different metabolic and contractile properties. This results in a patchwork of cells expressing different enzyme isoforms, more particularly myosin heavy chain (MHC) isoforms [
39]. We thus hypothesized that the presence/absence of E2E1 in the cytosol of muscle fibers might be related to the metabolic and contractile properties of the cells, i.e., fiber type, and we sought to verify this hypothesis. First, to better visualize the labeling of E2E1 in the cytoplasm, we eliminated the nuclei of muscle cross-sections (cf. “material and methods” section). Second, using antibodies specific of each MHC (see “material and methods” section), we identified each fiber type. Due to the absence of a specific antibody, note that type IIX fibers were identified by their lack of reactivity when using anti-MHCIIa and MHCIIb antibodies. In other words, they were negative to any of the MHC antibody we used. Using serial cross-sections, we found that cytoplasmic E2E1 was restricted to the intermediate type IIA fibers (and to a lesser extent to type IIX) and was not detectable in fast-twitch type IIB fibers (
Figure 1B,C). This reflected an expression directly correlated to the contractile and metabolic properties of muscle fibers. The T. anterior is devoid of type I fibers (slow-twitch fibers), we thus used human biopsies both for confirming the fiber specificity of E2E1 in another organism and for identifying the potential presence of E2E1 in type I fibers. We confirmed that E2E1 is expressed in human type IIA fibers (
Figure 1D) and is faintly present in type IIX. It should be noticed that type IIB fibers are absent in humans like in all the muscles from large mammals. Interestingly, we found that E2E1 was also strongly expressed in the cytoplasm of type I fibers, indicating that the E2E1 cytoplasmic location is restricted to slow and intermediate fibers. Although the expression levels were only qualitative, the combined mouse and human data suggest a gradual expression of E2E1 within each fiber type, with I > IIA >> IIX >>> IIB. The simplest hypothesis is that the E2E1 substrates are present in the cytoplasm of slow-twitch and intermediate fibers, which may confer to E2E1 a specific role in skeletal muscle cells expressing a peculiar set of proteins, e.g., myosin heavy chains (MHCI or MHCIIa). As MuRF1 targets different sarcomeric proteins including MHCs, we thus decided to explore whether known substrates of MuRF1 that are expressed either in any fiber type (α-actin [
16]) or in specific muscle cells (MHCs [
13]) could be potential targets of E2E1.
3.2. The MuRF1-E2E1 Couple Is Able to Target α-Actin But Not MHCIIa for Degradation in HEK293T Cells
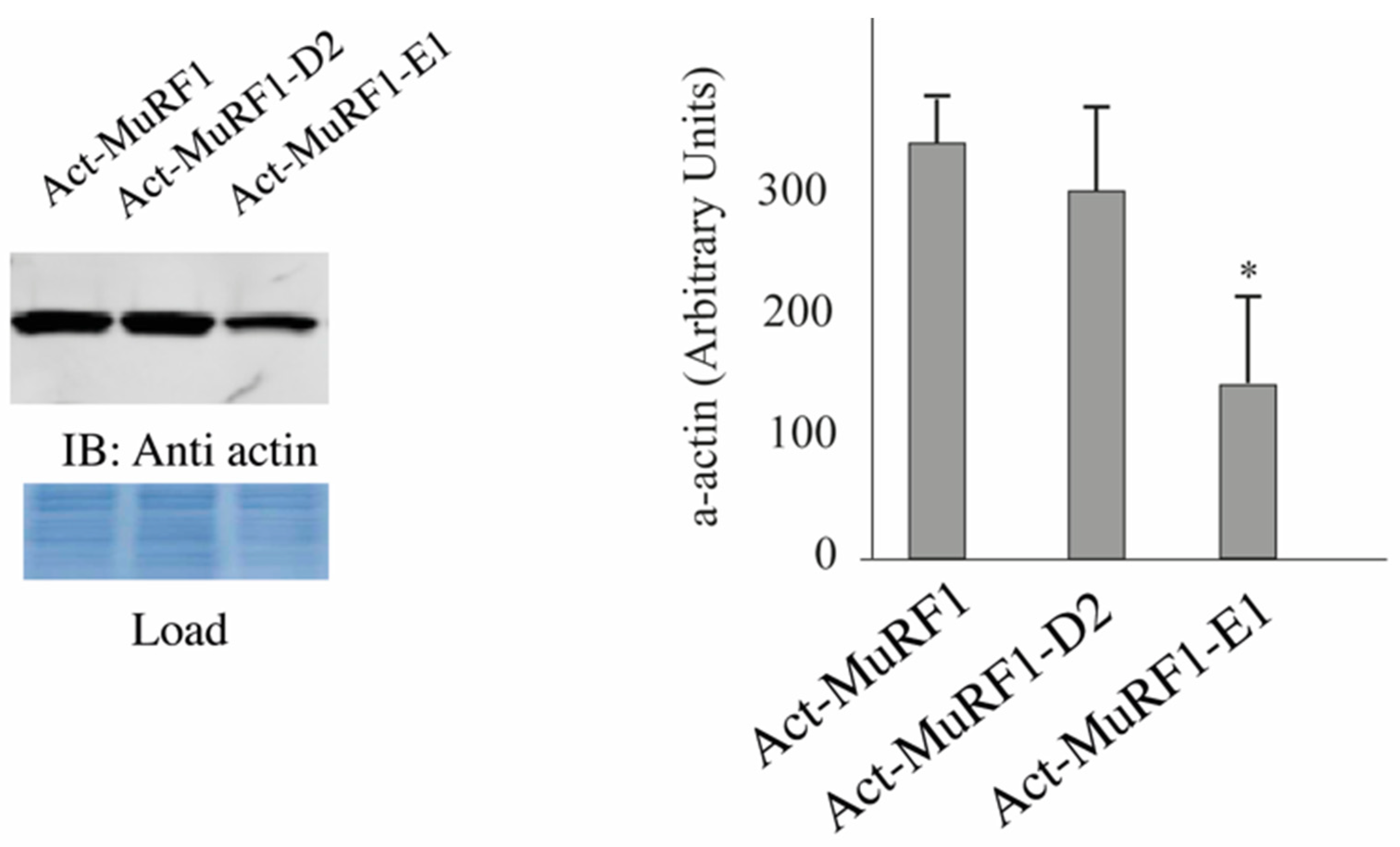
Due to the high abundance of α-actin and MHCs in muscle cells, detecting modifications of these proteins may be pretty tricky and we thus decided to start our investigations by using heterologous cells that lack α-actin [
32] and MHCIIa. We co-transfected HEK293T cells with plasmids encoding for α-actin or MHCIIa and MuRF1 with or without E2E1. As a negative control, we used E2D2 that is not able to catalyze α-actin degradation [
32]. We already used this model and demonstrated that α-actin is partially degraded in the presence of MuRF1 thanks to endogenous E2 enzymes present in HEK293T cells [
32]. We observed a 58% depression of α-actin levels when E2E1 was co-expressed with MuRF1, while no effect was detected in the presence of E2D2 (
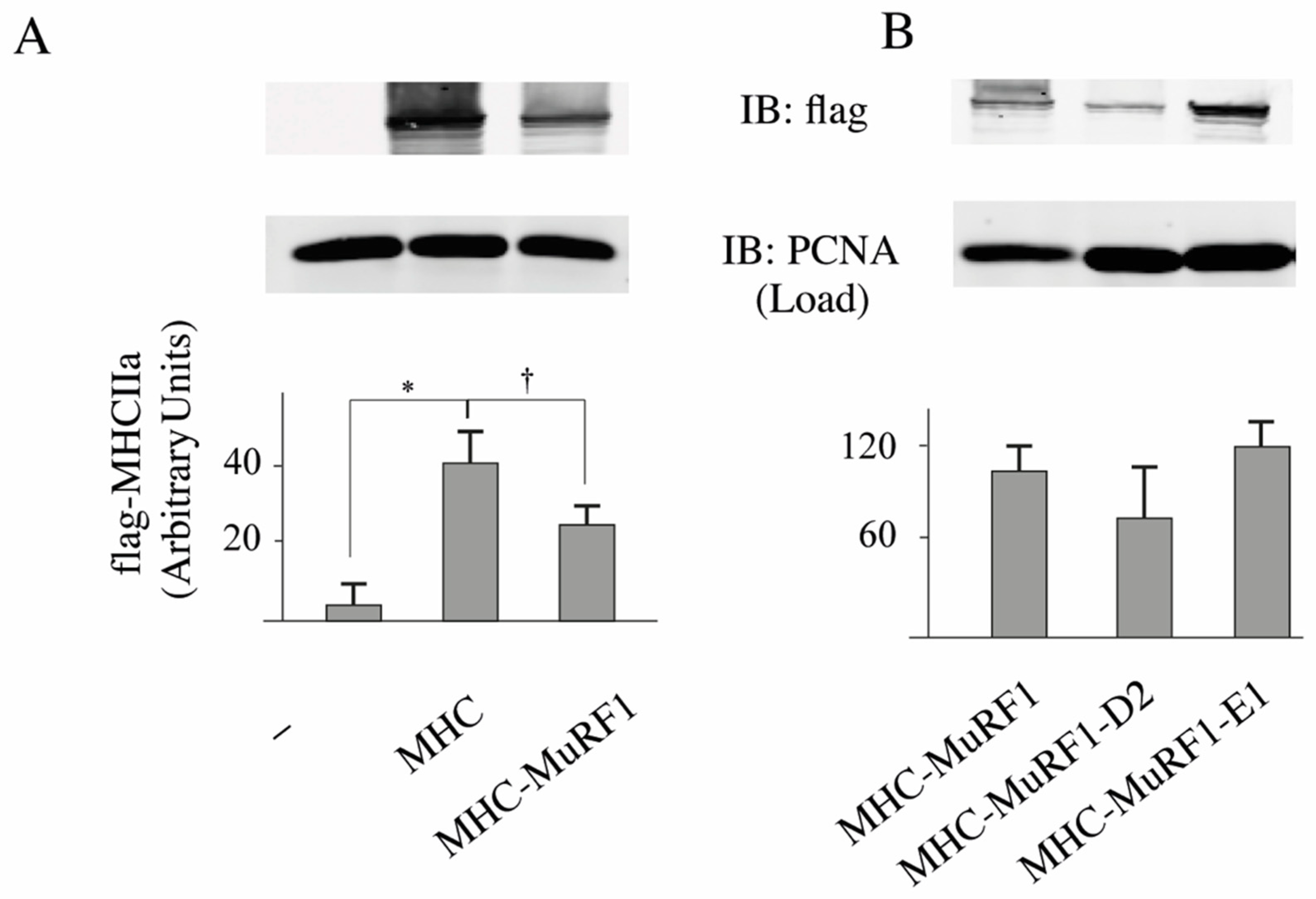
Figure 2). We used a similar approach for MHCIIa and first verified that MuRF1 alone was able to target MHCIIa for degradation (−36%,
p < 0.01;
Figure 3A). However, we were not able to detect MHCIIa degradation when E2E1 was over expressed with MuRF1 in HEK293T cells (
Figure 3B). Altogether, these data were surprising as E2E1 was able to target a protein present in any type of muscle fiber (α-actin) but not a protein specific of type IIA fibers (MHCIIa), which did not match with the fiber type specificity we observed for E2E1. However, the lack of effect of E2E1 on MHCIIa may be explained by 2 hypotheses: (1) E2E1 is not the E2 enzyme implicated in MHCIIa degradation; (2) E2E1 is lacking a co-factor not present in heterologous HEK293T cells. As a corollary of the latter hypothesis, the co-factor may be present only in atrophying muscle cells. Thus, we moved to a model of cultured skeletal muscle cells (C2C12 myotubes) that can be submitted to catabolic stimuli.
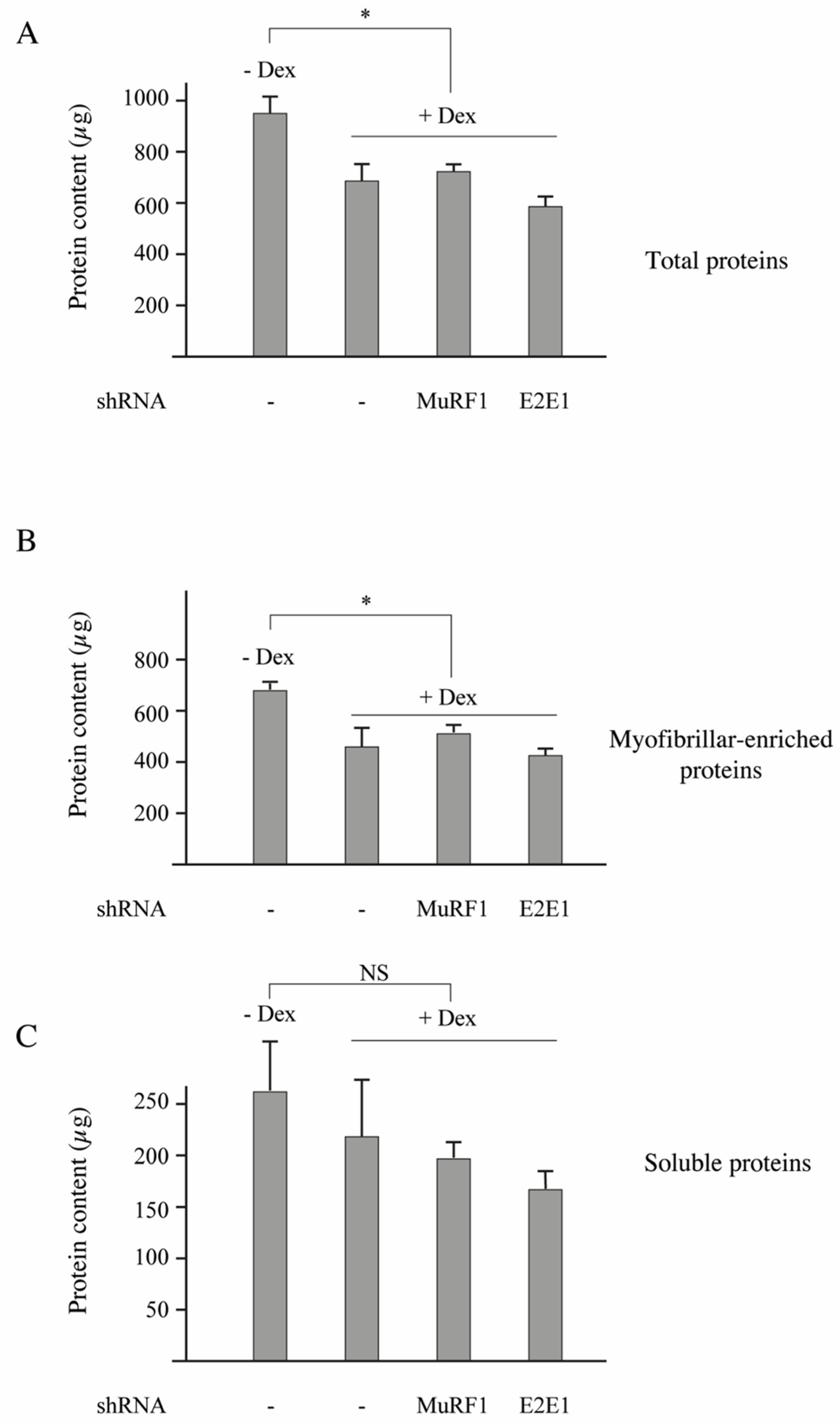
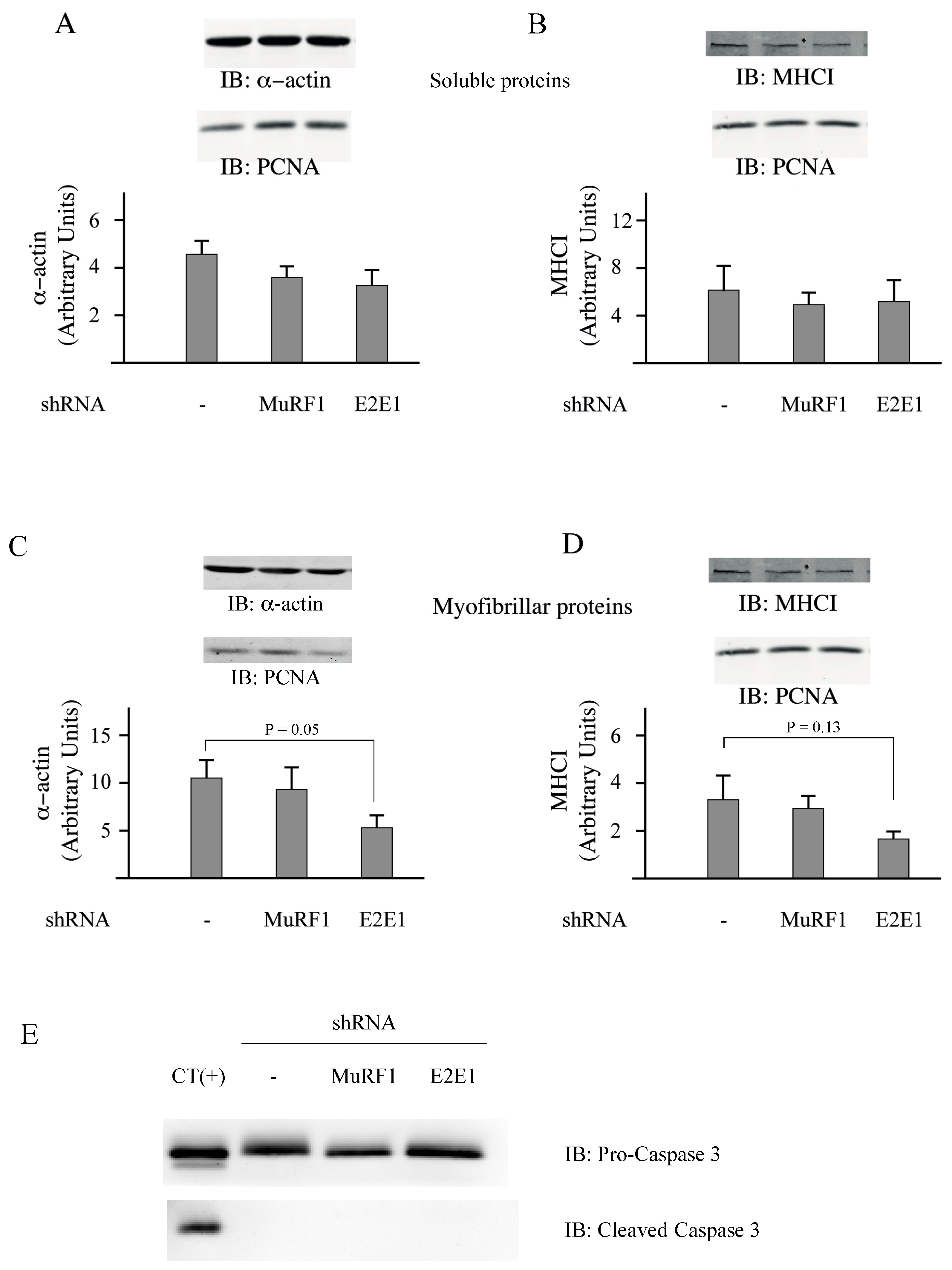
3.3. E2E1 Repression Decreased α-Actin Levels and Tended to Depress MHCI Levels in Catabolic C2C12 Myotubes
C2C12 myotubes were treated or not treated with dexamethasone (Dex, 1 µM) for inducing a catabolic situation. Dex-treatment induced a significant decrease of the total protein content (−33%,
p < 0,05;
Figure 4A) and transfection with shRNAs directed against either MuRF1 or E2E1 did not further modify protein levels (
Figure 4A). The total protein decrease was due to a depression of the myofibrillar-enriched fraction with no significant modification of the soluble fraction (
Figure 4B,C).
Previous work showed that Dex decreased the C2C12 myotube’s diameter (a hallmark of atrophy) by increasing protein degradation, notably through increased levels of MuRF1 and MAFbx [
40]. The lack of effect of the shRNAs directed against MuRF1 and E2E1 was not surprising for 2 reasons: (1) contractile proteins represent “only” around 10% of the total proteins in C2C12 myotubes (unpublished data); and (2) in our conditions, we just analyzed a fraction enriched in myofibrillar proteins (total minus soluble proteins). We then addressed the impact of E2E1 knockdown on individual sarcomeric proteins in Dex-treated C2C12 myotubes and MuRF1 knockdown was used as a control. α-actin or MHCI levels were not significantly impacted by the MuRF1 knockdown even though these proteins are MuRF1 targets (
Figure 5A–D). This is in accordance with previous studies indicating that the MuRF1 targeting of α-actin can be visualized only in peculiar conditions (e.g., use of chimeric α-actin) because of α-actin abundance [
16] and that MHCI targeting depends on both MuRF1 and MuRF3 due to at least a partial redundancy of these E3 ligases [
13]. We found that the E2E1 knockdown did not impact α-actin and MHCI levels in the soluble fraction (
Figure 5A,B). However, the E2E1 knockdown tended to depress α-actin (−47%,
p = 0.05) and MHCI content (−50%,
p = 0.13) in the myofibrillar-enriched fraction (
Figure 5C,D). Altogether, the MuRF1-E2E1 couple was able to target α-actin for the degradation in heterologous HEK293T cells but E2E1 was clearly not involved in the enhanced degradation of myofibrillar proteins in catabolic C2C12 myotubes. By contrast, the E2E1 knockdown was even deleterious as it depressed contractile protein levels.
The knockdown of E2E1 might have indirectly impacted C2C12 myotubes by promoting apoptosis. Indeed, during apoptotic situations (e.g., staurosporine treatment), actin (both the α and the β type) is cleaved by caspase 3 and the fragments are further degraded by the UPS [
41,
42]. We thus addressed a potential increase of caspase 3 levels and more particularly the cleaved caspase 3, which is the active form. We used PERK
−/− MEF cells treated with resveratrol as a positive control of caspase 3 induction [
43]. In these cells, a substantial amount of cleaved-caspase 3 (the active form) was detected (
Figure 5E, CT(+)), indicating an activation of apoptosis. By contrast, no activated caspase 3 was detected in C2C12 myotubes and the levels of total caspase 3 were similar in control, MuRF1 and E2E1 knocked down cells (
Figure 5E). This suggests that the modest reduction in myofibrillar protein levels observed upon E2E1 knockdown was not due to exacerbated apoptosis.
The protection of α-actin and potentially some MHC isoforms by E2E1 may be either direct or indirect and it remains to be determined whether the impact of E2E1 on contractile proteins is due to the manipulation of protein synthesis or degradation. Indeed, the capacity of E2E1 to modulate gene expression in HeLa cells through histone ubiquitylation makes possible an indirect effect of E2E1 on myofibrillar proteins [
31]. To our knowledge, no study has addressed the potential control of contractile proteins through histone ubiquitylation and, in this work, we could not attribute the protective effect of E2E1 on catabolic muscle cells to either the nuclear or the cytoplasmic pool of E2E1. This point should be addressed in future investigations but another difficulty resides in the ability of E2E1 to translocate from the cytoplasm to the nucleus, which complicates data interpretation. Indeed, E2E1 may regulate its targets mainly through re-localization rather than
de novo transcription or translation. The use of E2E1 mutants unable to translocate may help to identify the mechanisms implicated in contractile protein preservation.
Nevertheless, the data obtained in C2C12 myotubes were quite surprising as they suggested that the presence of E2E1 was needed for lowering the effects of Dex and thus protecting the contractile apparatus during a catabolic situation. This was in contradiction with the capacity of E2E1 to drive α-actin degradation in heterologous cells (see
Figure 2). However, the data obtained in HEK293T cells may not be representative of the E2E1 role in muscle cells or may only represent one of its multiple roles. For clarifying these discrepant results, we decided to address the impact of E2E1 in catabolic skeletal muscles from mice treated with Dex.
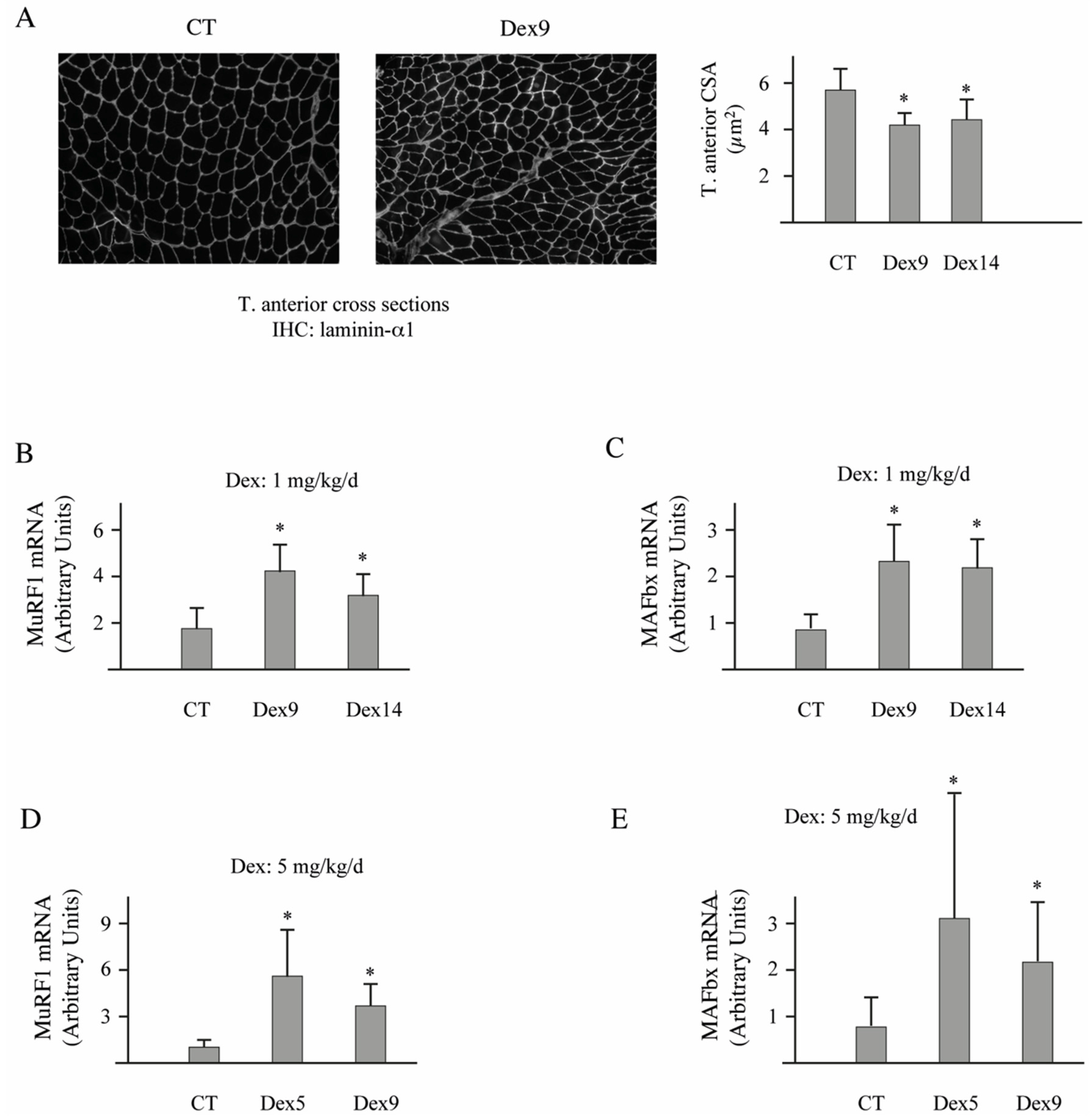
3.4. Dex-Treatment Induces Muscle Atrophy and Activates the UPS in Mice
Dex treatment induces muscle atrophy in humans and rodents [
16,
44,
45]. However, the concentration and the timing of Dex that induce both muscle atrophy and increased activation of proteolytic systems in mice greatly varies in the literature from 1 to 25 mg/kg/day with different injection modes (intraperitoneally, gavage, etc.) [
44,
46,
47]. In addition, few studies addressed both muscle atrophy and the activation of the UPS. Furthermore, corticoids like Dex may alter food intake in mice by increasing the appetite [
48,
49], which may modify proteostasis. Thus, we first set up the best conditions that induce skeletal muscle atrophy and activate the UPS, with a special focus on the E3 ligase involved in contractile protein targeting, i.e., MuRF1.
Mice food intake was individually monitored for 1 week before initiating the experiment. Thereafter, Dex-treated animals were pair-fed according to control levels. However, we did not notice any detectable variation of food intake (not shown), which indicated that nutrient availability could not interfere with the Dex-treatment. We tested two doses of Dex (1 and 5 mg/d/kg) dissolved in drinking water for 5, 9, or 14 days. Both doses of Dex were efficient whatever the time of experiment and induced a significant muscle atrophy that paralleled the total body weight loss of animals (
Table 1). However, atrophy was slightly higher for the four muscles tested (gastrocnemius, soleus, T. anterior, and EDL) for 5-days of treatment at 5 mg/kg/d. Accordingly, muscle weight loss was accompanied by decreased cross-sectional areas in the T. anterior muscle (
Figure 6A, −22/−24%,
p < 0.05).
The two E3 ligases MuRF1 and MAFbx are considered as the best markers of muscle atrophy even in moderate atrophying conditions [
10,
50]. We observed that both doses of Dex efficiently up-regulated MuRF1 and MAFbx in the T. anterior muscle for any of the time points tested (
Figure 6B–E). However, MuRF1 mRNA levels increased more with the dose of 5 mg/kg/d (5-days of treatment, +560%,
p < 0.01; 9-days of treatment, +360%,
p < 0.01) than with 1 mg/kg/d (9-days of treatment, +246%,
p < 0.01; 14-days of treatment, +85%,
p < 0.01). As Dex treatment at 5 mg/kg/d for 5 days induced higher levels of atrophy and MuRF1 up regulation, we used this condition for addressing the potential role of E2E1 in vivo in skeletal muscles.
3.5. E2E1 Knockdown Aggravates Muscle Atrophy in Dex-Treated Mice
Another series of mice were transfected with scramble siRNA (scr-siRNA, negative control) or with a mixture of siRNA directed against E2E1 (siE2E1, knockdown). The same animal was used for both transfections, with the left leg being the control (scr-siRNA) and the right leg being the knocked down (siE2E1). After 4 days, the mice were treated with Dex (5 mg/d/kg) for 5 days. The plasmids encoding for the siRNAs also expressed emGFP, which allowed identifying the transfected cells. The average efficiency of cell transfection was 55 and 56% in scr-siRNA and siE2E1 transfected muscles respectively and 100–160 fibers per leg were analyzed (total fibers, 1449). We took advantage of the presence of both transfected (GFP-positive) and non-transfected (GFP-negative) cells within the same muscle to compare the fiber size between the 2 populations. We found that the scr-siRNA did not affect the fiber cross-sectional area (
Figure 7A left panel and
Figure 7B). By contrast, the knockdown of E2E1 induced a global decrease in cell size (−32%,
p < 0.001,
Figure 7A right panel and
Figure 7B), which showed that the E2E1 knockdown negatively impacted the catabolic T. anterior muscle. The decrease in fiber size homogenously impacted the muscle as indicated by the global shift of fiber cross-sectional area towards lower sizes (
Figure 7C). As cytoplasmic E2E1 is present in type IIA fibers, the latter may be more sensitive to E2E1 knockdown. Thus, we further analyzed whether the E2E1 knockdown may have impacted differentially the fibers depending on their metabolic and contractile properties. No significant difference was detected, indicating that the decrease of fiber size affected all the fibers (not shown). In addition, E2E1 knockdown did not modify α-actin and MHCIIa levels (
Supplementary Figure S2), indicating that muscle atrophy was homogeneous.
Based on the effect of MuRF1-E2E1 on telethonin, we first hypothesized that these ubiquitinating enzymes could be responsible for the degradation of contractile proteins in catabolic skeletal muscles. However, like MuRF1, telethonin is present not only in the contractile apparatus where it interacts with the giant protein titin but also as a free protein either in the cytoplasm or in the nucleus [
38,
51]. Indeed, telethonin possesses pleiotropic functions including regulatory ones and the MuRF1-dependent degradation of telethonin may not be related to the sarcomeric function of telethonin. We show in this work that E2E1 is both cytosolic and nuclear and that the main in vivo function of E2E1 is not to promote muscle cell atrophy. In contrast, E2E1 is necessary for avoiding excessive muscle loss in Dex-treated mice. The dual location of E2E1 suggests different functions with probably different E3 ligases and future investigations will have to identify which pool of E2E1 is crucial for avoiding excessive muscle atrophy. The importance of E2E1 for maintaining skeletal muscle cell size is in accordance with the deleterious effect of E2E1 knockdown observed in cultured C2C12 myotubes, i.e., depressed levels of α-actin and a tendency to reduce MHC content (see
Figure 5).
An interesting question is whether the 2 other isoforms of the E2E sub-family are also located and/or expressed differentially among the different fiber types. It is nevertheless tempting to hypothesize at least partially divergent roles for the E2E isoforms on the basis of what is already known about E2 enzymes. Indeed, a single amino acid modification is able to change the catalytic activity of an E2 (see Reference [
17] for a review). This means that the highly divergent N-terminal extensions may drastically orientate the enzymes to very different fate. This is highlighted by the already known implication of E2E enzymes in diseases, with E2E1 involved in cancer and Sjogren’s syndrome, E2E2 in diabetes and E2E3 in Liddle’s syndrome [
52].

 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}