Synthesis and Biological Evaluation of Novel Triple-Modified Colchicine Derivatives as Potent Tubulin-Targeting Anticancer Agents

 ,
,
Abstract
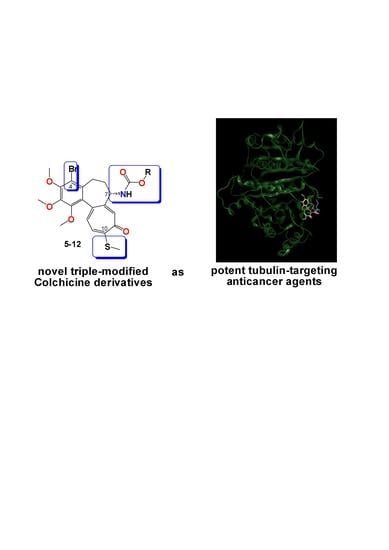
:
1. Introduction
2. Materials and Methods
2.1. General
2.2. Spectroscopic Measurements
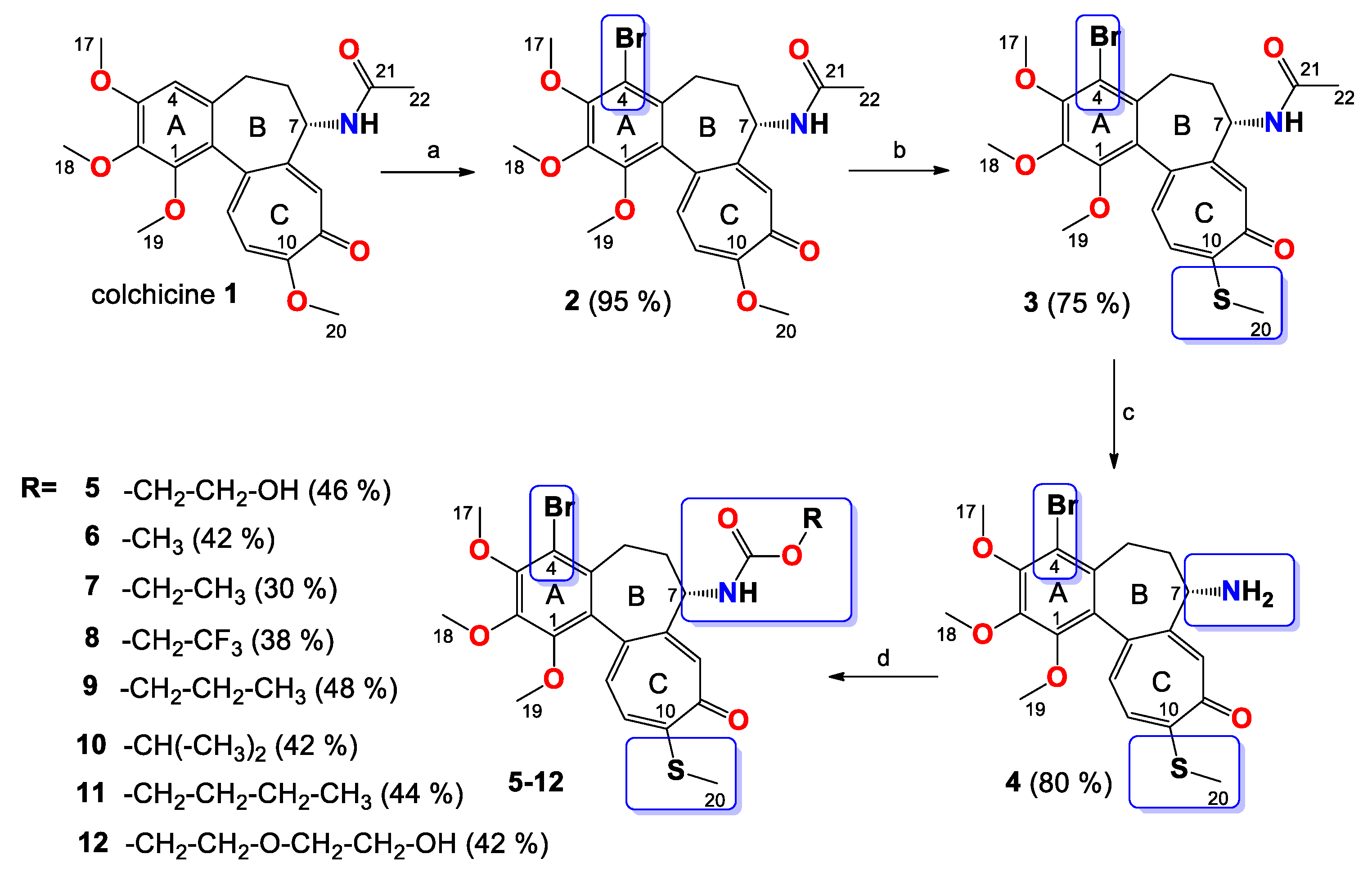
2.3. Synthesis
2.3.1. Synthesis of 2
2.3.2. Synthesis of 3
2.3.3. Synthesis of 4
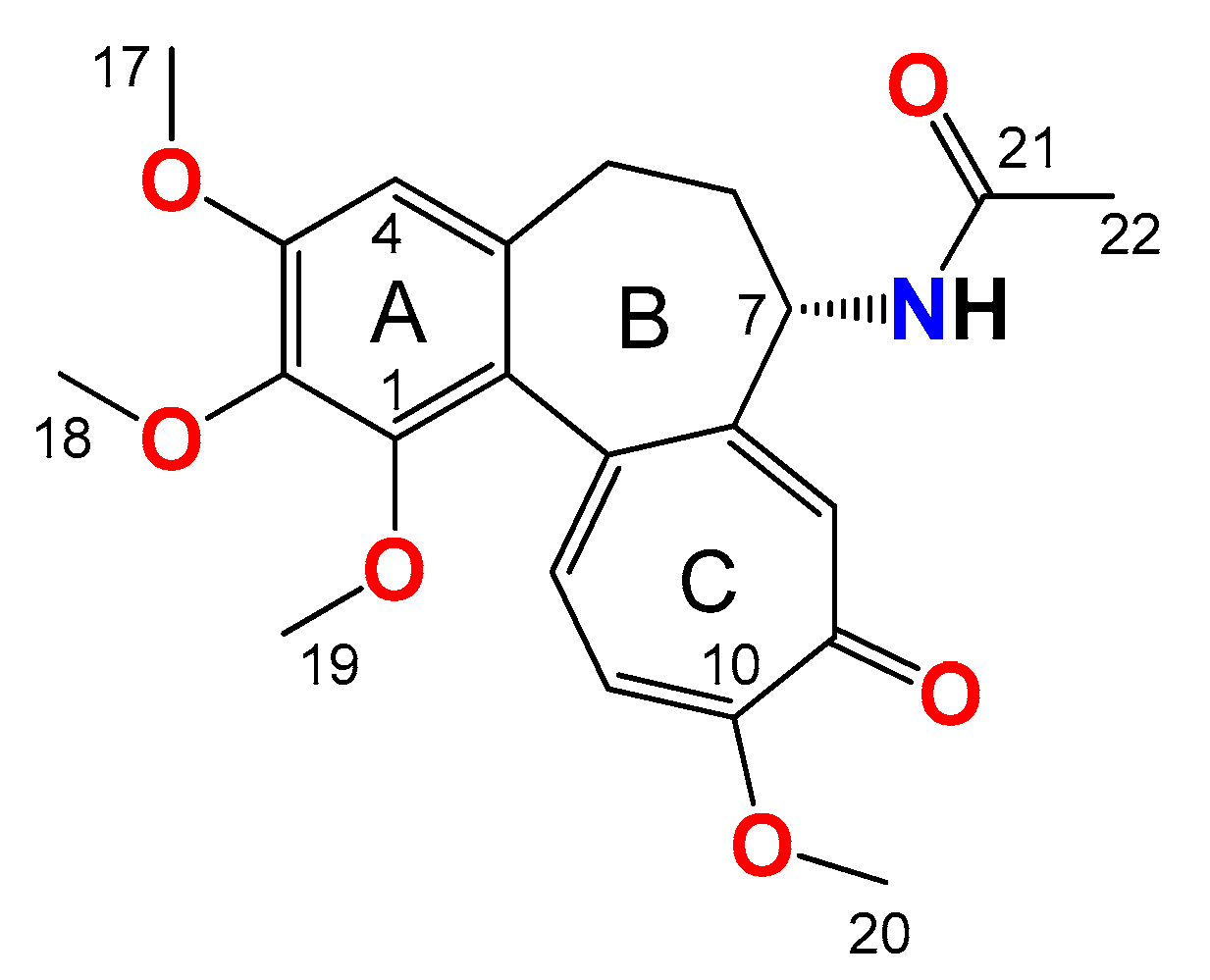
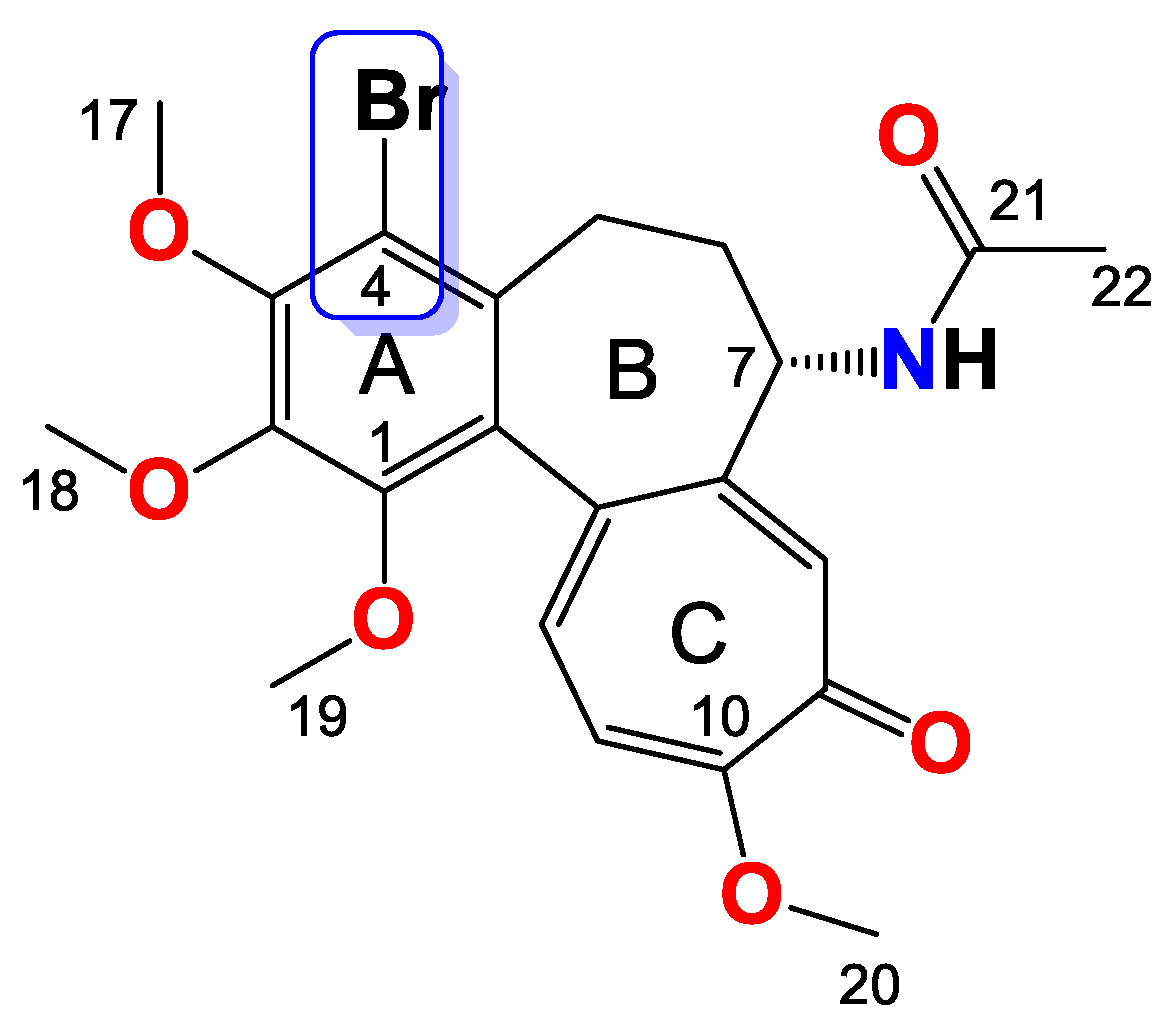
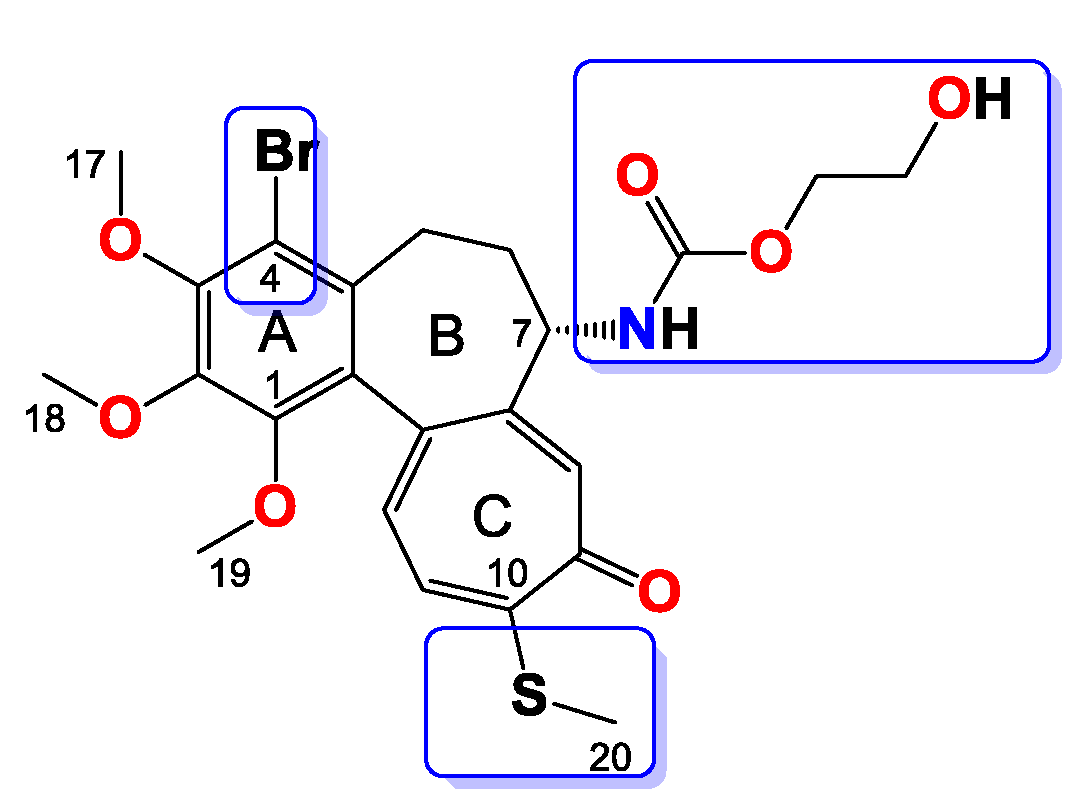
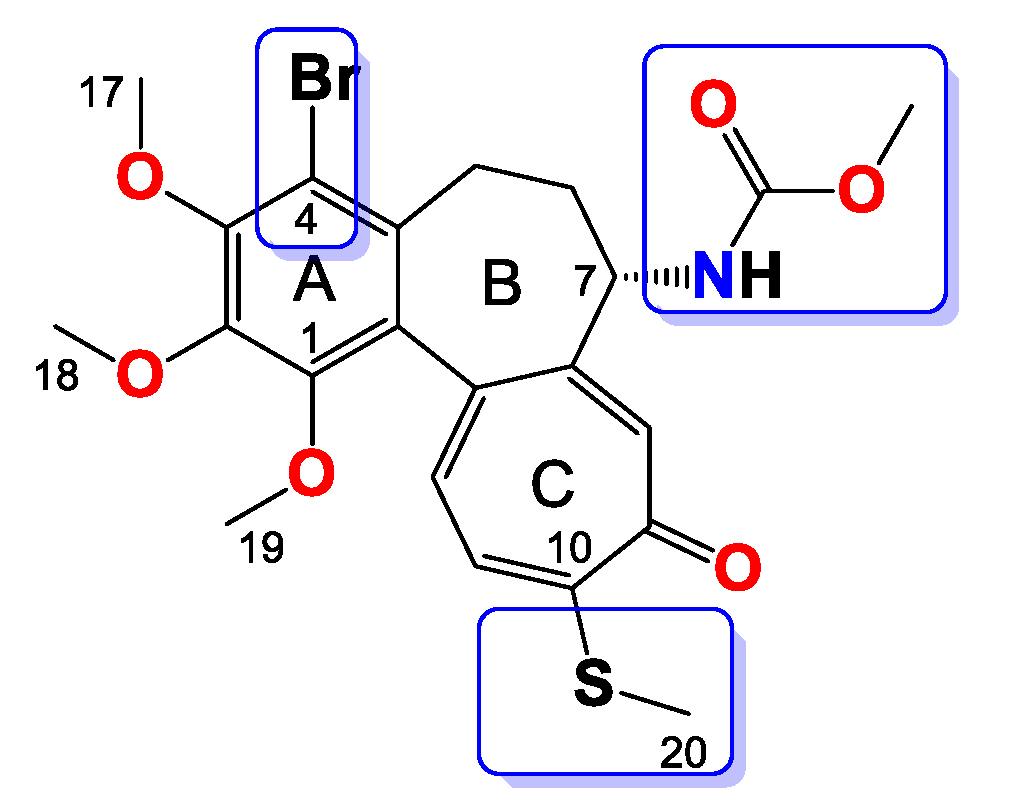
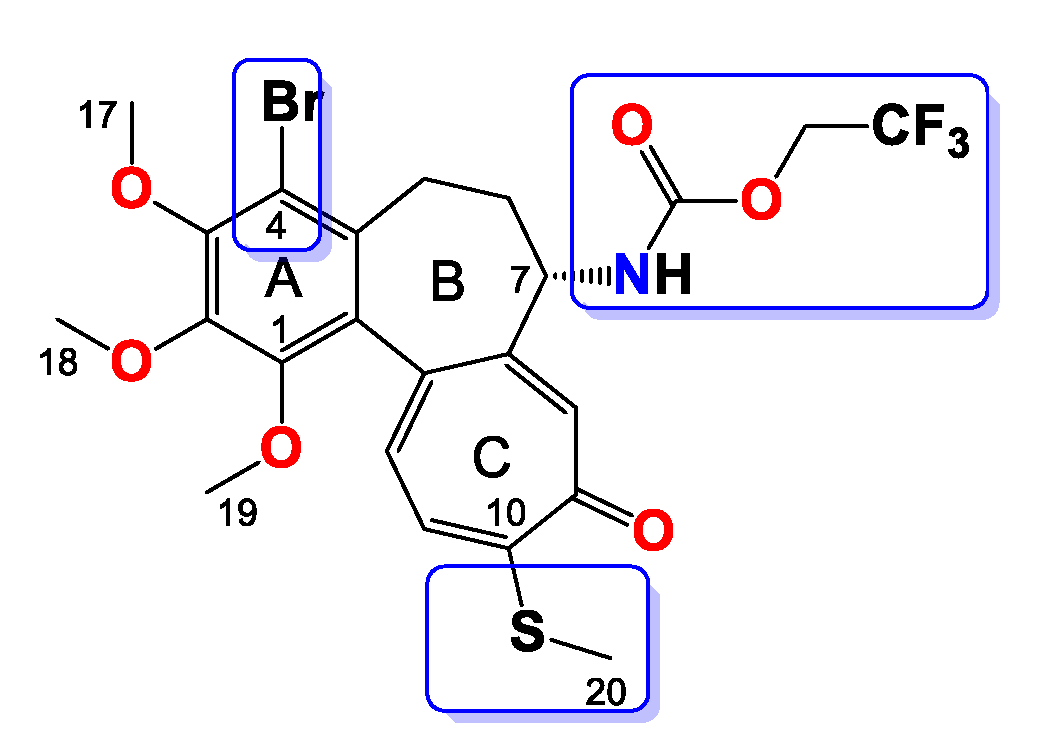
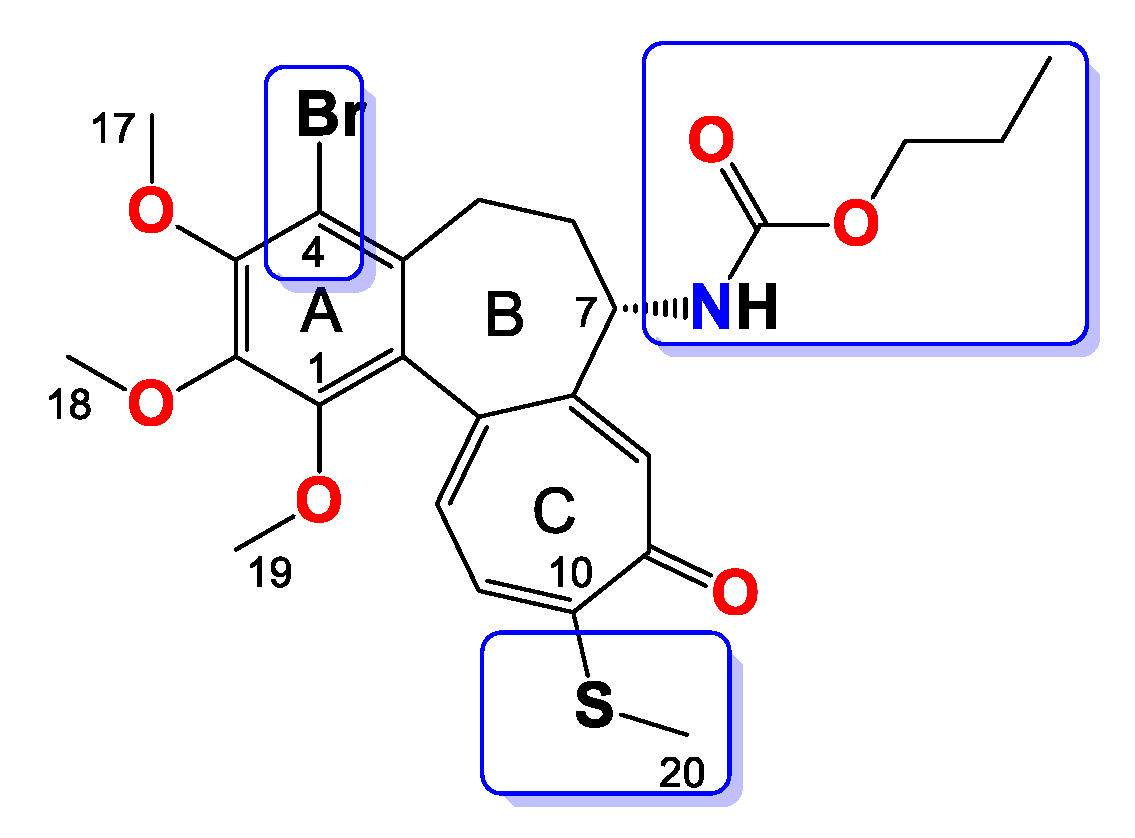
2.3.4. General Procedure for the Synthesis of Colchicine Derivatives (5–12)
Compound 5
Compound 6
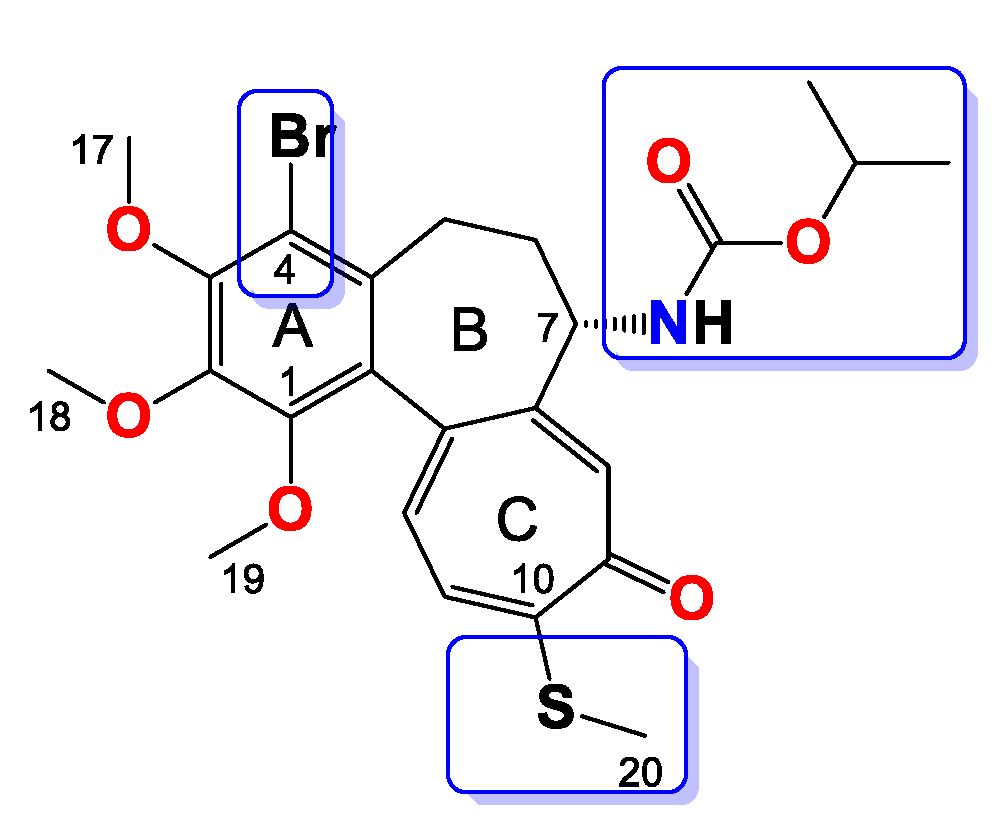
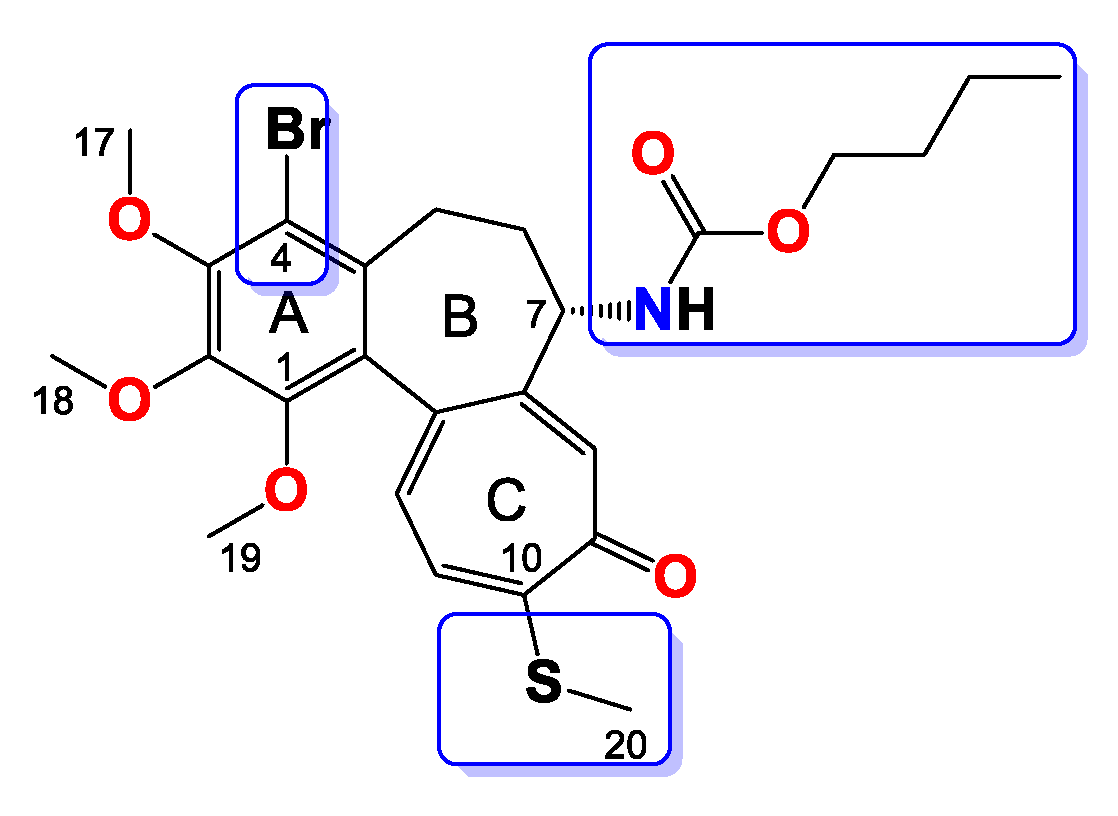
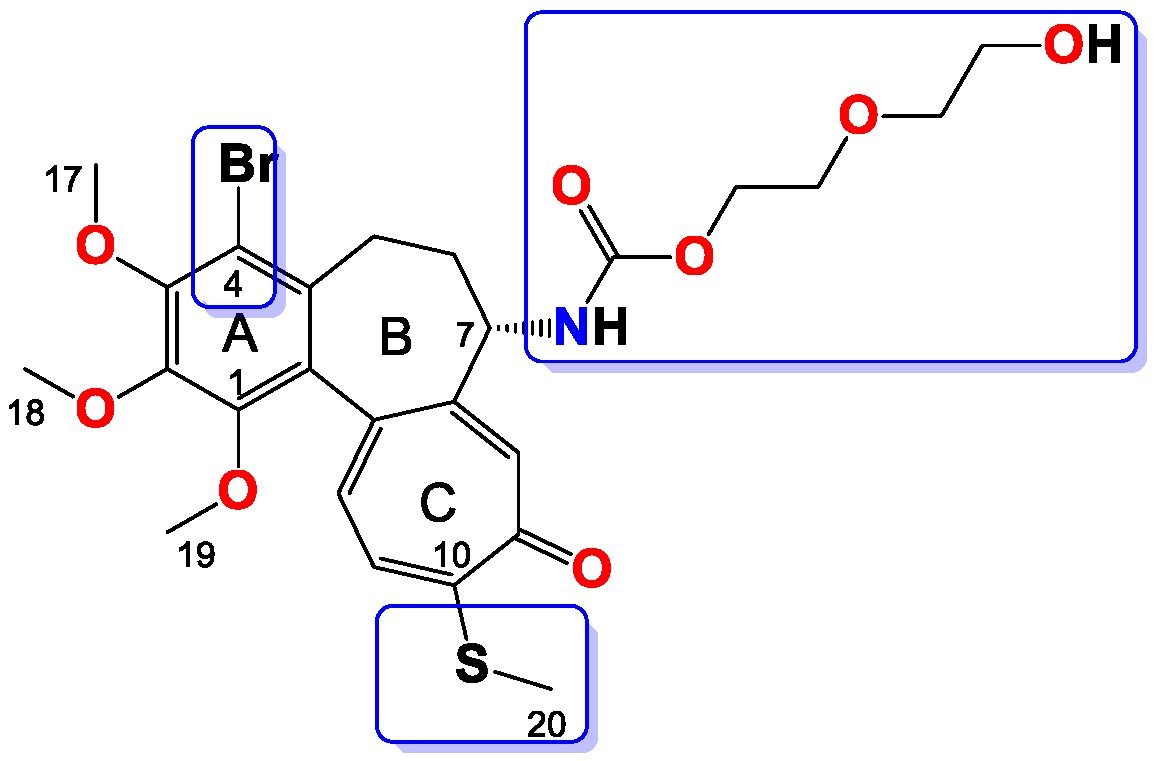
Compound 7
Compound 8
Compound 9
Compound 10
Compound 11
Compound 12
2.4. Antiproliferative Activity of Colchicine and Its Derivatives
2.4.1. The Antiproliferative Assays In Vitro
2.4.2. SRB
2.5. Molecular Docking Simulations
3. Results
3.1. Chemistry
3.2. In Vitro Determination of Drug-Induced Inhibition of Human Cancer Cell Line Growth
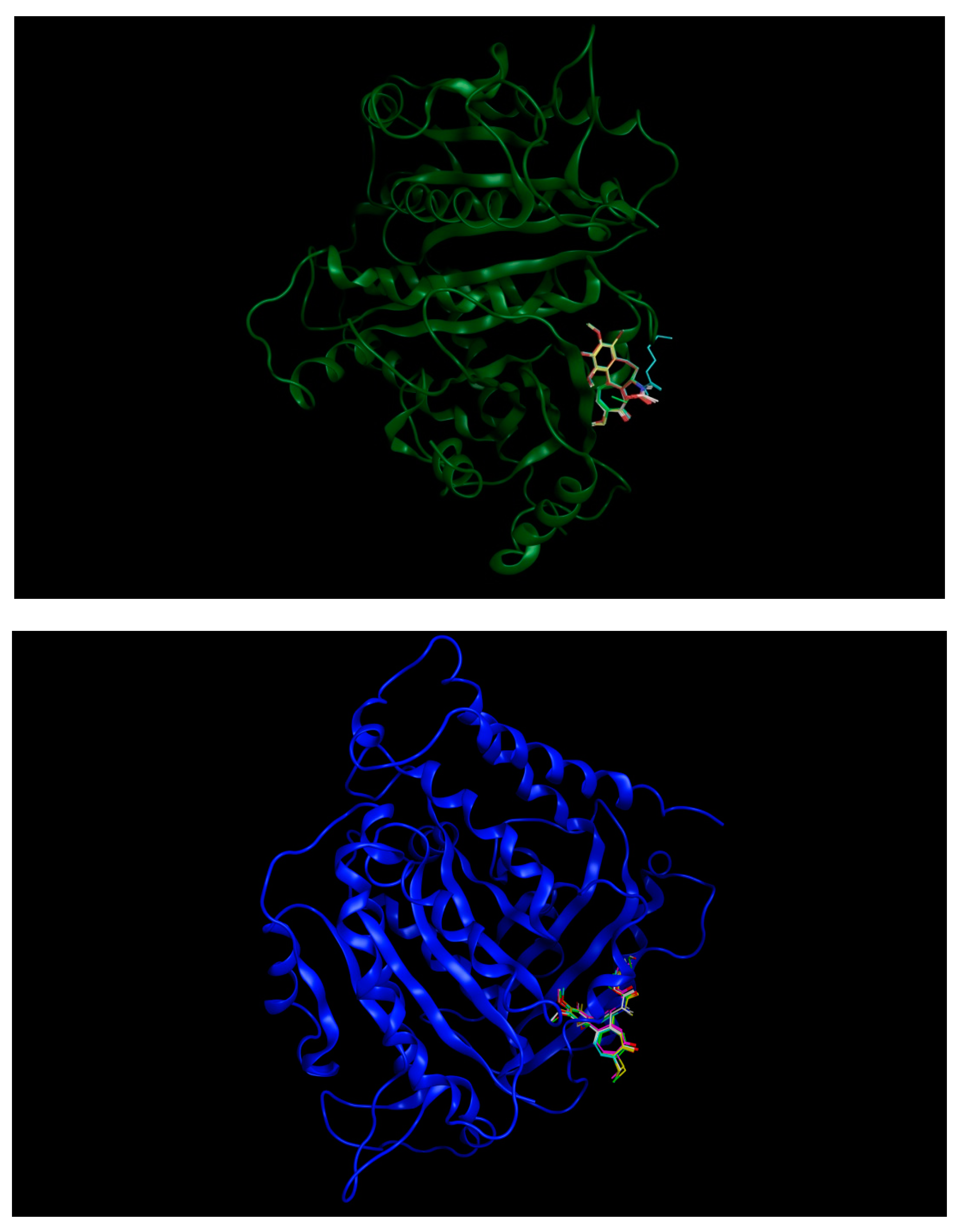
3.3. Molecular Docking: In Silico Determination of Drug-Induced Inhibition of βІ Tubulin
3.4. Linear Regression with Two Independent Variables
3.5. MM/PBSA and MM/GBSA: In Silico Determination of Drug-Induced Inhibition of β Tubulin Isotypes
3.6. Linear Regression with Two Independent Variables
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Slobodnick, A.; Shah, B.; Pillinger, M.H.; Krasnokutsky, S. Colchicine: Old and new. Am. J. Med. 2015, 128, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Shchegravina, E.S.; Knyazev, D.I.; Beletskaya, I.P.; Svirshchevskaya, E.V.; Schmalz, H.G.; Fedorov, A.Y. Synthesis of nonracemic pyrrolo-allocolchicinoids exhibiting potent cytotoxic activity. Eur. J. Org. Chem. 2016, 2016, 5620–5623. [Google Scholar] [CrossRef]
- Zemer, D.; Livneh, A.; Danon, Y.L.; Pras, M.; Sohar, E. Long-term colchicine treatment in children with familial Mediterranean fever. Arthritis Rheumatol. 1991, 34, 973–977. [Google Scholar] [CrossRef]
- Ben-Chetrit, E.; Levy, M. Colchicine prophylaxis in familial mediterranean fever: Reappraisal after 15 years. Semin. Arthritis Rheumatol. 1991, 20, 241–246. [Google Scholar] [CrossRef]
- Cerquaglia, C.; Diaco, M.; Nucera, G.; La Regina, M.; Montalto, M.; Manna, R. Pharmacological and clinical basis of treatment of Familial Mediterranean Fever (FMF) with colchicine or analogues: An update. Curr. Drug Targets Inflamm. Allergy 2005, 4, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Nakajima, A.; Urayama, A.; Nakae, K.; Kogure, M.; Inaba, G. Double-masked trial of cyclosporin versus colchicine and long-term open study of cyclosporin in Behçet’s disease. Lancet 1989, 1, 1093–1096. [Google Scholar] [CrossRef]
- Kaplan, M.M. New strategies needed for treatment of primary biliary cirrhosis? Gastroenterology 1993, 104, 651–653. [Google Scholar] [CrossRef]
- Gong, Y.; Gluud, C. Colchicine for primary biliary cirrhosis: A cochrane hepato-biliary group systematic review of randomized clinical trials. Am. J. Gastroenterol. J. Gastroenterol. 2005, 100, 1876–1885. [Google Scholar] [CrossRef] [PubMed]
- McKendry, R.J.; Kraag, G.; Seigel, S.; al-Awadhi, A. Therapeutic value of colchicine in the treatment of patients with psoriatic arthritis. Ann. Rheum. Dis. 1993, 52, 826–828. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Gertz, M.A.; Greipp, P.R.; Witzig, T.E.; Lust, J.A.; Lacy, M.Q.; Therneau, T.M. A Trial of three regimens for primary amyloidosis: Colchicine alone, melphalan and prednisone, and melphalan, prednisone, and colchicine. N. Engl. J. Med. 1997, 336, 1202–1207. [Google Scholar] [CrossRef] [PubMed]
- Imazio, M.; Gaita, F. Colchicine for cardiovascular medicine. Future Cardiol. 2016, 12, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Imazio, M.; Brucato, A.; Cemin, R.; Ferrua, S.; Belli, R.; Maestroni, S.; Trinchero, R.; Spodick, D.H.; Adler, Y. Colchicine for recurrent pericarditis (CORP). Ann. Intern. Med. 2011, 155, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Seligmann, J.; Twelves, C. Tubulin: An example of targeted chemotherapy. Future Med. Chem. 2013, 5, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Katsetos, C.D.; Dráber, P. Tubulins as therapeutic targets in cancer: From bench to bedside. Curr. Pharm. Des. 2012, 18, 2778–2792. [Google Scholar] [CrossRef] [PubMed]
- Avendaño, C.; Menéndez, J.C. Medicinal Chemistry of Anticancer Drugs; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Kozaka, T.; Nakagawa-Goto, K.; Shi, Q.; Lai, C.Y.; Hamel, E.; Bastow, K.F.; Brossi, A.; Lee, K.H. Antitumor agents 273. Design and synthesis of N-alkyl-thiocolchicinoids as potential antitumor agents. Bioorg. Med. Chem. Lett. 2010, 20, 4091–4094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raspaglio, G.; Ferlini, C.; Mozzetti, S.; Prislei, S.; Gallo, D.; Das, N.; Scambia, G. Thiocolchicine dimers: A novel class of topoisomerase-I inhibitors. Biochem. Pharmacol. 2005, 69, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Bartusik, D.; Tomanek, B.; Lattová, E.; Perreault, H.; Tuszynski, J.; Fallone, G. Derivatives of thiocolchicine and its applications to CEM cells treatment using 19F Magnetic Resonance ex vivo. Bioorg. Chem. 2010, 38, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prajapati, P.B.; Bodiwala, K.B.; Marolia, B.P. Oxidative degradation kinetic study of thiocolchicoside using stability indicating high performance thin layer chromatographic method. Pharm. Methods 2014, 5, 1–10. [Google Scholar] [CrossRef]
- Banerjee, A.; Kasmala, L.T.; Hamel, E.; Sun, L.; Lee, K.-H. Interaction of novel thiocolchicine analogs with the tubulin isoforms from bovine brain 1. Biochem. Biophys. Res. Commun. 1999, 254, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Marangon, J.; Christodoulou, M.S.; Casagrande, F.V.M.; Tiana, G.; Dalla Via, L.; Aliverti, A.; Passarella, D.; Cappelletti, G.; Ricagno, S. Tools for the rational design of bivalent microtubule-targeting drugs. Biochem. Biophys. Res. Commun. 2016, 479, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Yasobu, N.; Kitajima, M.; Kogure, N.; Shishido, Y.; Matsuzaki, T.; Nagaoka, M.; Takayama, H. Design, synthesis, and antitumor activity of 4-halocolchicines and their pro-drugs activated by cathepsin B. ACS Med. Chem. Lett. 2011, 2, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Majcher, U.; Urbaniak, A.; Maj, E.; Moshari, M.; Delgado, M.; Wietrzyk, J.; Bartl, F.; Chambers, T.C.; Tuszynski, J.A.; Huczyński, A. Synthesis, antiproliferative activity and molecular docking of thiocolchicine urethanes. Bioorg. Chem. 2018, 81, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Verdier-Pinard, P.; Brossi, A.; Hamel, E.; Lee, K.H. Antitumor agents-CLXXV. Anti-tubulin action of (+)-thiocolchicine prepared by partial synthesis. Bioorg. Med. Chem. 1997, 5, 2277–2282. [Google Scholar] [CrossRef]
- Kerkes, P.; Sharma, P.N.; Brossi, A.; Chignell, C.F.; Quinn, F.R. Synthesis and biological effects of novel thiocolchicines. 3. evaluation of N-acyldeacetylthiocolchicines, N-(alkoxycarbonyl)deacetylthiocolchicines, and O-ethyldemethylthiocolchicines. New synthesis of thiodemecolcine and antileukemic effects of 2-demethyl- and 3-demethylthiocolchicine. J. Med. Chem. 1985, 28, 1204–1208. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Nevozhay, D. Cheburator software for automatically calculating drug Inhibitory concentrations from in vitro screening assays. PLoS ONE 2014, 9, e106186. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leandro-García, L.J.; Leskelä, S.; Landa, I.; Montero-Conde, C.; López-Jiménez, E.; Letón, R.; Cascón, A.; Robledo, M.; Rodríguez-Antona, C. Tumoral and tissue-specific expression of the major human β-tubulin isotypes. Cytoskeleton 2010, 67, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Devalapally, H.; Chakilam, A.; Amiji, M.M. Role of nanotechnology in pharmaceutical product development. J. Pharm. Sci. 2007, 96, 2547–2565. [Google Scholar] [CrossRef] [PubMed]
- Niu, M.M.; Qin, J.Y.; Tian, C.P.; Yan, X.F.; Dong, F.G.; Cheng, Z.Q.; Fida, G.; Yang, M.; Chen, H.; Gu, Y.Q. Tubulin inhibitors: Pharmacophore modeling, virtual screening and molecular docking. Acta Pharmacol. Sin. 2014, 35, 967–979. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. Update on activities at the universal protein resource (UniProt) in 2013. Nucleic Acids Res. 2013, 41, D43–D47. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, R.B.G.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef] [PubMed]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2009, 31. [Google Scholar] [CrossRef] [PubMed]
- Preto, J.; Gentile, P.; Winter, P.; Churchill, C.; Omar, S.; Tuszynski, J.A. Molecular dynamics and related computational methods with applications to drug discovery. In Workshop on Coupled Mathematical Models for Physical and Nanoscale Systems and Their Applications; Springer: Cham, Switzerland, 2016; pp. 267–285. [Google Scholar]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Spasevska, I.; Ayoub, A.T.; Winter, P.; Preto, J.; Wong, G.K.S.; Dumontet, C.; Tuszynski, J.A. Modeling the Colchicum autumnale tubulin and a comparison of its interaction with colchicine to human tubulin. Int. J. Mol. Sci. 2017, 18, 1676. [Google Scholar] [CrossRef] [PubMed]
- Onufriev, A.; Bashford, D.; Case, D.A. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins Struct. Funct. Bioinform. 2004, 55, 383–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Cells | Normal Cells | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | A549 | MCF-7 | LoVo | LoVo/DX | BALB/3T3 | |||||
| IC50 (μM) | SI | IC50 (μM) | SI | IC50 (μM) | SI | IC50 (μM) | SI | RI | IC50 (μM) | |
| 1 | 0.125 ± 0.013 | 1.11 | 0.054 ± 0.028 | 2.57 | 0.108 ± 0.025 | 1.29 | 1.69 ± 0.28 | 0.08 | 15.65 | 0.139 ± 0.073 |
| 2 | 0.105 ± 0.008 | 1.35 | 0.027 ± 0.008 | 5.26 | 0.084 ± 0.021 | 1.69 | 1.550 ± 0.170 | 0.09 | 18.45 | 0.142 ± 0.073 |
| 3 | 0.010 ± 0.0001 | 10.3 | 0.015 ± 0.002 | 6.87 | 0.014 ± 0.004 | 7.36 | 0.135 ± 0.012 | 0.76 | 9.64 | 0.103 ± 0.089 |
| 4 | 0.115 ± 0.007 | 8.13 | 0.178 ± 0.020 | 7.08 | 0.125 ± 0.044 | 10.08 | 0.700 ± 0.088 | 1.80 | 5.60 | 1.260 ± 0.796 |
| 5 | 0.074 ± 0.009 | 1.41 | 0.057 ± 0.011 | 1.82 | 0.074 ± 0.019 | 1.41 | 1.010 ± 0.020 | 0.10 | 13.65 | 0.104 ± 0.043 |
| 6 | 0.010 ± 0.0001 | 6.60 | 0.013 ± 0.002 | 5.08 | 0.007 ± 0.002 | 9.43 | 0.050 ± 0.010 | 1.32 | 7.14 | 0.066 ± 0.031 |
| 7 | 0.012 ± 0.004 | 8.50 | 0.018 ± 0.002 | 5.67 | 0.011 ± 0.004 | 9.27 | 0.071 ± 0.010 | 1.44 | 6.45 | 0.102 ± 0.063 |
| 8 | 0.030 ± 0.021 | 4.60 | 0.055 ± 0.026 | 2.51 | 0.018 ± 0.010 | 7.67 | 0.074 ± 0.007 | 1.86 | 4.11 | 0.138 ± 0.010 |
| 9 | 0.012 ± 0.004 | 9.67 | 0.027 ± 0.007 | 4.30 | 0.011 ± 0.0001 | 10.55 | 0.072 ± 0.011 | 1.61 | 6.55 | 0.116 ± 0.009 |
| 10 | 0.089 ± 0.020 | 1.94 | 0.132 ± 0.017 | 1.31 | 0.054 ± 0.017 | 3.20 | 0.089 ± 0.026 | 1.94 | 1.65 | 0.173 ± 0.108 |
| 11 | 0.095 ± 0.005 | 1.54 | 0.125 ± 0.014 | 1.17 | 0.062 ± 0.013 | 2.35 | 0.091 ± 0.009 | 1.60 | 1.47 | 0.146 ± 0.014 |
| 12 | 0.093 ± 0.014 | 2.14 | 0.125 ± 0.015 | 1.08 | 0.281 ± 0.185 | 0.48 | 4.240 ± 1.330 | 0.03 | 15.09 | 0.135 ± 0.015 |
| Doxorubicin | 0.258 ± 0.044 | 0.64 | 0.386 ± 0.118 | 0.43 | 0.092 ± 0.018 | 1.80 | 4.75 ± 0.99 | 0.035 | 51.60 | 0.166 ± 0.074 |
| Cisplatin | 6.367 ± 1.413 | 0.61 | 10.70 ± 0.753 | 0.36 | 4.37 ± 0.73 | 0.89 | 5.70 ± 0.63 | 0.68 | 1.3 | 3.90 ± 1.50 |
| Compound | Binding Energy (kcal/mol) | Interactions | MW | MlogP | Active Residues |
|---|---|---|---|---|---|
| 1 | −8.09 |  | 399.44 | 1.37 | Asn258 |
| 2 | −8.40 |  | 481.37 | 1.93 | Met259 Asn258 Lys352 |
| 3 | −8.60 |  | 497.44 | 2.69 | Met259 Lys352 Val315 |
| 4 | −8.12 |  | 455.40 | 2.71 | Asn349 Met259 Lys352 Cys241 |
| 5 | −7.68 |  | 543.46 | 2.20 | Cys241 Ala317 Val315 Met259 |
| 6 | −8.99 |  | 513.43 | 2.74 | Met259 Lys352 Val315 |
| 7 | −9.20 |  | 527.46 | 2.95 | Met259 Val315 |
| 8 | −7.61 |  | 581.43 | 3.26 | Cys241 Asn258 Ala317 |
| 9 | −8.21 |  | 541.49 | 3.16 | Cys241 Ala317 |
| 10 | −8.06 |  | 541.49 | 3.16 | Cys241 Ala317 |
| 11 | −9.25 |  | 528.51 | 2.30 | Lys352 Asn258 Val315 |
| 12 | −7.92 |  | 587.52 | 1.87 | Ala317 Asn258 Met259 Cys241 |
| MCF-7 | LoVo | LoVo/DX | A549 | BALB/3T3 | ||
| Linear regression of colchicine derivatives (R2) | βI | 0.11 | 0.66 | 0.84 | 0.43 | 0.02 |
| βIIa | 0.01 | 0.53 | 0.68 | 0.30 | 0.07 | |
| βIIb | 0.15 | 0.69 | 0.76 | 0.42 | 0.0004 | |
| βIII | 0.01 | 0.57 | 0.71 | 0.36 | 0.09 | |
| βIVa | 0.16 | 0.65 | 0.74 | 0.51 | 0.31 | |
| βIVb | 0.02 | 0.72 | 0.80 | 0.43 | 0.01 | |
| βVI | 0.03 | 0.56 | 0.66 | 0.38 | 0.09 |
| Ligand | GBSA βI | PBSA βI | GBSA βIIa | PBSA βIIa | GBSA βIII | PBSA βIII | GBSA βIVb | PBSA βIVb | GBSA βVI | PBSA βVI |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | −40.45 | −40.20 | −40.83 | −45.40 | −33.53 | −32.05 | −39.53 | −44.63 | −43.07 | −35.61 |
| 2 | −36.57 | −44.26 | −46.99 | −31.05 | −33.09 | −32.52 | −44.79 | −36.88 | −46.81 | −44.01 |
| 3 | −33.97 | −37.17 | −33.04 | −29.80 | −27.66 | −28.16 | −34.17 | −30.91 | −33.5 | −37.88 |
| 4 | −37.12 | −43.09 | −36.51 | −41.35 | −34.78 | −31.90 | −40.18 | −43.70 | −39.14 | −33.84 |
| 5 | −36.80 | −44.14 | −33.28 | −42.68 | −29.91 | −22.86 | −34.98 | −39.89 | −37.87 | −39.19 |
| 6 | −37.20 | −45.72 | −38.78 | −38.70 | −29.35 | −29.16 | −30.74 | −22.96 | −34.44 | −31.81 |
| 7 | −40.29 | −44.06 | −39.28 | −45.47 | −28.69 | −26.87 | −40.99 | −49.02 | −41.90 | −42.59 |
| 8 | −48.15 | −51.01 | −40.07 | −25.4 | −45.89 | −50.04 | −47.43 | −50.29 | −50.68 | −45.55 |
| 9 | −54.12 | −39.54 | −58.30 | −54.17 | −42.75 | −41.12 | −41.59 | −32.40 | −61.45 | −51.74 |
| 10 | −58.57 | −48.74 | −60.92 | −37.23 | −61.92 | −55.56 | −58.22 | −55.78 | −59.39 | −49.74 |
| 11 | −46.77 | −49.84 | −41.34 | −41.52 | −29.28 | −19.52 | −43.05 | −23.88 | −45.26 | −35.5 |
| 12 | −52.71 | −45.46 | −55.36 | −34.82 | −38.95 | −26.10 | −53.97 | −32.11 | −50.28 | −46.36 |
| MCF-7 | LoVo | LoVo/DX | A549 | BALB/3T3 | ||
| Linear regression of colchicine derivatives (R2) | βI | 0.18 | 0.65 | 0.54 | 0.48 | 0.0006 |
| βIIa | 0.09 | 0.57 | 0.56 | 0.41 | 0.0006 | |
| βIII | 0.31 | 0.61 | 0.40 | 0.62 | 0.03 | |
| βIVb | 0.22 | 0.78 | 0.67 | 0.58 | 0.03 | |
| βVI | 0.17 | 0.64 | 0.67 | 0.47 | 0.0005 |
| MCF-7 | LoVo | LoVo/DX | A549 | BALB/3T3 | ||
| Linear regression of colchicine derivatives (R2) | βI | 0.21 | 0.60 | 0.65 | 0.55 | 0.0005 |
| βIIa | 0.02 | 0.54 | 0.68 | 0.36 | 0.0004 | |
| βIII | 0.08 | 0.57 | 0.59 | 0.46 | 0.013 | |
| βIVb | 0.12 | 0.66 | 0.71 | 0.51 | 0.095 | |
| βVI | 0.04 | 0.58 | 0.70 | 0.37 | 0.013 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majcher, U.; Klejborowska, G.; Kaik, M.; Maj, E.; Wietrzyk, J.; Moshari, M.; Preto, J.; Tuszynski, J.A.; Huczyński, A. Synthesis and Biological Evaluation of Novel Triple-Modified Colchicine Derivatives as Potent Tubulin-Targeting Anticancer Agents. Cells 2018, 7, 216. https://0-doi-org.brum.beds.ac.uk/10.3390/cells7110216
Majcher U, Klejborowska G, Kaik M, Maj E, Wietrzyk J, Moshari M, Preto J, Tuszynski JA, Huczyński A. Synthesis and Biological Evaluation of Novel Triple-Modified Colchicine Derivatives as Potent Tubulin-Targeting Anticancer Agents. Cells. 2018; 7(11):216. https://0-doi-org.brum.beds.ac.uk/10.3390/cells7110216
Chicago/Turabian StyleMajcher, Urszula, Greta Klejborowska, Magdalena Kaik, Ewa Maj, Joanna Wietrzyk, Mahshad Moshari, Jordane Preto, Jack A. Tuszynski, and Adam Huczyński. 2018. "Synthesis and Biological Evaluation of Novel Triple-Modified Colchicine Derivatives as Potent Tubulin-Targeting Anticancer Agents" Cells 7, no. 11: 216. https://0-doi-org.brum.beds.ac.uk/10.3390/cells7110216