Mitochondrial Fatty Acid Oxidation Disorders Associated with Short-Chain Enoyl-CoA Hydratase (ECHS1) Deficiency


1
Department of Biochemistry and Molecular Biology, Monash Biomedicine Discovery Institute, Monash University, 3800 Melbourne, Australia
2
Centre for Innate Immunity and Infectious Diseases, Hudson Institute of Medical Research, 3168 Melbourne, Australia
3
Department of Molecular and Translational Science, Monash University, 3168 Melbourne, Australia
*
Author to whom correspondence should be addressed.
Cells 2018, 7(6), 46; https://0-doi-org.brum.beds.ac.uk/10.3390/cells7060046
Submission received: 24 April 2018
/
Revised: 15 May 2018
/
Accepted: 16 May 2018
/
Published: 23 May 2018
(This article belongs to the Special Issue Mitochondrial Biology in Health and Disease)
Abstract
:Mitochondrial fatty acid β-oxidation (FAO) is the primary pathway for fatty acid metabolism in humans, performing a key role in liver, heart and skeletal muscle energy homeostasis. FAO is particularly important during times of fasting when glucose supply is limited, providing energy for many organs and tissues, including the heart, liver and brain. Deficiencies in FAO can cause life-threatening metabolic disorders in early childhood that present with liver dysfunction, hypoglycemia, dilated hypertrophic cardiomyopathy and Reye-like Syndrome. Alternatively, FAO defects can also cause ‘milder’ adult-onset disease with exercise-induced myopathy and rhabdomyolysis. Short-chain enoyl-CoA hydratase (ECHS1) is a key FAO enzyme involved in the metabolism of fatty acyl-CoA esters. ECHS1 deficiency (ECHS1D) also causes human disease; however, the clinical manifestation is unlike most other FAO disorders. ECHS1D patients commonly present with Leigh syndrome, a lethal form of subacute necrotizing encephalomyelopathy traditionally associated with defects in oxidative phosphorylation (OXPHOS). In this article, we review the clinical, biochemical and genetic features of the ESHS1D patients described to date, and discuss the significance of the secondary OXPHOS defects associated with ECHS1D and their contribution to overall disease pathogenesis.
1. Mitochondrial Metabolism
Mitochondria are the ‘powerhouses’ of the cell, producing 95% of all cellular energy in the form of adenosine triphosphate (ATP) [1]. ATP is expended for essential processes such as growth, reproduction, cell signaling and differentiation [2]. Additionally, mitochondria are involved in other important cellular functions including apoptosis, calcium homeostasis, biosynthesis of heme and iron-sulfur clusters, as well as innate immune responses [2,3].
Under aerobic conditions, mitochondria produce ATP via three key biochemical pathways: the tricarboxylic acid (TCA) cycle, oxidative phosphorylation (OXPHOS) and fatty acid β-oxidation (FAO). Acetyl-coenzyme A (acetyl-CoA) derived from sugars, fats and proteins is oxidized via the TCA cycle to generate the reducing equivalents nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) [4]. NADH and FADH2 are then oxidized by complex I (NADH: ubiquinone oxidoreductase) and complex II (succinate: ubiquinone oxidoreductase) respectively of the respiratory chain (RC) to drive ATP generation by OXPHOS. The electrons liberated from NADH and FADH2 are transferred via ubiquinone to complex III (ubiquinol: ferricytochrome c oxidoreductase), then cytochrome c, and finally complex IV (cytochrome c oxidase) which reduces O2 to generate H2O. This electron transfer facilitates the pumping of protons out of the mitochondrial matrix by complexes I, III and IV, which establishes an electrochemical membrane potential (ΔΨm) across the inner mitochondrial membrane (IMM) [5]. ΔΨm drives protons back into the mitochondrial matrix through complex V (F1F0-ATP synthase), resulting in the phosphorylation of adenosine diphosphate (ADP) to generate ATP [5].
2. Fatty Acid β-Oxidation (FAO)
Fatty acids are the main energy source during fasting when glucose is not available, and the preferred substrates for catabolic metabolism in the heart, liver and skeletal muscle [6]. Free fatty acids are activated in the cytosol by acyl-CoA synthetases to form fatty acyl-CoA esters. These are subsequently transported into the mitochondria via the carnitine shuttle system. Carnitine O-palmitoyltransferase 1 (CPT1) catalyzes the addition of carnitine to fatty acyl-CoA esters to form acylcarnitines, which are transported across the IMM via carnitine acylcarnitine translocase (CACT). Once inside the mitochondrial matrix, carnitine is removed by carnitine O-palmitoyltransferase 2 (CPT2) to regenerate fatty acyl-CoA esters and free carnitine, which is recycled back across the IMM by CACT (Figure 1).
Through a series of four enzymatic reactions, dehydrogenation, hydration, a second dehydrogenation and thiolysis, fatty acyl-CoA chains within the mitochondria are processed to yield one acetyl-CoA molecule, two electrons and a fatty acyl-CoA shortened by two carbons. This series of reactions is then performed repeatedly until only two acetyl-CoA molecules remain (commonly termed the β-oxidation spiral) (Figure 1). Both dehydrogenation steps are cofactor-dependent, resulting in the reduction of NAD+ and FAD to NADH and FADH2, which are subsequently oxidized by OXPHOS complexes I and II respectively [7].
The enzymes involved in FAO exhibit chain length specificity [8]. Very long-, medium-, and short-chain acyl-CoA dehydrogenases (VLCAD, MCAD, SCAD) catalyze the first dehydrogenation step of C24-C12, C12-C6 and C6-C4 carbon chain length fatty acyl-CoAs respectively. For the remaining three reactions, longer acyl-CoA chains (C16-C8) are catalyzed by the multi-domain mitochondrial trifunctional protein (MTP), which harbors long-chain enoyl-CoA hydratase, long-chain 3-hydroxyacyl-CoA dehydrogenase and 3-ketoacyl-CoA thiolase activities [9]. For medium- and short-chain fatty acids, the last three steps of FAO are catalyzed by short-chain enoyl-CoA hydratase (ECHS1), hydroxyacyl-CoA dehydrogenase (HADH) and 3-ketoacyl-CoA thiolase (KAT) (Figure 1).
3. ECHS1 is a Multifunctional Enzyme
Short-chain enoyl-CoA hydratase (ECHS1; EC 4.2.1.17) is responsible for the second step of FAO. ECHS1 activity was first observed by Stern and Del Campillo [10] in ox heart and liver, with human ECHS1 cDNA clones first isolated in 1993 [10]. The 11 kb ECHS1 gene locus was subsequently mapped to chromosome 10q26.2–q26.3 by fluorescence in situ hybridization, encoding eight exons with the 5′ and 3′ untranslated regions contained within exons I and VIII respectively [11].
ECHS1 is transcribed as a single 1.4 kb mRNA, with expression observed in hepatocytes, fibroblasts and myocytes [10]. The translated 290 amino acid precursor protein contains a 27-amino-acid N-terminal mitochondrial targeting signal that is cleaved upon entry into the mitochondrial matrix [12], where the resulting 28.3-kDa mature protein forms an active 188 kDa homohexamer composed of a ‘dimer of trimers’ [13,14] (Figure 2A).
ECHS1 catalyzes the conversion of trans-Δ2-enoyl-CoA thioesters to 3-l-hydroxyacyl-CoA thioesters by stereospecific hydration of the trans double bond between carbons two and three [15] (Figure 2B). ECHS1 has strongest substrate affinity for the 4-carbon crotonyl-CoA, but can bind enoyl-CoA chains up to 10 carbon atoms in length [14]. While ECHS1 has considerably higher specificity for straight-chain enoyl-CoA thioesters as part of the FAO pathway, it also exhibits moderate activity for degrading methacrylyl-CoA (valine pathway), 3-methylcronytyl-CoA (leucine pathway) and tiglyl-CoA (isoleucine pathway) [16,17]. Interestingly, only metabolites of the valine pathway have been detected in plasma and urine of patients with ECHS1 deficiency (ECHS1D), suggesting that ECHS1 is vital for valine metabolism, but not leucine or isoleucine metabolism [16,17].
4. FAO Disease
Defects in FAO were first described in the 1970′s in patients with carnitine O-palmitoyltransferase deficiencies [18,19,20], with the first pathogenic mutations identified in ACADM (which encodes the medium-chain acyl-CoA dehydrogenase) [21,22,23]. Pathogenic mutations have now been identified in at least 22 different FAO genes, and can affect up to 1 in 10,000 individuals in certain populations [1]. Patients can present in early childhood with severe (often lethal) liver dysfunction, hypoglycemia and Reye-like syndrome (a combination of encephalopathy due to acute brain swelling and liver dysfunction caused by fat accumulation) [24]. Cardiac symptoms, such as dilated hypertrophic cardiomyopathy and arrhythmias, are also common [25,26]. Alternatively, ‘milder’ adult-onset disease, presenting with exercise-induced myopathy and rhabdomyolysis (breakdown of muscle fibers), has also been reported [27].
Disease presentation may not be persistent, with patients showing no symptoms or biochemical deficiencies until an episode of metabolic crisis. These episodes can be triggered by various circumstances, including prolonged fasting, exercise, infection, exposure to cold, or a fat-rich diet [7,28,29]. Treatment options are limited, focusing on restricting dietary long-chain fatty acids that cannot be metabolized (and which become toxic), as well as maintaining blood glucose levels.
FAO deficiencies are also believed to cause about 1–3% of unexplained sudden infant deaths [30], with deficiencies in MCAD and long-chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD) reported [31,32]. In addition, mutations in the genes encoding MCAD, SCAD, LCHAD and CPT2 are associated with acute fatty liver of pregnancy (AFLP) and hemolysis, elevated liver enzymes and low platelets (HELLP) syndrome, both of which carry significant neonatal and maternal morbidity and mortality during pregnancy [33,34].
5. ECHS1 Deficiency (ECHS1D)
ECHS1D onset is usually at birth or in early childhood, with death occurring within the first two days of life in some cases [35]. The clinical presentation of ECHS1D is typified by Leigh syndrome (subacute necrotizing encephalomyelopathy) or Leigh-like syndrome, with symptoms including (but not limited to) developmental delay, dystonia, metabolic acidosis, cardiomyopathy and apnea. Leigh syndrome is a progressive neurodegenerative disease characterized by bilateral symmetric brain lesions and psychomotor regression [36], and is not typically observed in other FAO disorders [37]. Leigh syndrome has been associated with more than 75 genes, mostly involved in OXPHOS complex I structure and assembly [38]. Brain magnetic resonance imaging (MRI) findings have revealed T2 bilateral hyperintensities, a hallmark of Leigh syndrome, in almost all reported cases of ECHS1D. Despite this, there are disparities between the clinical presentation in ECHS1D and the classical features of Leigh syndrome (as defined in Rahman, et al. [39]). Indeed, Haack, et al. [40] proposed that ECHS1D is a distinct form of Leigh-like syndrome associated with severe progressive encephalopathy, accompanied by bilateral brain lesions and mitochondrial dysfunction.
More recently, three patients were identified with clinical symptoms that expand the phenotypic spectrum of ECHS1D [41,42,43]. One patient harboring ECHS1 variants previously associated with Leigh-like syndrome also presented with cutis laxa, a connective tissue disorder characterized by loose, inelastic skin [43]. Another two patients displayed symptoms of paroxysmal exercise-induced dyskinesia (PED) [41,42]. PED is a much milder form of ECHS1D that offers a more optimistic prognosis, being characterized by recurrent attacks of abnormal dystonic movement that is triggered by prolonged exercise [44]. While these PED patients were unrelated, they shared a common ECHS1 mutation (c.518C > T; p.Ala173Val), suggesting an association between this variant and PED. Interestingly, T2 hyperintensities were still observed in these patients, but Leigh-like symptoms were absent. Conversely, a sibling of one of these patients did suffer from Leigh-like syndrome with severe generalized dystonia, further exemplifying the substantial clinical heterogeneity of ECHS1D [42].
6. Pathogenic Mutations in ECHS1
Forty-two patients (from 33 families) with ECHS1D have been described to date (Table 1). As with almost all primary disorders of FAO, ECHS1D follows an autosomal recessive pattern of inheritance. Human mutations in ECHS1 were first described in two infant siblings [45], with all subsequent cases exhibiting compound heterozygosity or homozygosity due to consanguinity. Twenty-seven of the 30 different ECHS1 mutations identified are missense, suggesting that null mutations may be incompatible with life [40,46]. Other noteworthy variants include nonsense mutations, frameshift mutations, and duplications resulting in protein truncation [35,40,42]. The only incidence of homozygosity for a truncating mutation was reported in two siblings with a very severe phenotype and death within 48 h, establishing some evidence for a genotype-phenotype relationship [35].
While most ECHS1 mutations are novel, the c.476A > G; p.Gln159Arg variant has been identified in seven unrelated ECHS1D patients with diverse racial backgrounds [17,40,43,47,48]. Furthermore, this variant may also represent a common founder mutation, as it has been independently reported in two families of Pakistani origin [40,48]. A second possible founder mutation in ECHS1 (c.538A > G; p.Thr180Ala) has also been identified in four French-Canadian patients [47]. Interestingly, this mutation has also been identified in an Irish Traveler family with a haplotype shared with the French-Canadian patients, suggesting an Irish ancestral origin with subsequent migration to Canada [48].
7. Biochemical and Metabolic Characterization of ECHS1D
ECHS1 may play a redundant role in FAO, as exemplified by the unremarkable acylcarnitine profiles in most patient cells [16,17,35,40,47,48,49,50,52]. However, extremely high acylcarnitine levels (of C6 and C4 chain lengths) were reported in one patient who died within one day of birth [46]. As such, elevated acylcarnitines may act as an indicator of disease in only the most severe cases of ECHS1D.
Loss of ECHS1 activity for valine metabolism also appears to play a role in disease pathology. ECHS1 acts on both methacrylyl-CoA from the valine pathway and acryloyl-CoA from an alternate pathway of odd-chain FAO that feeds into the valine pathway [54]. In ECHS1D, these two intermediates accumulate, becoming toxic by spontaneous reaction with thiol groups and other mitochondrial cysteine residues, resulting in impaired ATP production and metabolic acidosis [55]. Specifically, methacrylyl-CoA and acryloyl-CoA react with lipoyl domains of the E2 subunit of the pyruvate dehydrogenase complex (PDC), inhibiting its function [17]. Indeed, reduced PDC activity has been observed in many ECHS1D patients, explaining the commonly observed symptom of lactic acidosis [17,35,40,43,45,47,48,51,52]. Notably, an inhibitory effect on two other lipoyl domain-containing mitochondrial enzymes was not detected [17], suggesting inhibition is specific to the PDC (via an unknown mechanism).
These findings have led to tentative correlations between ECHS1D severity, degree of lactic acidosis and reduced PDC activity. In cases where ECHS1D caused early death, lactate levels were high and PDC activity low [35]. Conversely, in mild cases of ECHS1D, lactic acidosis was absent [16,41,42].
Other metabolic markers of ECHS1D, such as increased metabolites of methacrylyl-CoA and acryloyl-CoA in patient urine, have also been identified [45]. In particular, large peaks of 2-methyl-2,3-dihydroxybutyrate have been detected in many ECHS1D cases [40,45,48,50,51]. While the origin of 2-methyl-2,3-dihydroxybutyrate is not known, evidence suggests it is a derivative of acryloyl-CoA [45]. Despite these findings, 2-methyl-2,3-dihydroxybutyrate concentrations have repeatedly measured within the normal range in milder ECHS1D cases [16,17,40,42]. Conversely, N-acetyl-S-(2-carboxypropyl)cysteine (produced from methacrylyl-CoA) is the only known metabolite that is elevated in the mildest cases of ECHS1D, making it the most useful biomarker for ECHS1D diagnosis [16,42,52].
8. Secondary OXPHOS Defects in ECHS1D
FAO and OXPHOS are tightly linked biochemically, with the oxidation of fatty acids generating NADH and FADH2 for oxidation by OXPHOS complexes I and II. In addition, studies have also reported physical interactions between FAO and OXPHOS proteins: OXPHOS complex I can bind the FAO protein LCHAD [56], while OXPHOS complex III can be purified in complexes with the FAO electron transfer flavoprotein (ETF) [57]. More recently, the FAO proteins VLCAD, MCAD, LCHAD, and ETF were shown to co-migrate with the OXPHOS supercomplex in a metabolically active super-structure that can oxidize fatty acids [58].
Interestingly, patients with primary deficiencies in LCHAD can exhibit significant OXPHOS enzyme defects [26,59]. These secondary OXPHOS defects were historically attributed to the accumulation of inhibitory fatty-acyl CoA intermediates. However, it now appears that more complex mechanisms are involved. Indeed, we recently showed that loss of the FAO protein MCAD can disrupt OXPHOS complex assembly and stability, resulting in reduced mitochondrial respiration and increased ROS generation in the presence of OXPHOS inhibitors [60].
Secondary OXPHOS defects have also been identified in ECHS1D. These range from isolated defects in complex I, complex III or complex IV to multiple complex I/III/IV or complex I/IV/V defects [40,47,48,49,53]. Furthermore, blue native polyacrylamide gel electrophoresis (BN-PAGE) has identified reduced steady-state levels of mature complex IV in one patient [47]. Conversely, no differences in complex IV levels were detected in three other ECHS1D patients [17,49].
While it is not clear what causes these secondary OXPHOS defects in ECHS1D, disruption of complex I activity may be explained via its interaction with the pyruvate dehydrogenase complex (PDC). As discussed above, toxic metabolites of the valine pathway can accumulate in ECHS1D and inhibit PDC activity [17]. This inhibition may disrupt the binding of complex I to the PDC [56], with a potential loss of both complex I stability and NADH dehydrogenase activity.
Overall, it is difficult to predict the effect of ECHS1D on OXPHOS. However, it is apparent that patients with secondary OXPHOS defects generally exhibit more severe Leigh-like symptoms (Table 1).
9. Concluding Remarks
Forty patients have been described with pathogenic mutations in ECHS1 since the identification of the first two patients with ECHS1D in 2014. Notably, most ECHS1D patients present with Leigh syndrome or Leigh-like syndrome, a severe disorder traditionally associated with defects in OXPHOS complex I activity. While loss of ECHS1 disrupts both FAO and valine metabolism, secondary OXPHOS defects have also been identified in some patients with ECHS1D. Furthermore, these secondary OXPHOS defects are associated with a more severe clinical phenotype, suggesting that they also contribute to disease pathology.
Like other FAO deficiencies, secondary OXPHOS defects in ECHS1D may be due to the accumulation of toxic fatty acid and/or valine metabolites, which can directly inhibit OXPHOS complex activity. Alternatively, the effects of these metabolites may be indirect by disrupting the interaction between the pyruvate dehydrogenase complex (PDC) and OXPHOS complex I. Whichever mechanism is involved, further research is required to clarify the relationship between ECHS1D and OXPHOS dysfunction, and to determine if the stability and/or biogenesis of the OXPHOS complexes is also disrupted by the loss of ECHS1. This knowledge will be invaluable for our understanding of the complex interactions between the FAO and OXPHOS pathways, and will help to advance the diagnosis and treatment of mitochondrial disorders such as ECHS1D.
Author Contributions
A.J.S. and M.M. researched the relevant literature, prepared the figures and tables and wrote the manuscript.
Acknowledgments
This work was supported by the Australian Research Council Future Fellowship Scheme (FT120100459), the William Buckland Foundation, the Australian Mitochondrial Disease Foundation, Monash University and the Hudson Institute of Medical Research. The Hudson Institute of Medical Research receives support from the Victorian Government Infrastructure Program.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nsiah-Sefaa, A.; McKenzie, M. Combined defects in oxidative phosphorylation and fatty acid beta-oxidation in mitochondrial disease. Biosci. Rep. 2016, 36, e00313. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.H.; Distelmaier, F.; Smeitink, J.A.M.; Willems, P.H.G.M. OXPHOS mutations and neurodegeneration. EMBO J. 2013, 32, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Smeitink, J.; van den Heuvel, L.; DiMauro, S. The genetics and pathology of oxidative phosphorylation. Nat. Rev. Genet. 2001, 2, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Wajner, M.; Amaral, A.U. Mitochondrial dysfunction in fatty acid oxidation disorders: Insights from human and animal studies. Biosci. Rep. 2016, 36, e00281. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, K.; Eaton, S. Mitochondrial beta-oxidation. Eur. J. Biochem. 2004, 271, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Van Eunen, K.; Volker-Touw, C.M.L.; Gerding, A.; Bleeker, A.; Wolters, J.C.; van Rijt, W.J.; Martines, A.-C.M.F.; Niezen-Koning, K.E.; Heiner, R.M.; Permentier, H.; et al. Living on the edge: Substrate competition explains loss of robustness in mitochondrial fatty-acid oxidation disorders. BMC Biol. 2016, 14, 107. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, K.; Pollitt, R.J.; Middleton, B. Human liver long-chain 3-hydroxyacyl-coenzyme a dehydrogenase is a multifunctional membrane-bound beta-oxidation enzyme of mitochondria. Biochem. Biophys. Res. Commun. 1992, 183, 443–448. [Google Scholar] [CrossRef]
- Kanazawa, M.; Ohtake, A.; Abe, H.; Yamamoto, S.; Satoh, Y.; Takayanagi, M.; Niimi, H.; Mori, M.; Hashimoto, T. Molecular cloning and sequence analysis of the cDNA for human mitochondrial short-chain enoyl-CoA hydratase. Enzyme Protein 1993, 47, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Janßen, U.; Davis, E.M.; Le Beau, M.M.; Stoffel, W. Human mitochondrial enoyl-CoA hydratase gene (ECHS1): Structural organization and assignment to chromosome 10q26. 2–q26. 3. Genomics 1997, 40, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Vaca Jacome, A.S.; Rabilloud, T.; Schaeffer-Reiss, C.; Rompais, M.; Ayoub, D.; Lane, L.; Bairoch, A.; Van Dorsselaer, A.; Carapito, C. N-terminome analysis of the human mitochondrial proteome. Proteomics 2015, 15, 2519–2524. [Google Scholar] [CrossRef] [PubMed]
- Hass, G.M.; Hill, R.L. The subunit structure of crotonase. J. Biol. Chem. 1969, 244, 6080–6086. [Google Scholar] [PubMed]
- Fong, J.C.; Schulz, H. Purification and properties of pig heart crotonase and the presence of short chain and long chain enoyl coenzyme A hydratases in pig and guinea pig tissues. J. Biol. Chem. 1977, 252, 542–547. [Google Scholar] [PubMed]
- Stern, J.R.; Del Campillo, A. Enzymatic reaction of crotonyl coenzyme A. J. Am. Chem. Soc. 1953, 75, 2277–2278. [Google Scholar] [CrossRef]
- Yamada, K.; Aiba, K.; Kitaura, Y.; Kondo, Y.; Nomura, N.; Nakamura, Y.; Fukushi, D.; Murayama, K.; Shimomura, Y.; Pitt, J.; et al. Clinical, biochemical and metabolic characterisation of a mild form of human short-chain enoyl-CoA hydratase deficiency: Significance of increased N-acetyl-S-(2-carboxypropyl)cysteine excretion. J. Med. Genet. 2015, 52, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandusse, S.; Friederich, M.W.; Burlina, A.; Ruiter, J.P.N.; Coughlin, C.R.; Dishop, M.K.; Gallagher, R.C.; Bedoyan, J.K.; Vaz, F.M.; Waterham, H.R.; et al. Clinical and biochemical characterization of four patients with mutations in ECHS1. Orphanet J. Rare Dis. 2015, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S.; DiMauro, P.M. Muscle carnitine palmityltransferase deficiency and myoglobinuria. Science 1973, 182, 929–931. [Google Scholar] [CrossRef] [PubMed]
- Karpati, G.; Carpenter, S.; Engel, A.G.; Watters, G.; Allen, J.; Rothman, S.; Klassen, G.; Mamer, O.A. The syndrome of systemic carnitine deficiency. Clinical, morphologic, biochemical, and pathophysiologic features. Neurology 1975, 25, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, N.; Lauritzen, R.; Rasmussen, K. Suberylglycine excretion in the urine from a patient with dicarboxylic aciduria. Clin. Chim. Acta 1976, 70, 417–425. [Google Scholar] [CrossRef]
- Kelly, D.P.; Whelan, A.J.; Ogden, M.L.; Alpers, R.; Zhang, Z.F.; Bellus, G.; Gregersen, N.; Dorland, L.; Strauss, A.W. Molecular characterization of inherited medium-chain acyl-CoA dehydrogenase deficiency. Proc. Natl. Acad. Sci. USA 1990, 87, 9236–9240. [Google Scholar] [CrossRef] [PubMed]
- Yokota, I.; Indo, Y.; Coates, P.M.; Tanaka, K. Molecular basis of medium chain acyl-coenzyme A dehydrogenase deficiency. An A to G transition at position 985 that causes a lysine-304 to glutamate substitution in the mature protein is the single prevalent mutation. J. Clin. Investig. 1990, 86, 1000–1003. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, Y.; Narisawa, K.; Miyabayashi, S.; Tada, K.; Coates, P.M. Molecular lesion in patients with medium-chain acyl-CoA dehydrogenase deficiency. Lancet 1990, 335, 1589. [Google Scholar] [CrossRef]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J. The Biochemistry and Physiology of Mitochondrial Fatty Acid beta-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Kompare, M.; Rizzo, W.B. Mitochondrial fatty-acid oxidation disorders. Semin. Pediatr. Neurol. 2008, 15, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Tyni, T.; Pihko, H. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Acta Paediatr. 1999, 88, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Wanders, R.J. A general introduction to the biochemistry of mitochondrial fatty acid beta-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef] [PubMed]
- De Lonlay, P.; Giurgea, I.; Touati, G.; Saudubray, J.M. Neonatal hypoglycaemia: Aetiologies. Semin. Neonatol. 2004, 9, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Kottlors, M.; Jaksch, M.; Ketelsen, U.P.; Weiner, S.; Glocker, F.X.; Lucking, C.H. Valproic acid triggers acute rhabdomyolysis in a patient with carnitine palmitoyltransferase type II deficiency. Neuromuscul. Disord. 2001, 11, 757–759. [Google Scholar] [CrossRef]
- Moczulski, D.; Majak, I.; Mamczur, D. An overview of beta-oxidation disorders. Postepy Hig. Med. Dosw. 2009, 63, 266–277. [Google Scholar]
- Emery, J.L.; Howat, A.J.; Variend, S.; Vawter, G.F. Investigation of inborn errors of metabolism in unexpected infant deaths. Lancet 1988, 2, 29–31. [Google Scholar] [CrossRef]
- Wanders, R.J.; Duran, M.; Ijlst, L.; de Jager, J.P.; van Gennip, A.H.; Jakobs, C.; Dorland, L.; van Sprang, F.J. Sudden infant death and long-chain 3-hydroxyacyl-CoA dehydrogenase. Lancet 1989, 2, 52–53. [Google Scholar] [CrossRef]
- Sim, K.G.; Hammond, J.; Wilcken, B. Strategies for the diagnosis of mitochondrial fatty acid beta-oxidation disorders. Clin. Chim. Acta 2002, 323, 37–58. [Google Scholar] [CrossRef]
- Mansouri, A.; Fromenty, B.; Durand, F.; Degott, C.; Bernuau, J.; Pessayre, D. Assessment of the prevalence of genetic metabolic defects in acute fatty liver of pregnancy. J. Hepatol. 1996, 25, 781. [Google Scholar] [CrossRef]
- Al Mutairi, F.; Shamseldin, H.E.; Alfadhel, M.; Rodenburg, R.J.; Alkuraya, F.S. A lethal neonatal phenotype of mitochondrial short-chain enoyl-CoA hydratase-1 deficiency. Clin. Genet. 2017, 91, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Aulbert, W.; Weigt-Usinger, K.; Thiels, C.; Köhler, C.; Vorgerd, M.; Schreiner, A.; Hoffjan, S.; Rothoeft, T.; Wortmann, S.B.; Heyer, C.M.; et al. Long survival in Leigh syndrome: New cases and review of literature. Neuropediatrics 2014, 45, 346–353. [Google Scholar] [PubMed]
- Saudubray, J.M.; Martin, D.; de Lonlay, P.; Touati, G.; Poggi-Travert, F.; Bonnet, D.; Jouvet, P.; Boutron, M.; Slama, A.; Vianey-Saban, C.; et al. Recognition and management of fatty acid oxidation defects: A series of 107 patients. J. Inherit. Metab. Dis. 1999, 22, 488–502. [Google Scholar] [CrossRef] [PubMed]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Blok, R.B.; Dahl, H.H.M.; Danks, D.M.; Kirby, D.M.; Chow, C.W.; Christodoulou, J.; Thorburn, D.R. Leigh syndrome: Clinical features and biochemical and DNA abnormalities. Ann. Neurol. 1996, 39, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Jackson, C.B.; Murayama, K.; Kremer, L.S.; Schaller, A.; Kotzaeridou, U.; de Vries, M.C.; Schottmann, G.; Santra, S.; Buchner, B.; et al. Deficiency of ECHS1 causes mitochondrial encephalopathy with cardiac involvement. Ann. Clin. Transl. Neurol. 2015, 2, 492–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahajan, A.; Constantinou, J.; Sidiropoulos, C. ECHS1 deficiency-associated paroxysmal exercise-induced dyskinesias: Case presentation and initial benefit of intervention. J. Neurol. 2017, 264, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Olgiati, S.; Skorvanek, M.; Quadri, M.; Minneboo, M.; Graafland, J.; Breedveld, G.J.; Bonte, R.; Ozgur, Z.; van den Hout, M.C.G.N.; Schoonderwoerd, K.; et al. Paroxysmal exercise-induced dystonia within the phenotypic spectrum of ECHS1 deficiency. Mov. Disord. 2016, 31, 1041–1048. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniam, S.; Riley, L.G.; Bratkovic, D.; Ketteridge, D.; Manton, N.; Cowley, M.J.; Gayevskiy, V.; Roscioli, T.; Mohamed, M.; Gardeitchik, T.; et al. Unique presentation of cutis laxa with Leigh-like syndrome due to ECHS1 deficiency. J. Inherit. Metab. Dis. 2017, 40, 745–747. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, K.P. Paroxysmal dyskinesias. Mov. Disord. 2011, 26, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Peters, H.; Buck, N.; Wanders, R.; Ruiter, J.; Waterham, H.; Koster, J.; Yaplito-Lee, J.; Ferdinandusse, S.; Pitt, J. ECHS1 mutations in Leigh disease: A new inborn error of metabolism affecting valine metabolism. Brain 2014, 137, 2903–2908. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.; Hamzeh, A.R.; Mohamed, M.; Malik, E.M.; Al-Ali, M.T.; Bastaki, F. Novel ECHS1 mutation in an Emirati neonate with severe metabolic acidosis. Metab. Brain Dis. 2016, 31, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Tetreault, M.; Fahiminiya, S.; Antonicka, H.; Mitchell, G.A.; Geraghty, M.T.; Lines, M.; Boycott, K.M.; Shoubridge, E.A.; Mitchell, J.J.; Michaud, J.L.; et al. Whole-exome sequencing identifies novel ECHS1 mutations in Leigh syndrome. Hum. Genet. 2015, 134, 981–991. [Google Scholar] [CrossRef] [PubMed]
- Fitzsimons, P.E.; Alston, C.L.; Bonnen, P.E.; Hughes, J.; Crushell, E.; Geraghty, M.T.; Tetreault, M.; O’Reilly, P.; Twomey, E.; Sheikh, Y.; et al. Clinical, biochemical, and genetic features of four patients with short-chain enoyl-CoA hydratase (ECHS1) deficiency. Am. J. Med. Genet. A 2018, 176, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Sakai, C.; Yamaguchi, S.; Sasaki, M.; Miyamoto, Y.; Matsushima, Y.; Goto, Y.I. ECHS1 mutations cause combined respiratory chain deficiency resulting in leigh syndrome. Hum. Mutat. 2015, 36, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Ganetzky, R.D.; Bloom, K.; Ahrens-Nicklas, R.; Edmondson, A.; Deardorff, M.A.; Bennett, M.J.; Ficicioglu, C. ECHS1 Deficiency as a Cause of Severe Neonatal Lactic Acidosis. JIMD Rep. 2016, 30, 33–37. [Google Scholar] [PubMed]
- Bedoyan, J.K.; Yang, S.P.; Ferdinandusse, S.; Jack, R.M.; Miron, A.; Grahame, G.; DeBrosse, S.D.; Hoppel, C.L.; Kerr, D.S.; Wanders, R.J.A. Lethal neonatal case and review of primary short-chain enoyl-CoA hydratase (SCEH) deficiency associated with secondary lymphocyte pyruvate dehydrogenase complex (PDC) deficiency. Mol. Genet. Metab. 2017, 120, 342–359. [Google Scholar] [CrossRef] [PubMed]
- Huffnagel, I.C.; Redeker, E.J.W.; Reneman, L.; Vaz, F.M.; Ferdinandusse, S.; Poll-The, B.T. Mitochondrial Encephalopathy and Transient 3-Methylglutaconic Aciduria in ECHS1 Deficiency: Long-Term Follow-Up. JIMD Rep. 2017, 39, 83–87. [Google Scholar] [PubMed]
- Ogawa, E.; Shimura, M.; Fushimi, T.; Tajika, M.; Ichimoto, K.; Matsunaga, A.; Tsuruoka, T.; Ishige, M.; Fuchigami, T.; Yamazaki, T.; et al. Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: A study of 106 Japanese patients. J. Inherit. Metab. Dis. 2017, 40, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Peters, H.; Ferdinandusse, S.; Ruiter, J.P.; Wanders, R.J.A.; Boneh, A.; Pitt, J. Metabolite studies in HIBCH and ECHS1 defects: Implications for screening. Mol. Genet. Metab. 2015, 115, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.K.; Hunt, S.M.; Scholem, R.; Fowler, K.; Grimes, A.; Mercer, J.F.; Truscott, R.M.; Cotton, R.G.; Rogers, J.G.; Danks, D.M. β-Hydroxyisobutyryl coenzyme A deacylase deficiency: A defect in valine metabolism associated with physical malformations. Pediatrics 1982, 70, 532–538. [Google Scholar] [PubMed]
- Sumegi, B.; Srere, P.A. Complex I binds several mitochondrial NAD-coupled dehydrogenases. J. Biol. Chem. 1984, 259, 15040–15045. [Google Scholar] [PubMed]
- Parker, A.; Engel, P.C. Preliminary evidence for the existence of specific functional assemblies between enzymes of the beta-oxidation pathway and the respiratory chain. Biochem. J. 2000, 345 Pt 3, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mohsen, A.W.; Mihalik, S.J.; Goetzman, E.S.; Vockley, J. Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J. Biol. Chem. 2010, 285, 29834–29841. [Google Scholar] [CrossRef] [PubMed]
- Das, A.M.; Fingerhut, R.; Wanders, R.J.; Ullrich, K. Secondary respiratory chain defect in a boy with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: Possible diagnostic pitfalls. Eur. J. Pediatr. 2000, 159, 243–246. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.C.; Tajika, M.; Shimura, M.; Carey, K.T.; Stroud, D.A.; Murayama, K.; Ohtake, A.; McKenzie, M. Loss of the Mitochondrial Fatty Acid β-Oxidation Protein Medium-Chain Acyl-Coenzyme A Dehydrogenase Disrupts Oxidative Phosphorylation Protein Complex Stability and Function. Sci. Rep. 2018, 8, 153. [Google Scholar] [CrossRef] [PubMed]
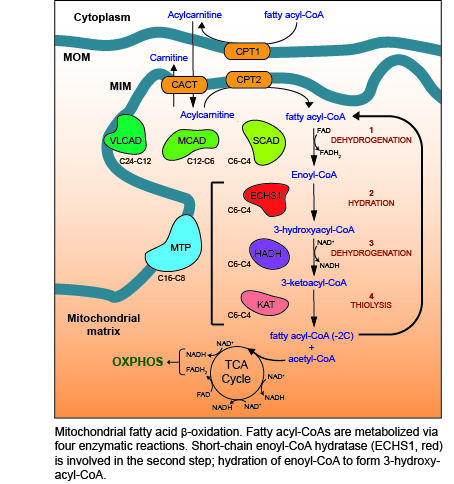
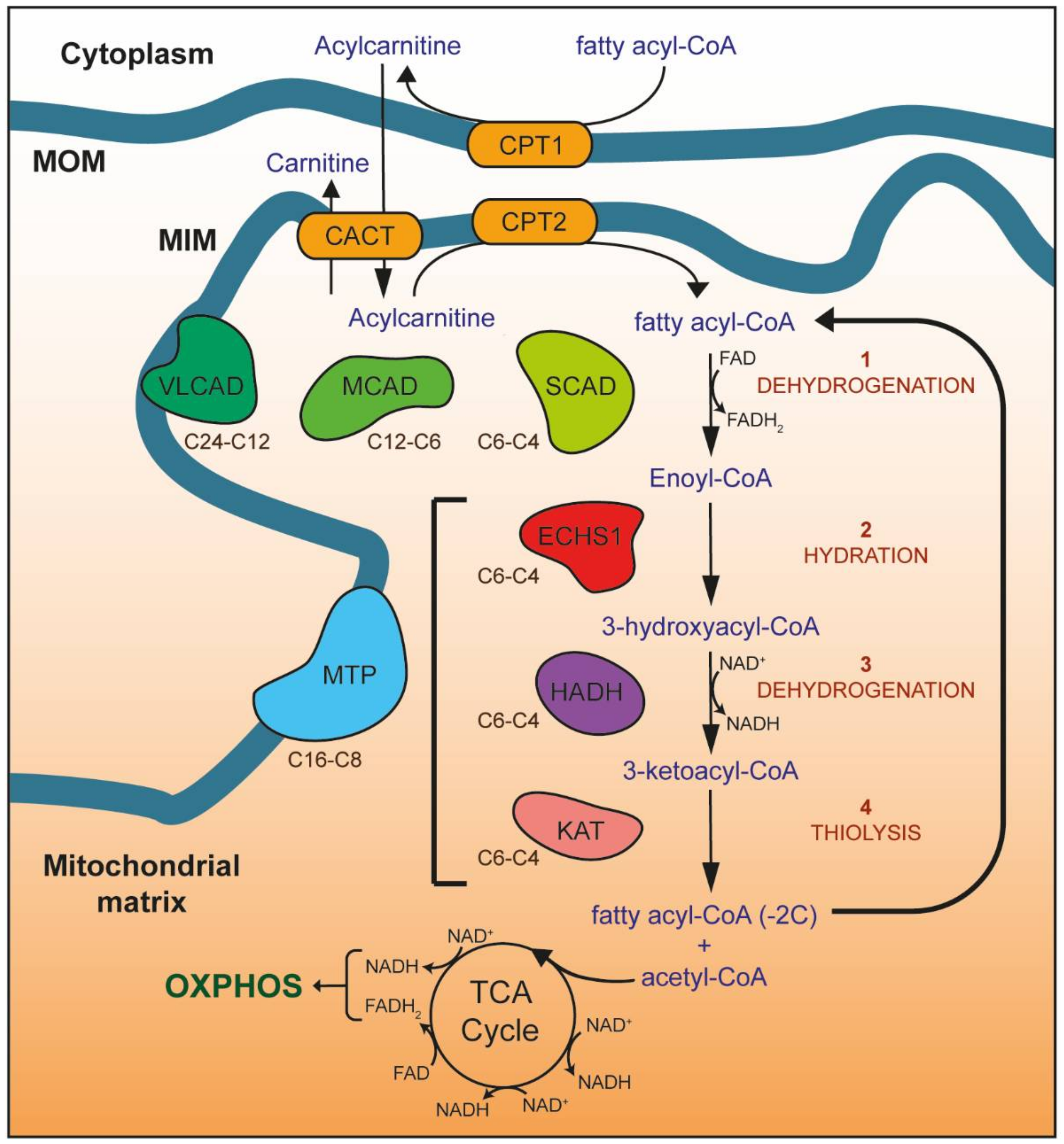
Figure 1.
Mitochondrial fatty acid β-oxidation (FAO). Enzymes of the carnitine shuttle system (yellow) are responsible for transporting fatty acyl-CoA esters into the mitochondrial matrix as acylcarnitines. Carnitine is added to fatty acyl-CoAs by carnitine O-palmitoyltransferase 1 (CPT1), forming acylcarnitines that are transported into the mitochondrial matrix by the carnitine acylcarnitine translocase (CACT). Once inside the mitochondrial matrix, carnitine O-palmitoyltransferase 2 (CPT2) removes the carnitine to regenerate the fatty acyl-CoA ester. Four reactions (1–4) then occur for each round of FAO, catalyzed by enzymes with different carbon chain length specificities (as shown): 1—dehydrogenation of fatty acyl-CoA esters by very long-chain (VLCAD), medium-chain (MCAD), and short-chain (SCAD) acyl-CoA dehydrogenases (shown in green) to form enoyl-CoA, 2—hydration of enoyl-CoA by the mitochondrial trifunctional protein (MTP, blue) or short-chain enoyl-CoA hydratase (ECHS1, red) to form 3-hydroxyacyl-CoA, 3—dehydrogenation of 3-hydroxyacyl-CoA by MTP or hydroxyacyl-CoA dehydrogenase (HADH, purple) to form 3-ketoacyl-CoA, 4—thiolysis of 3-ketoacyl-CoA by MTP or 3-ketoacyl-CoA thiolase (KAT, pink). The resulting fatty acyl-CoA is shortened by two carbons, with the generation of acetyl-CoA, NADH and FADH2. NADH and FADH2 provide electrons for OXPHOS, while acetyl-CoA enters the TCA cycle to generate further NADH and FADH2. The shortened fatty acyl-CoA undergoes further rounds of FAO until only two acetyl-CoA molecules remain. MOM, mitochondrial outer membrane; MIM, mitochondrial inner membrane.
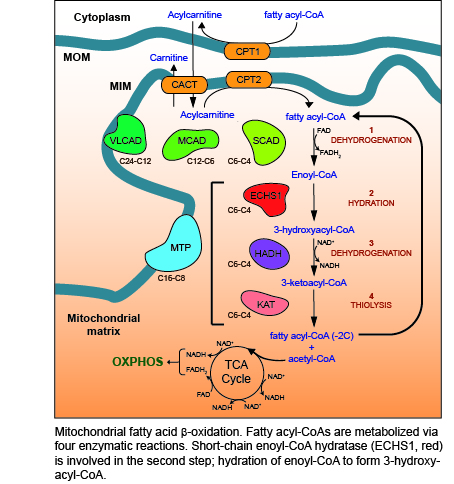
Figure 1.
Mitochondrial fatty acid β-oxidation (FAO). Enzymes of the carnitine shuttle system (yellow) are responsible for transporting fatty acyl-CoA esters into the mitochondrial matrix as acylcarnitines. Carnitine is added to fatty acyl-CoAs by carnitine O-palmitoyltransferase 1 (CPT1), forming acylcarnitines that are transported into the mitochondrial matrix by the carnitine acylcarnitine translocase (CACT). Once inside the mitochondrial matrix, carnitine O-palmitoyltransferase 2 (CPT2) removes the carnitine to regenerate the fatty acyl-CoA ester. Four reactions (1–4) then occur for each round of FAO, catalyzed by enzymes with different carbon chain length specificities (as shown): 1—dehydrogenation of fatty acyl-CoA esters by very long-chain (VLCAD), medium-chain (MCAD), and short-chain (SCAD) acyl-CoA dehydrogenases (shown in green) to form enoyl-CoA, 2—hydration of enoyl-CoA by the mitochondrial trifunctional protein (MTP, blue) or short-chain enoyl-CoA hydratase (ECHS1, red) to form 3-hydroxyacyl-CoA, 3—dehydrogenation of 3-hydroxyacyl-CoA by MTP or hydroxyacyl-CoA dehydrogenase (HADH, purple) to form 3-ketoacyl-CoA, 4—thiolysis of 3-ketoacyl-CoA by MTP or 3-ketoacyl-CoA thiolase (KAT, pink). The resulting fatty acyl-CoA is shortened by two carbons, with the generation of acetyl-CoA, NADH and FADH2. NADH and FADH2 provide electrons for OXPHOS, while acetyl-CoA enters the TCA cycle to generate further NADH and FADH2. The shortened fatty acyl-CoA undergoes further rounds of FAO until only two acetyl-CoA molecules remain. MOM, mitochondrial outer membrane; MIM, mitochondrial inner membrane.

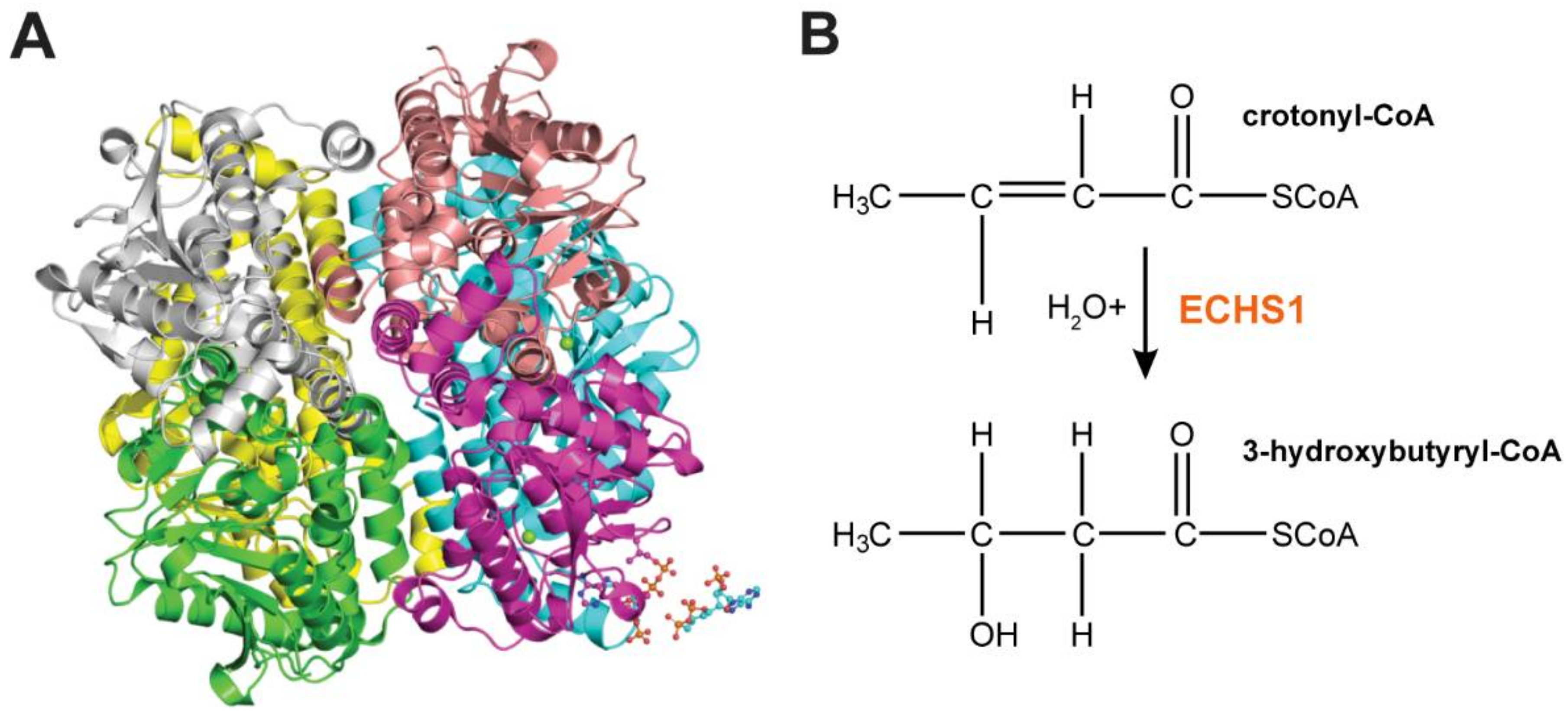
Figure 2.
ECHS1 Structure and Function. (A) Homohexameric ECHS1 crystal structure at 2.55 Å resolution (PDB: 2hw5), showing six ECHS1 units colored by chain. Two copies of the 4-carbon substrate crotonyl-CoA are shown (bottom right hand corner). (B) ECHS1 catalyzes the conversion of trans-Δ2-enoyl-CoA thioesters to 3-l-hydroxyacyl-CoA thioesters by stereospecific hydration of the trans double bond between carbons two and three. Hydration of crotonyl-CoA to 3-hydroxybutyryl-CoA is shown.
Figure 2.
ECHS1 Structure and Function. (A) Homohexameric ECHS1 crystal structure at 2.55 Å resolution (PDB: 2hw5), showing six ECHS1 units colored by chain. Two copies of the 4-carbon substrate crotonyl-CoA are shown (bottom right hand corner). (B) ECHS1 catalyzes the conversion of trans-Δ2-enoyl-CoA thioesters to 3-l-hydroxyacyl-CoA thioesters by stereospecific hydration of the trans double bond between carbons two and three. Hydration of crotonyl-CoA to 3-hydroxybutyryl-CoA is shown.


{kind=link}
{kind=link}
{kind=link}
Table 1.
The clinical, biochemical and metabolic features of all reported ECHS1D patients.
| Reference | Peters et al. 2014 [45] | Sakai et al. 2015 [49] | Haack et al. 2015 [40] | |||||
| Patient ID | Patient 1 | Patient 2 | Patient 1 | FI, II:2 | F2: II:1 | F3. II:6 | F4; II:1 | |
| Age at presentation | Birth | 3 months | 2 months | Birth | Birth | Birth | Birth | |
| Death | 4 months | 8 months | NL | 4 months | 11 months | 3 years | 7.5 years | |
| Parental consanguinity | No | No | No | No | No | Yes | No | |
| Mutation 1 (genetic level; protein effect) | c.473C > A; p.Ala158Asp | c.473C > A; p.Ala158Asp | c.2T > G; p.Met1Arg | c.176A > G; p.Asn59Ser | c.197T > C; p.Ile66Thr | c.476A > G; p.Gln159Arg | c.161G > A; p.Arg54His | |
| Mutation 2 (genetic level; protein effect) | c.414 + 3G > C; splicing | c.414 + 3G > C; splicing | c.5C > T; p.Ala2Val | c.476A > G; p.Gln159Arg | c.449A > G; p.Asp150Gly | c.476A > G; p.Gln159Arg | c.817A > G; p.Lys273Glu | |
| T2 hyperintensity | Yes | NL | Yes | Yes | Yes | Yes | NL | |
| Acylcarnitine profile | ND | ND | Normal | Normal | Normal | Normal | NL | |
| PDC activity | Reduced | Reduced | ND | ND | Reduced | ND | ND | |
| OXPHOS activity | ND | ND | Reduced CI, CIII and CIV (patient cells), reduced CI, CIV and CV (immortalized myoblasts) | Reduced CI in liver, normal in heart and muscle | Normal | ND | Normal (but reduced overall ATP production) | |
| OXPHOS complex steady-state levels | ND | ND | Normal | ND | ND | ND | ND | |
| Reference | Haack et al. 2015 [40] (continued) | |||||||
| Patient ID | F5; II:3 | F6, II:1 | F7, II:2 | F8, II:1 | F9, II:2 | F10, II:1 | ||
| Age at presentation | Birth | Birth | 2 years | 1 year | Birth | 11 months | ||
| Death | Alive at 2.3 years | Alive at 3 years | Alive at 5 years | Alive at 8 years | Alive at 16 years | Alive at 31 years | ||
| Parental consanguinity | Yes | No | No | No | No | No | ||
| Mutation 1 (genetic level; protein effect) | c.673T > C; p.Cys225Arg | c.98T > C; p.Phe33Ser | c.268G > A, p.Gly90Arg | c.161G > A; p.Arg54His | c.161G > A; p.Arg54His | c.229G > C; p.Glu77Gln | ||
| Mutation 2 (genetic level; protein effect) | c.673T > C; p.Cys225Arg | c.176A > G; p.Asn59Ser | c.583G > A; p.Gly195Ser | c.394G > A; p.Ala132Thr | c.431dup; p.Leu145Alafs*6 | c.476A > G; p.Gln159Arg | ||
| T2 hyperintensity | Yes | Yes | Yes | ND | Yes | Yes | ||
| Acylcarnitine profile | Normal | Normal | NL | NL | NL | NL | ||
| PDC activity | ND | ND | ND | ND | Normal | ND | ||
| OXPHOS activity | Normal | Reduced CIV in muscle | Normal | ND | Normal | Normal | ||
| OXPHOS complex steady-state levels | ND | ND | ND | ND | ND | ND | ||
| Reference | Ferdinandusse et al. 2015 [17] | Tetreault et al. 2015 [47] | ||||||
| Patient ID | Patient 1 | Patient 2 | Patient 3 | Patient 4 | P1 | P2 | P3 | P4 |
| Age at presentation | Birth | Birth | Early infancy | 1 year | 2.5 months | 2.9 years | 10 months | 6 months |
| Death | 24 h | 2 days | Alive at 7 years | 3 years | 10 months | Alive at 18 years | Alive at 13 years | Alive at 12 years |
| Parental consanguinity | Yes | Yes | No | No | No | No | No | No |
| Mutation 1 (genetic level; protein effect) | c.817A > G; p.Lys273Glu | c.817 > G; p.Lys273Glu | c.433C > T; p.Leu145Phe | c.673T > C; p.Cys225Arg | c.538A > G; p.Thr180Ala | c.538A > G; p.Thr180Ala | c.538A > G; p.Thr180Ala | c.538A > G; p.Thr180Ala |
| Mutation 2 (genetic level; protein effect) | c.817A > G; p.Lys273Glu | c.817A > G; p.Lys273Glu | c.476A > G; p.Gln159Arg | c.674G > C; p.Cys225Ser | c.583G > A; p.Gly195Ser | c.713C > T; p.Ala238Val | c.713C > T; p.Ala238Val | c.476A > G; p.Gln159Arg |
| T2 hyperintensity | ND | Yes | NL | Yes | Yes | Yes | Yes | Yes |
| Acylcarnitine profile | Normal | Normal | Normal | Normal | Normal | Normal | Normal | Normal |
| PDC activity | Reduced | Reduced | ND | ND | Reduced | Normal | ND | Normal |
| OXPHOS activity | Normal | Normal | ND | ND | Mild reduction of CI and CIII in muscle | Normal | Normal | Normal |
| OXPHOS complex steady-state levels | Normal | Normal | ND | ND | ND | ND | ND | Reduced CIV in fibroblasts |
| Reference | Yamada et al. 2015 [16] | Ganetzky et al. 2016 [50] | Olgiati et al. 2016 [42] | Nair et al. 2016 [46] | ||||
| Patient ID | III-2 | III-3 | Patient 1 | Patient 2 | II-1 | II-2 | Patient 1 | |
| Age at presentation | 10 months | 7 months | Prenatal | Prenatal | 3.5 years | 4.5 years | Birth | |
| Death | Alive at 7 years | 5 years | 16 h | 24 h | Alive at 17 years | Alive at 15 years | 24 h | |
| Parental consanguinity | No | No | No | No | No | No | Yes | |
| Mutation 1 (genetic level; protein effect) | c.176A > G; p.Asn59Ser | c.176A > G; p.Asn59Ser | c.8C > A; p.Ala3Asp | c.8C > A; p.Ala3Asp | c.232G > T; p.Glu78Ter | c.232G > T; p.Glu78Ter | c.842A > G; p.Glu281Gly | |
| Mutation 2 (genetic level; protein effect) | c.413C > T; p.Ala138Val | c.413C > T; p.Ala138Val | c.389T > A; p.Val130Asp | c.389T > A; p.Val130Asp | c.518C > T; p.Ala173Val | c.518C > T; p.Ala173Val | c.842A > G; p.Glu281Gly | |
| T2 hyperintensity | Yes | Yes | ND | ND | Yes | Yes | ND | |
| Acylcarnitine profile | Normal | Normal | Mild C4 elevation | Mild C4 elevation | ND | ND | Elevated C4 and C6 | |
| PDC activity | ND | ND | ND | ND | ND | ND | ND | |
| OXPHOS activity | Normal | ND | ND | ND | ND | ND | ND | |
| OXPHOS complex steady-state levels | ND | ND | ND | ND | ND | ND | ND | |
| Reference | Mahajan et al. 2017 [41] | Al Mutairi et al. 2017 [35] | Balasubramaniam et al. 2017 [43] | Bedoyan et al. 2017 [51] | Huffnagel et al. 2017 [52] | |||
| Patient ID | Patient 1 | Patient 1 | Patient 2 | Patient 1 | Patient 1 | Patient 1 | ||
| Age at presentation | 8 years | Birth | Birth | 17 months | Birth | 6 weeks | ||
| Death | Alive at 8 years | 2 days | 8 ho | Alive at 4.5 years | 39 days | Alive at 26 years | ||
| Parental consanguinity | No | Yes | Yes | No | No | No | ||
| Mutation 1 (genetic level; protein effect) | c.518C > T; p.Ala173Val | c.88 + 5G > A; p.Ala31Glufs*23 | c.88 + 5G > A; p.Ala31Glufs*23 | c.476A > G; p.Gln159Arg | c.836T > C; p.Phe279Ser | c.229G > C p.Glu77Gln | ||
| Mutation 2 (genetic level; protein effect) | c.817A > G; p.Lys273Glu | c.88 + 5G > A; p.Ala31Glufs*23 | c.88 + 5G > A; p.Ala31Glufs*23 | c.538A > G; p.Thr180Ala | c.8C > A; p.Ala3Asp | c.563C > T p.Ala188Val | ||
| T2 hyperintensity | Yes | ND | ND | Yes | Yes | Yes | ||
| Acylcarnitine profile | ND | Mild C3, C4, C5 and C10 elevation | Normal | ND | ND | Normal | ||
| PDC activity | ND | Reduced | Normal | ND | Reduced | ND | ||
| OXPHOS activity | ND | ND | Normal | ND | Reduced | Normal | ||
| OXPHOS complex steady-state levels | ND | ND | ND | ND | ND | ND | ||
| Reference | Ogawa et al. 2017 [53] | Fitzsimons et al. 2018 [48] | ||||||
| Patient ID | Pt376 | Pt536 | Pt1038 | Pt1135 | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
| Age at presentation | NL | NL | NL | NL | 5 months | 3 months | 5 months | 2 weeks |
| Death | NL | NL | NL | NL | 3 years | 21 months | 28 months | 13 months |
| Parental consanguinity | No | No | No | No | Yes | Yes | Yes | Yes |
| Mutation 1 (genetic level; protein effect) | c.98T > C; p.Phe33Ser | c.5C > T; p.Ala2Val | c.5C > T; p.Ala2Val | c.5C > T; p.Ala2Val | c.476A > G; p.Gln159Arg | c.538A > G; p.Thr180Ala | c.538A > G; p.Thr180Ala | c.538A > G; p.Thr180Ala |
| Mutation 2 (genetic level; protein effect) | c.176A > G; p.Asn59Ser | c.1A > G; p.Met1Val | c.176A > G; p.Asn59Ser | c.176A > G; p.Asn59Ser | c.476A > G; p.Gln159Arg | c.538A > G; p.Thr180Ala | c.538A > G; p.Thr180Ala | c.538A > G; p.Thr180Ala |
| T2 hyperintensity | ND | ND | ND | ND | Yes | Yes | Yes | Yes |
| Acylcarnitine profile | ND | ND | ND | ND | Normal | ND | Mild reduction of free carnitine and long-chain acylcarnitines | Normal |
| PDC activity | ND | ND | ND | ND | Reduced | ND | Normal | ND |
| OXPHOS activity | Reduced CIV | Normal | Normal (but reduced oxygen consumption rate) | Reduced CI | Normal | ND | Reduced CIII in muscle | ND |
| OXPHOS complex steady-state levels | ND | ND | ND | ND | ND | ND | ND | ND |
T2 hyperintensity refers to regions of high intensity on T2 weighted magnetic resonance imaging scans of the brain. OXPHOS complex steady-state levels were determined by blue native polyacrylamide gel electrophoresis. PDC, pyruvate dehydrogenase complex; OXPHOS, oxidative phosphorylation; ND, not determined; NL, not listed; CI, complex I; CIII, complex III; CIV, complex IV; CV, complex V; C3DC, malonylcarnitine; C4, butyrylcarnitine; C5DC, glutarylcarnitine; C6, hexanoylcarnitine; C10, decanoylcarnitine.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sharpe, A.J.; McKenzie, M. Mitochondrial Fatty Acid Oxidation Disorders Associated with Short-Chain Enoyl-CoA Hydratase (ECHS1) Deficiency. Cells 2018, 7, 46. https://0-doi-org.brum.beds.ac.uk/10.3390/cells7060046
AMA Style
Sharpe AJ, McKenzie M. Mitochondrial Fatty Acid Oxidation Disorders Associated with Short-Chain Enoyl-CoA Hydratase (ECHS1) Deficiency. Cells. 2018; 7(6):46. https://0-doi-org.brum.beds.ac.uk/10.3390/cells7060046
Chicago/Turabian StyleSharpe, Alice J., and Matthew McKenzie. 2018. "Mitochondrial Fatty Acid Oxidation Disorders Associated with Short-Chain Enoyl-CoA Hydratase (ECHS1) Deficiency" Cells 7, no. 6: 46. https://0-doi-org.brum.beds.ac.uk/10.3390/cells7060046
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.