Retinoids Issued from Hepatic Stellate Cell Lipid Droplet Loss as Potential Signaling Molecules Orchestrating a Multicellular Liver Injury Response

 , , ,
, , ,
Abstract
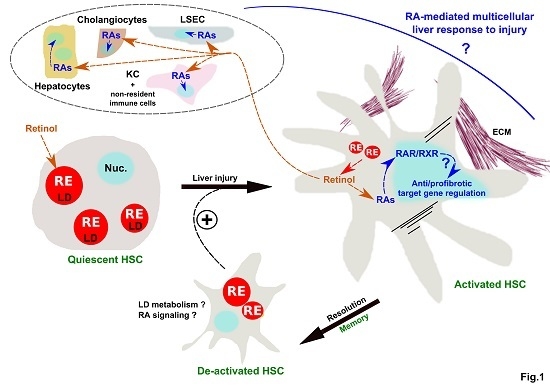
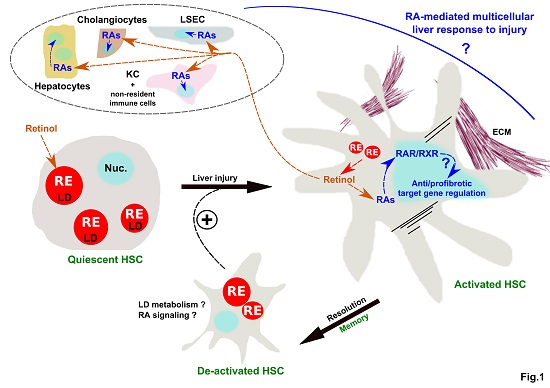
:
{kind=link}
{kind=link}
1. Introduction
2. Mechanisms of LD Loss and Requirement for HSC Activation
3. Control of HSC Activation through Retinoids Issued from LD Loss
4. Retinol Released by LD Loss as A Potential Cross-Talk Signal Between HSC and Other Hepatic Cell Populations in Liver (Patho) Physiology
5. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Asahina, K.; Tsai, S.Y.; Li, P.; Ishii, M.; Maxson, R.E.; Sucov, H.M.; Tsukamoto, H. Mesenchymal origin of hepatic stellate cells, submesothelial cells, and perivascular mesenchymal cells during mouse liver development. Hepatology 2009, 49, 998–1011. [Google Scholar] [CrossRef] [PubMed]
- Blaner, W.S.; O’Byrne, S.M.; Wongsiriroj, N.; Kluwe, J.; D’Ambrosio, D.M.; Jiang, H.; Schwabe, R.F.; Hillman, E.M.C.; Piantedosi, R.; Libien, J. Hepatic stellate cell lipid droplets: A specialized lipid droplet for retinoid storage. Biochim. Biophys. Acta 2009, 1791, 467–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Tuohetahuntila, M.; Molenaar, M.R.; Spee, B.; Brouwers, J.F.; Houweling, M.; Vaandrager, A.B.; Helms, J.B. ATGL and DGAT1 are involved in the turnover of newly synthesized triacylglycerols in hepatic stellate cells. J. Lipid Res. 2016, 57, 1162–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuohetahuntila, M.; Molenaar, M.R.; Spee, B.; Brouwers, J.F.; Wubbolts, R.; Houweling, M.; Yan, C.; Du, H.; VanderVen, B.C.; Vaandrager, A.B.; et al. Lysosome-mediated degradation of a distinct pool of lipid droplets during hepatic stellate cell activation. J. Biol. Chem. 2017, 292, 12436–12448. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, P.; Dongiovanni, P.; Motta, B.M.; Meroni, M.; Lepore, S.M.; Mancina, R.M.; Pelusi, S.; Russo, C.; Caddeo, A.; Rossi, G.; et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum. Mol. Genet. 2016, 25, 5212–5222. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, F.V.; Claudel, T.; Tardelli, M.; Caligiuri, A.; Stulnig, T.M.; Marra, F.; Trauner, M. The PNPLA3 I148M variant modulates the fibrogenic phenotype of human hepatic stellate cells. Hepatology 2017, 65, 1875–1890. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Li, S.; Wang, J.; Li, Y. In vitro inhibition of hepatic stellate cell activation by the autophagy-related lipid droplet protein ATG2A. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhao, S.; Yao, Z.; Wang, L.; Shao, J.; Chen, A.; Zhang, F.; Zheng, S. Autophagy regulates turnover of lipid droplets via ROS-dependent Rab25 activation in hepatic stellate cell. Redox Biol. 2017, 11, 322–334. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Gea, V.; Friedman, S.L. Autophagy fuels tissue fibrogenesis. Autophagy 2012, 8, 849–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jophlin, L.L.; Koutalos, Y.; Chen, C.; Shah, V.H.; Rockey, D.C. Hepatic stellate cells retain retinoid-laden lipid droplets after cellular transdifferentiation into activated myofibroblasts. Am. J. Physiol. Gastrointest. Liver Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Testerink, N.; Ajat, M.; Houweling, M.; Brouwers, J.F.; Pully, V.V.; van Manen, H.-J.; Otto, C.; Helms, J.B.; Vaandrager, A.B. Replacement of retinyl esters by polyunsaturated triacylglycerol species in lipid droplets of hepatic stellate cells during activation. PLoS ONE 2012, 7, e34945. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, M.R.; Vaandrager, A.B.; Helms, J.B. Some Lipid Droplets Are More Equal Than Others: Different Metabolic Lipid Droplet Pools in Hepatic Stellate Cells. Lipid Insights 2017, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Byrne, S.M.; Wongsiriroj, N.; Libien, J.; Vogel, S.; Goldberg, I.J.; Baehr, W.; Palczewski, K.; Blaner, W.S. Retinoid absorption and storage is impaired in mice lacking lecithin:retinol acyltransferase (LRAT). J. Biol. Chem. 2005, 280, 35647–35657. [Google Scholar] [CrossRef] [PubMed]
- Kluwe, J.; Wongsiriroj, N.; Troeger, J.S.; Gwak, G.-Y.; Dapito, D.H.; Pradere, J.-P.; Jiang, H.; Siddiqi, M.; Piantedosi, R.; O’Byrne, S.M.; et al. Absence of hepatic stellate cell retinoid lipid droplets does not enhance hepatic fibrosis but decreases hepatic carcinogenesis. Gut 2011, 60, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- O’Byrne, S.M.; Blaner, W.S. Retinol and retinyl esters: Biochemistry and physiology. J. Lipid Res. 2013, 54, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Saeed, A.; Dullaart, R.P.F.; Schreuder, T.C.M.A.; Blokzijl, H.; Faber, K.N. Disturbed Vitamin A Metabolism in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2017, 10. [Google Scholar] [CrossRef] [PubMed]
- Bitetto, D.; Bortolotti, N.; Falleti, E.; Vescovo, S.; Fabris, C.; Fattovich, G.; Cussigh, A.; Cmet, S.; Fornasiere, E.; Ceriani, E.; et al. A deficiency is associated with hepatitis C virus chronic infection and with unresponsiveness to interferon-based antiviral therapy. Hepatology 2013, 57, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Chaves, G.V.; Pereira, S.E.; Saboya, C.J.; Spitz, D.; Rodrigues, C.S.; Ramalho, A. Association between liver vitamin A reserves and severity of nonalcoholic fatty liver disease in the class III obese following bariatric surgery. Obes. Surg. 2014, 24, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-S.; Jeong, W.-I. Retinoic acids and hepatic stellate cells in liver disease. J. Gastroenterol. Hepatol. 2012, 27 (Suppl. S2), 75–79. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Kaji, T.; Shimono, R.; Hayashida, Y.; Matsufuji, H.; Tsuyama, S.; Maezono, R.; Kosai, K.; Takamatsu, H. Therapeutic effects of vitamin A on experimental cholestatic rats with hepatic fibrosis. Pediatr. Surg. Int. 2011, 27, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Hisamori, S.; Tabata, C.; Kadokawa, Y.; Okoshi, K.; Tabata, R.; Mori, A.; Nagayama, S.; Watanabe, G.; Kubo, H.; Sakai, Y. All-trans-retinoic acid ameliorates carbon tetrachloride-induced liver fibrosis in mice through modulating cytokine production. Liver Int. 2008, 28, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Cai, S.-Y.; Mennone, A.; Vig, P.; Boyer, J.L. Cenicriviroc, a cytokine receptor antagonist, potentiates all-trans retinoic acid in reducing liver injury in cholestatic rodents. Liver Int. 2018, 38, 1128–1138. [Google Scholar] [CrossRef] [PubMed]
- Hellemans, K.; Verbuyst, P.; Quartier, E.; Schuit, F.; Rombouts, K.; Chandraratna, R.A.S.; Schuppan, D.; Geerts, A. Differential modulation of rat hepatic stellate phenotype by natural and synthetic retinoids. Hepatology 2004, 39, 97–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fortuna, V.A.; Trugo, L.C.; Borojevic, R. Acyl-CoA: Retinol acyltransferase (ARAT) and lecithin:retinol acyltransferase (LRAT) activation during the lipocyte phenotype induction in hepatic stellate cells. J. Nutr. Biochem. 2001, 12, 610–621. [Google Scholar] [CrossRef]
- Yi, H.-S.; Lee, Y.-S.; Byun, J.-S.; Seo, W.; Jeong, J.-M.; Park, O.; Duester, G.; Haseba, T.; Kim, S.C.; Park, K.-G.; et al. Alcohol dehydrogenase III exacerbates liver fibrosis by enhancing stellate cell activation and suppressing natural killer cells in mice. Hepatology 2014, 60, 1044–1053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Troeger, J.S.; Mederacke, I.; Gwak, G.-Y.; Dapito, D.H.; Mu, X.; Hsu, C.C.; Pradere, J.-P.; Friedman, R.A.; Schwabe, R.F. Deactivation of hepatic stellate cells during liver fibrosis resolution in mice. Gastroenterology 2012, 143, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Cong, M.; Paik, Y.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, M.B. Hepatic stellate cells: Fibrogenic, regenerative or both? Heterogeneity and context are key. Hepatol. Int. 2016, 10, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, V.; Harris, E.N.; Kidambi, S. SECs (Sinusoidal Endothelial Cells), Liver Microenvironment, and Fibrosis. Biomed. Res. Int. 2017, 2017, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.J.; An, D.; Cai, Y.; Repa, J.J.; Hung-Po Chen, T.; Flores, M.; Postic, C.; Magnuson, M.A.; Chen, J.; Chien, K.R.; et al. Hepatocyte-specific mutation establishes retinoid X receptor alpha as a heterodimeric integrator of multiple physiological processes in the liver. Mol. Cell. Biol. 2000, 20, 4436–4444. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Tsuei, J.; Wan, Y.-J.Y. Biological functional annotation of retinoic acid alpha and beta in mouse liver based on genome-wide binding. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 307, G205–G218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boergesen, M.; Pedersen, T.Å.; Gross, B.; van Heeringen, S.J.; Hagenbeek, D.; Bindesbøll, C.; Caron, S.; Lalloyer, F.; Steffensen, K.R.; Nebb, H.I.; et al. Genome-wide profiling of liver X. receptor, retinoid X. receptor, and peroxisome proliferator-activated receptor α in mouse liver reveals extensive sharing of binding sites. Mol. Cell. Biol. 2012, 32, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Dubois-Chevalier, J.; Dubois, V.; Dehondt, H.; Mazrooei, P.; Mazuy, C.; Sérandour, A.A.; Gheeraert, C.; Guillaume, P.; Baugé, E.; Derudas, B.; et al. The logic of transcriptional regulator recruitment architecture at cis-regulatory modules controlling liver functions. Genome Res. 2017, 27, 985–996. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.-X.; Hu, Y.; Wan, Y.-J.Y. Microbiota and bile acid profiles in retinoic acid-primed mice that exhibit accelerated liver regeneration. Oncotarget 2016, 7, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Shmarakov, I.O.; Jiang, H.; Yang, K.J.Z.; Goldberg, I.J.; Blaner, W.S. Hepatic retinoid stores are required for normal liver regeneration. J. Lipid Res. 2013, 54, 893–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, T.; Jiang, M.; Kastner, P.; Chambon, P.; Metzger, D. Selective ablation of retinoid X receptor alpha in hepatocytes impairs their lifespan and regenerative capacity. Proc. Natl. Acad. Sci. USA. 2001, 98, 4581–4586. [Google Scholar] [CrossRef] [PubMed]
- Ohata, M.; Yamauchi, M.; Takeda, K.; Toda, G.; Kamimura, S.; Motomura, K.; Xiong, S.; Tsukamoto, H. RAR and RXR expression by Kupffer cells. Exp. Mol. Pathol. 2000, 68, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sanchez, E.; Firrincieli, D.; Housset, C.; Chignard, N. Expression patterns of nuclear receptors in parenchymal and non-parenchymal mouse liver cells and their modulation in cholestasis. Biochim. Biophys. Acta 2017, 1863, 1699–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Kruijt, J.K.; van der Sluis, R.J.; Van Berkel, T.J.C.; Hoekstra, M. Nuclear receptor atlas of female mouse liver parenchymal, endothelial, and Kupffer cells. Physiol. Genom. 2013, 45, 268–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchimura, K.; Nakamuta, M.; Enjoji, M.; Irie, T.; Sugimoto, R.; Muta, T.; Iwamoto, H.; Nawata, H. Activation of retinoic X receptor and peroxisome proliferator-activated receptor-gamma inhibits nitric oxide and tumor necrosis factor-alpha production in rat Kupffer cells. Hepatology 2001, 33, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Marzioni, M.; Saccomanno, S.; Agostinelli, L.; Rychlicki, C.; De Minicis, S.; Pierantonelli, I.; Trauner, M.; Fickert, P.; Müller, T.; Shanmukhappa, K.; et al. PDX-1/Hes-1 interactions determine cholangiocyte proliferative response to injury in rodents: Possible implications for sclerosing cholangitis. J. Hepatol. 2013, 58, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Neumann, K.; Kruse, N.; Szilagyi, B.; Erben, U.; Rudolph, C.; Flach, A.; Zeitz, M.; Hamann, A.; Klugewitz, K. Connecting liver and gut: Murine liver sinusoidal endothelium induces gut tropism of CD4+ T cells via retinoic acid. Hepatology 2012, 55, 1976–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiskirchen, R.; Tacke, F. Cellular and molecular functions of hepatic stellate cells in inflammatory responses and liver immunology. Hepatobiliary Surg. Nutr. 2014, 3, 344–363. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, S.; Mucida, D.; Tyznik, A.J.; Kronenberg, M.; Cheroutre, H. Hepatic stellate cells function as regulatory bystanders. J. Immunol. 2011, 186, 5549–5555. [Google Scholar] [CrossRef] [PubMed]
- D’Ambrosio, D.N.; Walewski, J.L.; Clugston, R.D.; Berk, P.D.; Rippe, R.A.; Blaner, W.S. Distinct populations of hepatic stellate cells in the mouse liver have different capacities for retinoid and lipid storage. PLoS ONE 2011, 6, e24993. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bobowski-Gerard, M.; Zummo, F.P.; Staels, B.; Lefebvre, P.; Eeckhoute, J. Retinoids Issued from Hepatic Stellate Cell Lipid Droplet Loss as Potential Signaling Molecules Orchestrating a Multicellular Liver Injury Response. Cells 2018, 7, 137. https://0-doi-org.brum.beds.ac.uk/10.3390/cells7090137
Bobowski-Gerard M, Zummo FP, Staels B, Lefebvre P, Eeckhoute J. Retinoids Issued from Hepatic Stellate Cell Lipid Droplet Loss as Potential Signaling Molecules Orchestrating a Multicellular Liver Injury Response. Cells. 2018; 7(9):137. https://0-doi-org.brum.beds.ac.uk/10.3390/cells7090137
Chicago/Turabian StyleBobowski-Gerard, Marie, Francesco Paolo Zummo, Bart Staels, Philippe Lefebvre, and Jérôme Eeckhoute. 2018. "Retinoids Issued from Hepatic Stellate Cell Lipid Droplet Loss as Potential Signaling Molecules Orchestrating a Multicellular Liver Injury Response" Cells 7, no. 9: 137. https://0-doi-org.brum.beds.ac.uk/10.3390/cells7090137