Role and Therapeutic Targeting of the PI3K/Akt/mTOR Signaling Pathway in Skin Cancer: A Review of Current Status and Future Trends on Natural and Synthetic Agents Therapy

 , , ,
, , ,
Abstract
:1. Introduction
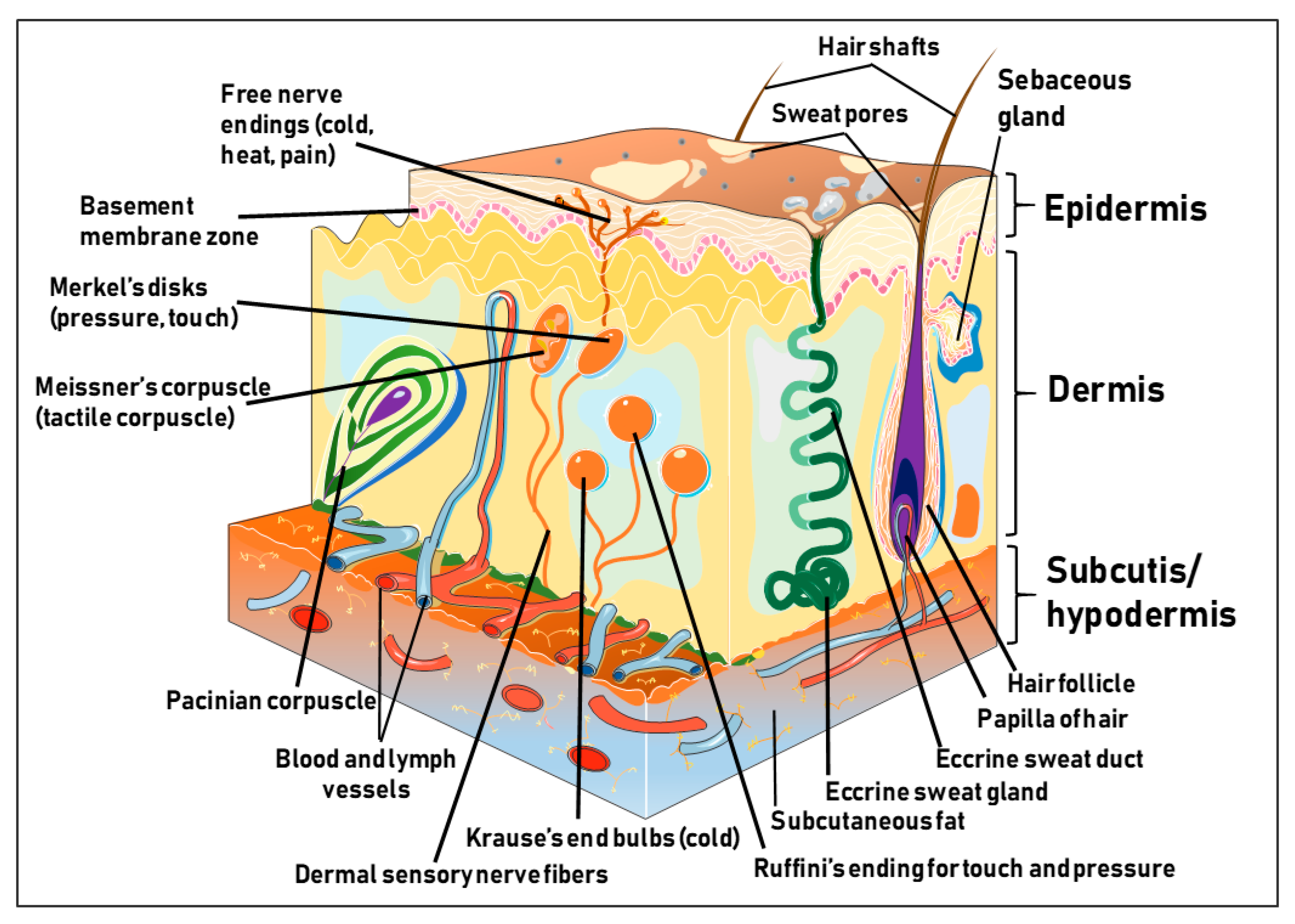
1.1. Structure and Function of the Human Skin
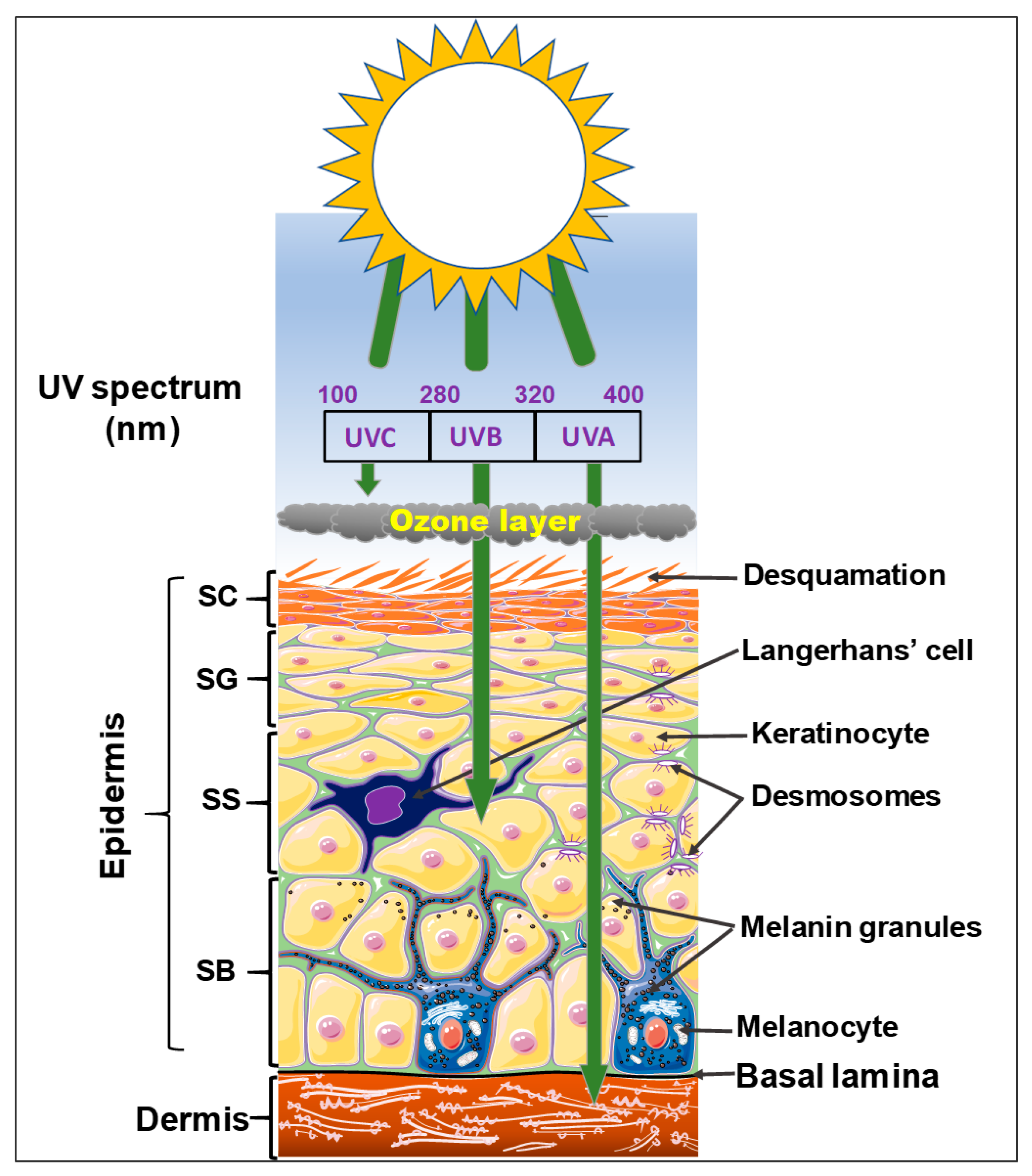
1.2. The Epidermis
1.3. The Dermis
1.4. The Hypodermis
2. Risk Factors Associated with Cutaneous Carcinogenesis
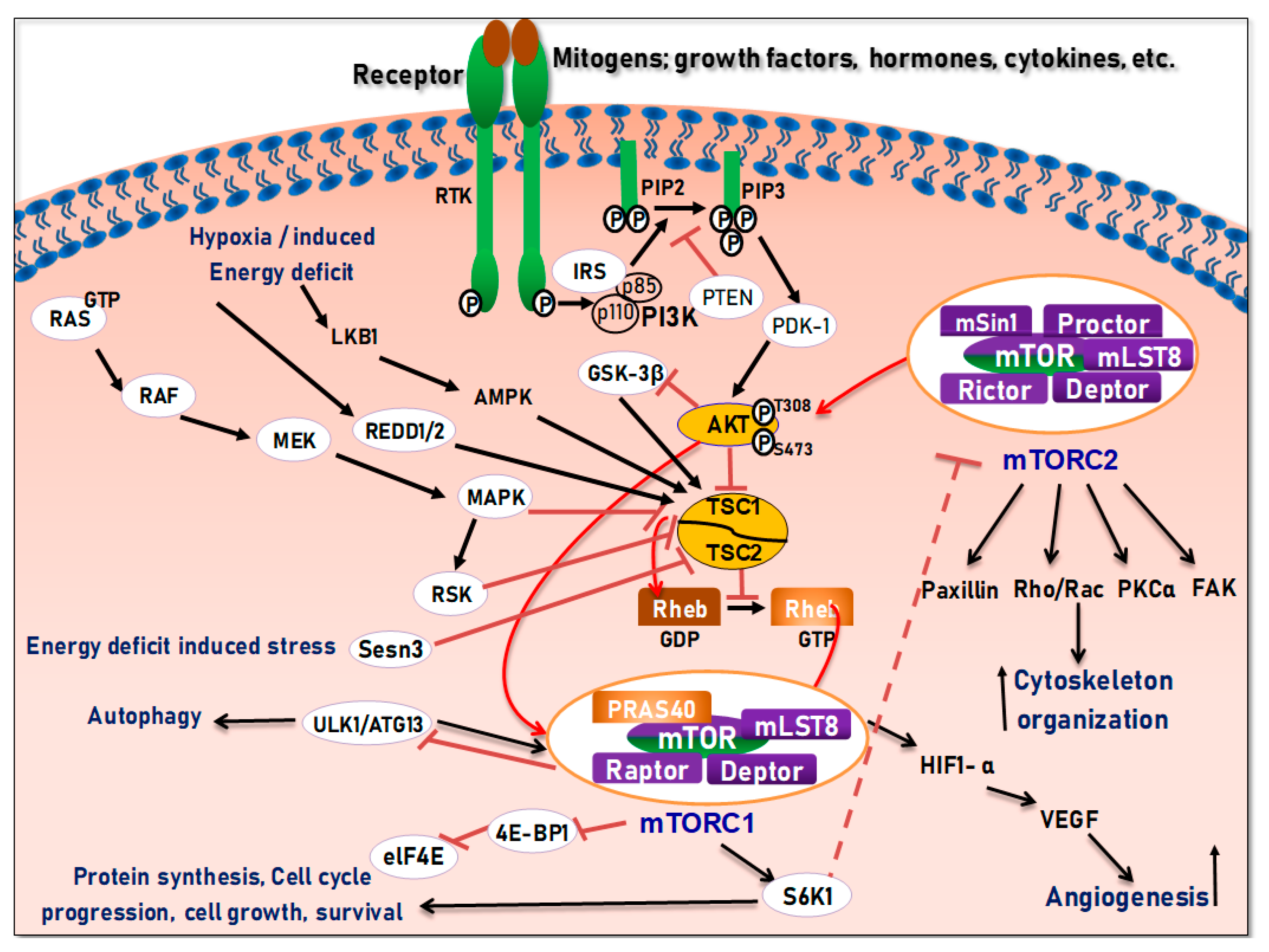
3. The PI3K/Akt/Mtor Signaling and Interrelations in Tissue Development
4. Structure and Function of the mTOR Pathway
5. Regulation of the PI3K/Akt/mTOR Pathways in Development and Carcinogenesis
6. Cutaneous Cancers Associated with Dysregulation of the PI3K/Akt/mTOR Pathways
6.1. Role of the PI3K/Akt/mTOR and their Targeting in Melanoma Skin Cancer
6.2. Targeting PI3K/Akt/mTOR and Associated Pathways with Chemotherapeutics, Biologic Drugs, Natural Products, and Synthetic Derivatives in Melanoma
6.2.1. Chemotherapeutic Small Molecules and Biologic Drugs
6.2.2. Natural Plant-Derived Extracts, and Phytochemicals and their Synthetic Derivatives
6.3. Targeting PI3K/Akt/mTOR for Treatment of Basal Cell Carcinoma
6.4. Targeting PI3K/Akt/mTOR for Treatment of Cutaneous Squamous Cell Carcinoma
6.5. Targeting PI3K/Akt/mTOR for Treatment of Merkel Cell Carcinoma
6.6. Targeting PI3K/Akt/mTOR for Treatment of Tuberous Sclerosis
7. Clinical Implications, Conclusions, and Future Prospects
8. Materials and Methods
Funding
Conflicts of Interest
Abbreviations
| PI3K | phosphatidyl-inositiol 3-kinase |
| BMZ | Bbaement membrane zone |
| mTOR | mammalian target of rapamycin |
| UV | ultraviolet |
| EGCG | Epigallocatechin-3-gallate |
| Akt | protein kinase B |
| NMSC | non-melanoma skin cancer |
| SCC | squamous cell carcinoma |
| BCC | basal cell carcinoma |
| MCC | Merkel cell carcinoma |
| K6 | keratin 6 |
| K14 | keratin 14 |
| EMT | epithelial-mesenchymal transition |
| CK15 | cytokeratin 15 |
| CK5 | cytokeratin |
References
- Chamcheu, J.C.; Adhami, V.M.; Siddiqui, I.V.; Mukhtar, H. Cutaneous Cell-and Gene-Based Therapies for Inherited and Acquired Skin Disorders. In Gene and Cell Therapy: Therapeutic Mechanisms and Strategies, 4th ed.; Templeton, N.S., Ed.; CRC Press, Taylor and Francis Group: Boca Raton, FL, USA, 2015. [Google Scholar]
- Gonzales, K.A.U.; Fuchs, E. Skin and Its Regenerative Powers: An Alliance between Stem Cells and Their Niche. Dev. Cell 2017, 43, 387–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magin, T.M.; Vijayaraj, P.; Leube, R.E. Structural and regulatory functions of keratins. Exp. Cell Res. 2007, 313, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Elias, P.M.; Eichenfield, L.F.; Fowler, J.F.; Horowitz, P.; McLeod, R.P. Update on the structure and function of the skin barrier: Atopic dermatitis as an exemplar of clinical implications. Semin. Cutan. Med. Surg. 2013, 32, S21–S24. [Google Scholar] [CrossRef] [PubMed]
- Pullar, J.M.; Carr, A.C.; Vissers, M.C.M. The Roles of Vitamin C in Skin Health. Nutrients 2017, 9, 866. [Google Scholar] [CrossRef] [PubMed]
- Segre, J.A. Epidermal barrier formation and recovery in skin disorders. J. Clin. Investig. 2006, 116, 1150–1158. [Google Scholar] [CrossRef]
- Chamcheu, J.C.; Siddiqui, I.A.; Syed, D.N.; Adhami, V.M.; Liovic, M.; Mukhtar, H. Keratin gene mutations in disorders of human skin and its appendages. Arch. Biochem. Biophys. 2011, 508, 123–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmets, C.A.; Katiyar, S. Green tea and skin cancer: Photoimmunology, angiogenesis and DNA repair. J. Nutr. Biochem. 2007, 18, 287–296. [Google Scholar]
- Eckhart, L.; Lippens, S.; Tschachler, E.; Declercq, W. Cell death by cornification. Biochim. Biophys. Acta 2013, 1833, 3471–3480. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Y.; Yang, X.; Wei, J.; Zhou, S.; Zhao, Z.; Cheng, J.; Duan, H.; Jia, T.; Lei, Q.; et al. Characterization of Th17 and FoxP3+ Treg Cells in Paediatric Psoriasis Patients. Scand. J. Immunol. 2016, 83, 174–180. [Google Scholar] [CrossRef]
- Ng, W.L.; Qi, J.T.Z.; Yeong, W.Y.; Naing, M.W. Proof-of-concept: 3D bioprinting of pigmented human skin constructs. Biofabrication 2018, 10, 25005. [Google Scholar] [CrossRef]
- Tharmarajah, G.; Eckhard, U.; Jain, F.; Marino, G.; Prudova, A.; Urtatiz, O.; Fuchs, H.; De Angelis, M.H.; Overall, C.M.; Van Raamsdonk, C.D. Melanocyte development in the mouse tail epidermis requires the Adamts9 metalloproteinase. Pigment. Cell Melanoma Res. 2018, 31, 693–707. [Google Scholar] [CrossRef]
- Serre, C.; Busuttil, V.; Botto, J.-M. Intrinsic and extrinsic regulation of human skin melanogenesis and pigmentation. Int. J. Cosmet. Sci. 2018, 40, 328–347. [Google Scholar] [CrossRef] [Green Version]
- Bertolesi, G.E.; McFarlane, S. Seeing the light to change colour: An evolutionary perspective on the role of melanopsin in neuroendocrine circuits regulating light-mediated skin pigmentation. Pigment. Cell Melanoma Res. 2018, 31, 354–373. [Google Scholar] [CrossRef] [Green Version]
- De Assis, L.V.M.; Moraes, M.N.; Magalhaes-Marques, K.K.; de Lauro Castrucci, A.M. Melanopsin and rhodopsin mediate UVA-induced immediate pigment darkening: Unravelling the photosensitive system of the skin. Eur. J. Cell Biol. 2018, 97, 150–162. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Hearing, V.J. Physiological factors that regulate skin pigmentation. BioFactors 2009, 35, 193–199. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Morita, A.; Maeda, A.; Hearing, V.J. Regulation of skin pigmentation and thickness by dickkopf 1 (DKK1). J. Investig. Dermatol. Symp. Proc. 2009, 14, 73–75. [Google Scholar] [CrossRef]
- Perez-Sanchez, A.; Barrajon-Catalan, E.; Herranz-Lopez, M.; Micol, V. Nutraceuticals for Skin Care: A Comprehensive Review of Human Clinical Studies. Nutrients 2018, 10, 403. [Google Scholar] [CrossRef]
- Sagi, Z.; Hieronymus, T. The Impact of the Epithelial–Mesenchymal Transition Regulator Hepatocyte Growth Factor Receptor/Met on Skin Immunity by Modulating Langerhans Cell Migration. Front. Immunol. 2018, 9, 517. [Google Scholar] [CrossRef]
- Deckers, J.; Hammad, H.; Hoste, E. Langerhans Cells: Sensing the Environment in Health and Disease. Front. Immunol. 2018, 9, 93. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, K.; Nümm, T.J.; Koch, S.; Herrmann, N.; Leib, N.; Bieber, T.; Stroisch, T.J. Langerhans and inflammatory dendritic epidermal cells in atopic dermatitis are tolerized toward TLR2 activation. Allergy 2018, 73, 2205–2213. [Google Scholar] [CrossRef]
- Petersson, M.; Niemann, C. Stem cell dynamics and heterogeneity: Implications for epidermal regeneration and skin cancer. Curr. Med. Chem. 2012, 19, 5984–5992. [Google Scholar] [CrossRef]
- Wang, L.-J.; Wang, Y.-L.; Yang, X. Progress in epidermal stem cells. Yi Chuan 2010, 32, 198–204. [Google Scholar] [CrossRef]
- Flores, E.R.; Halder, G. Stem cell proliferation in the skin: Alpha-catenin takes over the hippo pathway. Sci. Signal. 2011, 4, pe34. [Google Scholar] [CrossRef]
- Uzarska, M.; Porowińska, D.; Bajek, A.; Drewa, T. Epidermal stem cells—Biology and potential applications in regenerative medicine. Postępy Biochem. 2013, 59, 219–227. [Google Scholar]
- Shen, Q.; Jin, H.; Wang, X. Epidermal Stem Cells and Their Epigenetic Regulation. Int. J. Mol. Sci. 2013, 14, 17861–17880. [Google Scholar] [CrossRef] [Green Version]
- Lavker, R.M.; Sun, T.-T. Epidermal stem cells: Properties, markers, and location. Proc. Natl. Acad. Sci. USA 2000, 97, 13473–13475. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Amagai, M. Dissecting the formation, structure and barrier function of the stratum corneum. Int. Immunol. 2015, 27, 269–280. [Google Scholar] [CrossRef] [Green Version]
- Osawa, R.; Akiyama, M.; Shimizu, H. Filaggrin Gene Defects and the Risk of Developing Allergic Disorders. Allergol. Int. 2011, 60, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Sandilands, A.; Sutherland, C.; Irvine, A.D.; McLean, W.H.I. Filaggrin in the frontline: Role in skin barrier function and disease. J. Cell Sci. 2009, 122, 1285–1294. [Google Scholar] [CrossRef]
- Koster, M.I. Making an epidermis. Ann. N. Y. Acad. Sci. 2009, 1170, 7–10. [Google Scholar] [CrossRef]
- Labat-Robert, J. Information exchanges between cells and extracellular matrix. Influence of aging. Biol. Aujourd’hui 2012, 206, 103–109. [Google Scholar] [CrossRef]
- Quan, T.; Fisher, G.J. Role of Age-Associated Alterations of the Dermal Extracellular Matrix Microenvironment in Human Skin Aging: A Mini-Review. Gerontology 2015, 61, 427–434. [Google Scholar] [CrossRef]
- Watt, F.M. Mammalian skin cell biology: At the interface between laboratory and clinic. Science 2014, 346, 937–940. [Google Scholar] [CrossRef]
- Gilchrest, B.A.; Geller, A.C.; Yaar, M.; Eller, M.S. The Pathogenesis of Melanoma Induced by Ultraviolet Radiation. N. Engl. J. Med. 1999, 340, 1341–1348. [Google Scholar] [CrossRef]
- Watson, M.; Holman, D.M.; Maguire-Eisen, M. Ultraviolet Radiation Exposure and Its Impact on Skin Cancer Risk. Semin. Oncol. Nurs. 2016, 32, 241–254. [Google Scholar] [CrossRef] [Green Version]
- Miyamura, Y.; Coelho, S.G.; Schlenz, K.; Batzer, J.; Smuda, C.; Choi, W.; Brenner, M.; Passeron, T.; Zhang, G.; Kolbe, L.; et al. The deceptive nature of UVA tanning versus the modest protective effects of UVB tanning on human skin. Pigment Cell Melanoma Res. 2011, 24, 136–147. [Google Scholar] [CrossRef]
- D’Orazio, J.; Jarrett, S.; Amaro-Ortiz, A.; Scott, T. UV radiation and the skin. Int. J. Mol. Sci. 2013, 14, 12222–12248. [Google Scholar] [CrossRef]
- McKenzie, R.L.; Aucamp, P.J.; Madronich, S.; Tourpali, K.; Bais, A.; Bernhard, G.; Ilyas, M. Ozone depletion and climate change: Impacts on UV radiation. Photochem. Photobiol. Sci. 2015, 14, 19–52. [Google Scholar] [CrossRef]
- Holick, M.F. Sunlight, UV-radiation, vitamin D and skin cancer: How much sunlight do we need? Adv. Exp. Med. Biol. 2008, 624, 1–15. [Google Scholar]
- Juzeniene, A.; Moan, J. Beneficial effects of UV radiation other than via vitamin D production. Dermato-Endocrinology 2012, 4, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Gupta, A.; Avci, P.; Dai, T.; Huang, Y.-Y.; Hamblin, M.R. Ultraviolet Radiation in Wound Care: Sterilization and Stimulation. Adv. Wound Care 2013, 2, 422–437. [Google Scholar] [CrossRef] [Green Version]
- Slominski, A.; Wortsman, J. Neuroendocrinology of the skin. Endocr. Rev. 2000, 21, 457–487. [Google Scholar] [CrossRef]
- Diffey, B.L. Solar ultraviolet radiation effects on biological systems. Phys. Med. Boil. 1991, 36, 299–328. [Google Scholar] [CrossRef]
- Abeyama, K.; Eng, W.; Jester, J.V.; Vink, A.A.; Edelbaum, D.; Cockerell, C.J.; Bergstresser, P.R.; Takashima, A. A role for NF-kappaB-dependent gene transactivation in sunburn. J. Clin. Investig. 2000, 105, 1751–1759. [Google Scholar] [CrossRef]
- Leo, M.S.; Sivamani, R.K. Phytochemical modulation of the Akt/mTOR pathway and its potential use in cutaneous disease. Arch. Dermatol. Res. 2014, 306, 861–871. [Google Scholar] [CrossRef]
- Huang, S. Targeting mTOR signaling for cancer therapy. Curr. Opin. Pharmacol. 2003, 3, 371–377. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR Signaling in Growth and Metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Guertin, D.A.; Sabatini, D.M. Defining the Role of mTOR in Cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Vanhaesebroeck, B.; Stephens, L.; Hawkins, P. PI3K signalling: The path to discovery and understanding. Nat. Rev. Mol. Cell Boil. 2012, 13, 195–203. [Google Scholar] [CrossRef]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR Signalling Pathway in Human Cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Sehgal, S.N.; Baker, H.; Vezina, C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J. Antibiot. 1975, 28, 727–732. [Google Scholar] [CrossRef]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994, 78, 35–43. [Google Scholar] [CrossRef]
- Buerger, C. Epidermal mTORC1 Signaling Contributes to the Pathogenesis of Psoriasis and Could Serve as a Therapeutic Target. Front. Immunol. 2018, 9, 2786. [Google Scholar] [CrossRef] [Green Version]
- Vahidnezhad, H.; Youssefian, L.; Uitto, J.; Youssefian, L. Molecular Genetics of the PI3K-AKT-mTOR Pathway in Genodermatoses: Diagnostic Implications and Treatment Opportunities. J. Investig. Dermatol. 2016, 136, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Huang, T.; Lin, X.; Meng, X.; Lin, M. Phosphoinositide-3 Kinase/Protein Kinase-B/Mammalian Target of Rapamycin Pathway in Psoriasis Pathogenesis. A Potential Therapeutic Target? Acta Derm. Venereol. 2014, 94, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Kezić, A.; Popovic, L.; Lalic, K. mTOR Inhibitor Therapy and Metabolic Consequences: Where Do We Stand? Oxidative Med. Cell. Longev. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Corti, F.; Nichetti, F.; Raimondi, A.; Niger, M.; Prinzi, N.; Torchio, M.; Tamborini, E.; Perrone, F.; Pruneri, G.; Di Bartolomeo, M.; et al. Targeting the PI3K/AKT/mTOR pathway in biliary tract cancers: A review of current evidences and future perspectives. Cancer Treat. Rev. 2019, 72, 45–55. [Google Scholar] [CrossRef]
- Cragg, G.M.; Newman, D.J. Natural Products: A Continuing Source of Novel Drug Leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [Green Version]
- Strickland, L.R.; Pal, H.C.; Elmets, C.A.; Afaq, F. Targeting Drivers of Melanoma with Synthetic Small Molecules and Phytochemicals. Cancer Lett. 2015, 359, 20–35. [Google Scholar] [CrossRef]
- Cronin, K.A.; Lake, A.J.; Scott, S.; Sherman, R.L.; Noone, A.-M.; Howlader, N.; Henley, S.J.; Anderson, R.N.; Firth, A.U.; Ma, J.; et al. Annual Report to the Nation on the Status of Cancer, part I: National cancer statistics. Cancer 2018, 124, 2785–2800. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Wu, J.; Qin, H.; Xu, J. The Role of Autophagy in the Resistance to BRAF Inhibition in BRAF-Mutated Melanoma. Target. Oncol. 2018, 13, 437–446. [Google Scholar] [CrossRef]
- Wahid, M.; Jawed, A.; Mandal, R.K.; Dar, S.A.; Akhter, N.; Somvanshi, P.; Khan, F.; Lohani, M.; Areeshi, M.Y.; Haque, S. Recent developments and obstacles in the treatment of melanoma with BRAF and MEK inhibitors. Crit. Rev. Oncol. 2018, 125, 84–88. [Google Scholar] [CrossRef]
- Shao, Z.; Bao, Q.; Jiang, F.; Qian, H.; Fang, Q.; Hu, X. VS-5584, a Novel PI3K-mTOR Dual Inhibitor, Inhibits Melanoma Cell Growth In Vitro and In Vivo. PLoS ONE 2015, 10, e0132655. [Google Scholar] [CrossRef]
- Syed, D.N.; Chamcheu, J.-C.; Khan, M.I.; Sechi, M.; Lall, R.K.; Adhami, V.M.; Mukhtar, H. Fisetin inhibits human melanoma cell growth through direct binding to p70S6K and mTOR: Findings from 3-D melanoma skin equivalents and computational modeling. Biochem. Pharmacol. 2014, 89, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Bosbous, M.W.; Dzwierzynski, W.W.; Neuburg, M. Lentigo Maligna: Diagnosis and Treatment. Clin. Plast. Surg. 2010, 37, 35–46. [Google Scholar] [CrossRef]
- Broussard, L.; Howland, A.; Ryu, S.; Song, K.; Norris, D.; Armstrong, C.A.; Song, P.I. Melanoma Cell Death Mechanisms. Chonnam Med. J. 2018, 54, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Hersey, P.; Bastholt, L.; Chiarion-Sileni, V.; Cinat, G.; Dummer, R.; Eggermont, A.M.M.; Espinosa, E.; Hauschild, A.; Quirt, I.; Robert, C.; et al. Small molecules and targeted therapies in distant metastatic disease. Ann. Oncol. 2009, 20, vi35–vi40. [Google Scholar] [CrossRef]
- Yu, Y. A novel combination treatment against melanoma with NRAS mutation and therapy resistance. EMBO Mol. Med. 2018, 10, e8573. [Google Scholar] [CrossRef]
- Caenepeel, S.; Cooke, K.; Wadsworth, S.; Huang, G.; Robert, L.; Moreno, B.H.; Parisi, G.; Cajulis, E.; Kendall, R.; Beltran, P.; et al. MAPK pathway inhibition induces MET and GAB1 levels, priming BRAF mutant melanoma for rescue by hepatocyte growth factor. Oncotarget 2017, 8, 17795–17809. [Google Scholar] [CrossRef]
- Guo, B.; Zhang, Q.; Wang, H.; Chang, P.; Tao, K. KCNQ1OT1 promotes melanoma growth and metastasis. Aging 2018, 10, 632–644. [Google Scholar] [CrossRef] [Green Version]
- Luan, W.; Li, L.; Shi, Y.; Bu, X.; Xia, Y.; Wang, J.; Djangmah, H.S.; Liu, X.; You, Y.; Xu, B. Long non-coding RNA MALAT1 acts as a competing endogenous RNA to promote malignant melanoma growth and metastasis by sponging miR-22. Oncotarget 2016, 7, 63901–63912. [Google Scholar] [CrossRef] [Green Version]
- Luan, W.; Li, R.; Liu, L.; Ni, X.; Shi, Y.; Xia, Y.; Wang, J.; Lu, F.; Xu, B. Long non-coding RNA HOTAIR acts as a competing endogenous RNA to promote malignant melanoma progression by sponging miR-152-3p. Oncotarget 2017, 8, 85401–85414. [Google Scholar] [CrossRef] [Green Version]
- Karbowniczek, M.; Spittle, C.S.; Morrison, T.; Wu, H.; Henske, E.P. mTOR Is Activated in the Majority of Malignant Melanomas. J. Investig. Dermatol. 2008, 128, 980–987. [Google Scholar] [CrossRef] [Green Version]
- Molhoek, K.R.; Brautigan, D.L.; Slingluff, C.L. Synergistic inhibition of human melanoma proliferation by combination treatment with B-Raf inhibitor BAY43-9006 and mTOR inhibitor Rapamycin. J. Transl. Med. 2005, 3, 39. [Google Scholar] [CrossRef]
- Vera Aguilera, J.; Rao, R.D.; Allred, J.B.; Suman, V.J.; Windschitl, H.E.; Kaur, J.S.; Maples, W.J.; Lowe, V.J.; Creagan, E.T.; Erickson, L.A.; et al. Phase II Study of Everolimus in Metastatic Malignant Melanoma (NCCTG-N0377, Alliance). Oncologist 2018, 23, 887-e94. [Google Scholar] [CrossRef]
- Niessner, H.; Kosnopfel, C.; Sinnberg, T.; Beck, D.; Krieg, K.; Wanke, I.; Lasithiotakis, K.; Bonin, M.; Garbe, C.; Meier, F. Combined activity of temozolomide and the mTOR inhibitor temsirolimus in metastatic melanoma involves DKK1. Exp. Dermatol. 2017, 26, 598–606. [Google Scholar] [CrossRef]
- Rangwala, R.; Chang, Y.C.; Hu, J.; Algazy, K.M.; Evans, T.L.; Fecher, L.A.; Schuchter, L.M.; Torigian, D.A.; Panosian, J.T.; Troxel, A.B.; et al. Combined MTOR and autophagy inhibition: Phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014, 10, 1391–1402. [Google Scholar] [CrossRef]
- Hainsworth, J.D.; Infante, J.R.; Spigel, D.R.; Peyton, J.D.; Thompson, D.S.; Lane, C.M.; Clark, B.L.; Rubin, M.S.; Trent, D.F.; Burris, H.A., III. Bevacizumab and everolimus in the treatment of patients with metastatic melanoma: A phase 2 trial of the Sarah Cannon Oncology Research Consortium. Cancer 2010, 116, 4122–4129. [Google Scholar] [CrossRef]
- Kolev, V.N.; Wright, Q.G.; Vidal, C.M.; Ring, J.E.; Shapiro, I.M.; Ricono, J.; Weaver, D.T.; Padval, M.V.; Pachter, J.A.; Xu, Q. PI3K/mTOR dual inhibitor VS-5584 preferentially targets cancer stem cells. Cancer Res. 2015, 75, 446–455. [Google Scholar] [CrossRef]
- Hutchinson, L. Skin cancer. Golden age of melanoma therapy. Nat. Rev. Clin. Oncol. 2015, 12, 1. [Google Scholar] [CrossRef]
- Webster, R.M.; Mentzer, S.E. The malignant melanoma landscape. Nat. Rev. Drug Discov. Engl. 2014, 13, 491–492. [Google Scholar] [CrossRef]
- Schadendorf, D.; Hauschild, A. Melanoma in 2013: Melanoma—The run of success continues. Nat. Rev. Clin. Oncol. 2014, 11, 75–76. [Google Scholar] [CrossRef]
- Mi, C.; Ma, J.; Shi, H.; Li, J.; Wang, F.; Lee, J.J.; Jin, X. 4′,6-dihydroxy-4-methoxyisoaurone inhibits the HIF-1alpha pathway through inhibition of Akt/mTOR/p70S6K/4E-BP1 phosphorylation. J. Pharmacol. Sci. 2014, 125, 193–201. [Google Scholar] [CrossRef]
- Wang, J.; Yang, Z.; Wen, J.; Ma, F.; Wang, F.; Yu, K.; Tang, M.; Wu, W.; Dong, Y.; Cheng, X.; et al. SKLB-M8 induces apoptosis through the AKT/mTOR signaling pathway in melanoma models and inhibits angiogenesis with decrease of ERK1/2 phosphorylation. J. Pharmacol. Sci. 2014, 126, 198–207. [Google Scholar] [CrossRef]
- Oudart, J.-B.; Doue, M.; Vautrin, A.; Brassart, B.; Sellier, C.; Dupont-Deshorgue, A.; Monboisse, J.C.; Maquart, F.X.; Brassart-Pasco, S.; Ramont, L. The anti-tumor NC1 domain of collagen XIX inhibits the FAK/PI3K/Akt/mTOR signaling pathway through alphavbeta3 integrin interaction. Oncotarget 2016, 7, 1516–1528. [Google Scholar] [CrossRef]
- Lee, B.Y.; Timpson, P.; Horvath, L.G.; Daly, R.J. FAK signaling in human cancer as a target for therapeutics. Pharmacol. Ther. 2015, 146, 132–149. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: Mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef]
- Si, L.; Xu, X.; Kong, Y.; Flaherty, K.T.; Chi, Z.; Cui, C.; Sheng, X.; Li, S.; Dai, J.; Yu, W.; et al. Major Response to Everolimus in Melanoma With Acquired Imatinib Resistance. J. Clin. Oncol. 2012, 30, e37–e40. [Google Scholar] [CrossRef]
- Pierard-Franchimont, C.; Hermanns-Lê, T.; Paquet, P.; Herfs, M.; Delvenne, P.; Piérard, G.E. Hedgehog-and mTOR-targeted therapies for advanced basal cell carcinomas. Future Oncol. 2015, 11, 2997–3002. [Google Scholar] [CrossRef]
- Kapoor, V.; Zaharieva, M.M.; Das, S.N.; Berger, M.R. Erufosine simultaneously induces apoptosis and autophagy by modulating the Akt–mTOR signaling pathway in oral squamous cell carcinoma. Cancer Lett. 2012, 319, 39–48. [Google Scholar] [CrossRef]
- Ding, L.-T.; Zhao, P.; Yang, M.-L.; Lv, G.-Z.; Zhao, T.-L. GDC-0084 inhibits cutaneous squamous cell carcinoma cell growth. Biochem. Biophys. Res. Commun. 2018, 503, 1941–1948. [Google Scholar] [CrossRef]
- Nguyen, N.; Sharma, A.; Nguyen, N.; Sharma, A.K.; Desai, D.; Huh, S.J.; Amin, S.; Meyers, C.; Robertson, G.P. Melanoma chemoprevention in skin reconstructs and mouse xenografts using isoselenocyanate-4. Cancer Prev. Res. 2011, 4, 248–258. [Google Scholar] [CrossRef]
- Kannan, A.; Lin, Z.; Shao, Q.; Zhao, S.; Fang, B.; Moreno, M.A.; Vural, E.; Stack, B.C., Jr.; Suen, J.Y.; Kannan, K.; et al. Dual mTOR inhibitor MLN0128 suppresses Merkel cell carcinoma (MCC) xenograft tumor growth. Oncotarget 2016, 7, 6576–6592. [Google Scholar] [CrossRef]
- Cassler, N.M.; Merrill, D.; Bichakjian, C.K.; Brownell, I. Merkel Cell Carcinoma Therapeutic Update. Curr. Treat. Options Oncol. 2016, 17, 36. [Google Scholar] [CrossRef]
- Villani, A.; Fabbrocini, G.; Costa, C.; Annunziata, M.C.; Scalvenzi, M. Merkel Cell Carcinoma: Therapeutic Update and Emerging Therapies. Dermatol. Ther. 2019, 9, 209–222. [Google Scholar] [CrossRef]
- Calero, R.; Morchon, E.; Martinez-Argudo, I.; Serrano, R. Synergistic anti-tumor effect of 17AAG with the PI3K/mTOR inhibitor NVP-BEZ235 on human melanoma. Cancer Lett. 2017, 406, 1–11. [Google Scholar] [CrossRef]
- Lin, Z.; Mei, H.; Fan, J.; Yin, Z.; Wu, G. Effect of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 against human Merkel cell carcinoma MKL-1 cells. Oncol. Lett. 2015, 10, 3663–3667. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.-Y.; Madhunapantula, S.V.; Desai, D.; Amin, S.; Robertson, G.P. Melanoma prevention using topical PBISe. Cancer Prev. Res. 2011, 4, 935–948. [Google Scholar] [CrossRef]
- Hou, G.; Xue, L.; Lu, Z.; Fan, T.; Tian, F.; Xue, Y. An activated mTOR/p70S6K signaling pathway in esophageal squamous cell carcinoma cell lines and inhibition of the pathway by rapamycin and siRNA against mTOR. Cancer Lett. 2007, 253, 236–248. [Google Scholar] [CrossRef]
- Werzowa, J.; Koehrer, S.; Strommer, S.; Cejka, D.; Fuereder, T.; Zebedin, E.; Wacheck, V. Vertical Inhibition of the mTORC1/mTORC2/PI3K Pathway Shows Synergistic Effects against Melanoma In Vitro and In Vivo. J. Investig. Dermatol. 2011, 131, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Chong, K.; Daud, A.; Ortiz-Urda, S.; Arron, S.T. Cutting Edge in Medical Management of Cutaneous Oncology. Semin. Cutan. Med. Surg. 2012, 31, 140–149. [Google Scholar] [CrossRef] [Green Version]
- So, P.-L.; Wang, G.Y.; Wang, K.; Chuang, M.; Chiueh, V.C.; Kenny, P.A.; Epstein, E.H. PI3K-AKT signaling is a downstream effector of retinoid prevention of murine basal cell carcinogenesis. Cancer Prev. Res. 2014, 7, 407–417. [Google Scholar] [CrossRef]
- Wu, C.-S.; Chen, G.-S.; Lin, P.-Y.; Pan, I.-H.; Wang, S.-T.; Lin, S.H.; Yu, H.-S.; Lin, C.-C. Tazarotene Induces Apoptosis in Human Basal Cell Carcinoma via Activation of Caspase-8/t-Bid and the Reactive Oxygen Species-Dependent Mitochondrial Pathway. DNA Cell Boil. 2014, 33, 652–666. [Google Scholar] [CrossRef] [Green Version]
- Velho, T.R. Metastatic melanoma—A review of current and future drugs. Drugs Context 2012, 2012, 1–17. [Google Scholar] [CrossRef]
- Lin, Z.; McDermott, A.; Shao, L.; Kannan, A.; Morgan, M.; Stack, B.C., Jr.; Moreno, M.; Davis, D.A.; Cornelius, L.A.; Gao, L. Chronic mTOR activation promotes cell survival in Merkel cell carcinoma. Cancer Lett. 2014, 344, 272–281. [Google Scholar] [CrossRef]
- Liang, G.; Liu, M.; Wang, Q.; Shen, Y.; Mei, H.; Li, D.; Liu, W. Itraconazole exerts its anti-melanoma effect by suppressing Hedgehog, Wnt, and PI3K/mTOR signaling pathways. Oncotarget 2017, 8, 28510–28525. [Google Scholar] [CrossRef] [Green Version]
- Head, S.A.; Shi, W.; Zhao, L.; Gorshkov, K.; Pasunooti, K.; Chen, Y.; Deng, Z.; Li, R.-J.; Shim, J.S.; Tan, W.; et al. Antifungal drug itraconazole targets VDAC1 to modulate the AMPK/mTOR signaling axis in endothelial cells. Proc. Natl. Acad. Sci. USA 2015, 112, E7276–E7285. [Google Scholar] [CrossRef]
- Zou, Y.; Ge, M.; Wang, X. Targeting PI3K-AKT-mTOR by LY3023414 inhibits human skin squamous cell carcinoma cell growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 2017, 490, 385–392. [Google Scholar] [CrossRef]
- Sweetlove, M.; Wrightson, E.; Kolekar, S.; Rewcastle, G.W.; Baguley, B.C.; Shepherd, P.R.; Jamieson, S.M.F. Inhibitors of pan-PI3K Signaling Synergize with BRAF or MEK Inhibitors to Prevent BRAF-Mutant Melanoma Cell Growth. Front. Oncol. 2015, 5, 135. [Google Scholar] [CrossRef] [Green Version]
- Polini, B.; Carpi, S.; Romanini, A.; Breschi, M.C.; Nieri, P.; Podestà, A. Circulating cell-free microRNAs in cutaneous melanoma staging and recurrence or survival prognosis. Pigment. Cell Melanoma Res. 2019, 32, 486–499. [Google Scholar] [CrossRef]
- Li, J.; Liu, X.; Li, C.; Wang, W. miR-224-5p inhibits proliferation, migration, and invasion by targeting PIK3R3/AKT3 in uveal melanoma. J. Cell. Biochem. 2019, 120, 12412–12421. [Google Scholar] [CrossRef]
- Jiang, Q.-Q.; Liu, W.-B. miR-25 Promotes Melanoma Progression by regulating RNA binding motif protein 47. Med. Sci. 2018, 34, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Meng, F.; Zhang, Y.; Li, X.; Wang, J. Clinical significance of miR-138 in patients with malignant melanoma through targeting of PDK1 in the PI3K/AKT autophagy signaling pathway. Oncol. Rep. 2017, 38, 1655–1662. [Google Scholar] [CrossRef]
- Micevic, G.; Muthusamy, V.; Damsky, W.; Theodosakis, N.; Liu, X.; Meeth, K.; Wingrove, E.; Santhanakrishnan, M.; Bosenberg, M. DNMT3b modulates melanoma growth by controlling levels of mTORC2 component RICTOR. Cell Rep. 2016, 14, 2180–2192. [Google Scholar] [CrossRef]
- Schmidt, K.M.; Dietrich, P.; Hackl, C.; Guenzle, J.; Bronsert, P.; Wagner, C.; Fichtner-Feigl, S.; Schlitt, H.J.; Geissler, E.K.; Hellerbrand, C.; et al. Inhibition of mTORC2/RICTOR Impairs Melanoma Hepatic Metastasis12. Neoplasia 2018, 20, 1198–1208. [Google Scholar] [CrossRef]
- Damsky, W.; Micevic, G.; Meeth, K.; Muthusamy, V.; Curley, D.P.; Santhankrishnan, M.; Erdélyi, I.; Platt, J.T.; Huang, L.; Theodosakis, N.; et al. mTORC1 activation blocks BrafV600E-induced growth-arrest, but is insufficient for melanoma formation. Cancer Cell 2015, 27, 41–56. [Google Scholar] [CrossRef]
- Ciołczyk-Wierzbicka, D.; Gil, D.; Laidler, P. Treatment of melanoma with selected inhibitors of signaling kinases effectively reduces proliferation and induces expression of cell cycle inhibitors. Med. Oncol. 2017, 35, 7. [Google Scholar] [CrossRef]
- Sikora, A.G.; Gelbard, A.; Davies, M.A.; Sano, D.; Ekmekcioglu, S.; Kwon, J.; Hailemichael, Y.; Jayaraman, P.; Myers, J.N.; Grimm, E.A.; et al. Targeted inhibition of inducible nitric oxide synthase inhibits growth of human melanoma in vivo and synergizes with chemotherapy. Clin. Cancer Res. 2010, 16, 1834–1844. [Google Scholar] [CrossRef]
- Ernst, D.S.; Eisenhauer, E.; Wainman, N.; Davis, M.; Lohmann, R.; Baetz, T.; Bélanger, K.; Smylie, M. Phase II Study of Perifosine in Previously Untreated Patients with Metastatic Melanoma. Investig. New Drugs 2005, 23, 569–575. [Google Scholar] [CrossRef]
- Jung, S.K.; Kim, J.E.; Lee, S.-Y.; Lee, M.H.; Byun, S.; Kim, Y.A.; Lim, T.G.; Reddy, K.; Huang, Z.; Bode, A.M.; et al. The P110 subunit of PI3-K is a therapeutic target of acacetin in skin cancer. Carcinogenesis 2014, 35, 123–130. [Google Scholar] [CrossRef]
- Chien, S.-T.; Lin, S.-S.; Wang, C.-K.; Lee, Y.-B.; Chen, K.-S.; Fong, Y.; Shih, Y.W. Acacetin inhibits the invasion and migration of human non-small cell lung cancer A549 cells by suppressing the p38alpha MAPK signaling pathway. Mol. Cell. Biochem. 2011, 350, 135–148. [Google Scholar] [CrossRef]
- Shin, D.-H.; Kim, O.-H.; Jun, H.-S.; Kang, M.-K. Inhibitory effect of capsaicin on B16-F10 melanoma cell migration via the phosphatidylinositol 3-kinase/Akt/Rac1 signal pathway. Exp. Mol. Med. 2008, 40, 486–494. [Google Scholar] [CrossRef]
- Wang, C.; Li, S.; Wang, M. Evodiamine-induced human melanoma A375-S2 cell death was mediated by PI3K/Akt/caspase and Fas-L/NF-kappaB signaling pathways and augmented by ubiquitin-proteasome inhibition. Toxicol. In Vitro 2010, 24, 898–904. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Li, D.-F.; Han, J.-C.; Wang, B.; Dong, Z.-P.; Yu, L.-N.; Pan, Z.H.; Qu, C.J.; Chen, Y.; Sun, S.G.; et al. Reprogramming induced by isoliquiritigenin diminishes melanoma cachexia through mTORC2-AKT-GSK3beta signaling. Oncotarget 2017, 8, 34565–34575. [Google Scholar]
- Lim, H.N.; Baek, S.B.; Jung, H.J. Bee Venom and Its Peptide Component Melittin Suppress Growth and Migration of Melanoma Cells via Inhibition of PI3K/AKT/mTOR and MAPK Pathways. Molecules 2019, 24, 929. [Google Scholar] [CrossRef]
- Lai, S.-L.; Mustafa, M.R.; Wong, P.-F.; Mr, M. Panduratin A induces protective autophagy in melanoma via the AMPK and mTOR pathway. Phytomedicine 2018, 42, 144–151. [Google Scholar] [CrossRef]
- Zou, N.; Wei, Y.; Li, F.; Yang, Y.; Cheng, X.; Wang, C. The inhibitory effects of compound Muniziqi granule against B16 cells and harmine induced autophagy and apoptosis by inhibiting Akt/mTOR pathway. BMC Complement. Altern. Med. 2017, 17, 517. [Google Scholar] [CrossRef]
- Sun, Z.; Zheng, L.; Liu, X.; Xing, W.; Liu, X. Sinomenine inhibits the growth of melanoma by enhancement of autophagy via PI3K/AKT/mTOR inhibition. Drug Des. Dev. Ther. 2018, 12, 2413–2421. [Google Scholar] [CrossRef]
- Espona-Fiedler, M.; Soto-Cerrato, V.; Hosseini, A.; Lizcano, J.M.; Guallar, V.; Quesada, R.; Gao, T.; Pérez-Tomás, R. Identification of dual mTORC1 and mTORC2 inhibitors in melanoma cells: Prodigiosin vs. obatoclax. Biochem. Pharmacol. 2012, 83, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Liu-Smith, F.; Meyskens, F. Molecular mechanisms of flavonoids in melanin synthesis and the potential for the prevention and treatment of melanoma. Mol. Nutr. Food Res. 2016, 60, 1264–1274. [Google Scholar] [CrossRef] [Green Version]
- Pal, H.C.; Hunt, K.M.; Diamond, A.; Elmets, C.A.; Afaq, F. Phytochemicals for the Management of Melanoma. Mini-Rev. Med. Chem. 2016, 16, 953–979. [Google Scholar]
- Adhami, V.M.; Syed, D.N.; Khan, N.; Mukhtar, H. Dietary flavonoid fisetin: A novel dual inhibitor of PI3K/Akt and mTOR for prostate cancer management. Biochem. Pharmacol. 2012, 84, 1277–1281. [Google Scholar] [CrossRef] [Green Version]
- Syed, D.N.; Mukhtar, H. Botanicals for the prevention and treatment of cutaneous melanoma. Pigment. Cell Melanoma Res. 2011, 24, 688–702. [Google Scholar] [CrossRef] [Green Version]
- Govindarajan, B.; Sligh, J.E.; Vincent, B.J.; Li, M.; Canter, J.A.; Nickoloff, B.J.; Rodenburg, R.J.; Smeitink, J.A.; Oberley, L.; Zhang, Y.; et al. Overexpression of Akt converts radial growth melanoma to vertical growth melanoma. J. Clin. Investig. 2007, 117, 719–729. [Google Scholar] [CrossRef] [Green Version]
- Pearce, L.R.; Alton, G.R.; Richter, D.T.; Kath, J.C.; Lingardo, L.; Chapman, J.; Hwang, C.; Alessi, D.R. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem. J. 2010, 431, 245–255. [Google Scholar] [CrossRef] [Green Version]
- Sechi, M.; Lall, R.K.; Afolabi, S.O.; Singh, A.; Joshi, D.C.; Chiu, S.-Y.; Mukhtar, H.; Syed, D.N. Fisetin targets YB-1/RSK axis independent of its effect on ERK signaling: Insights from in vitro and in vivo melanoma models. Sci. Rep. 2018, 8, 15726. [Google Scholar] [CrossRef]
- Pal, H.C.; Baxter, R.D.; Hunt, K.M.; Agarwal, J.; Elmets, C.A.; Athar, M.; Afaq, F. Fisetin, a phytochemical, potentiates sorafenib-induced apoptosis and abrogates tumor growth in athymic nude mice implanted with BRAF-mutated melanoma cells. Oncotarget 2015, 6, 28296–28311. [Google Scholar] [CrossRef]
- Zhao, G.; Han, X.; Zheng, S.; Li, Z.; Sha, Y.; Ni, J.; Sun, Z.; Qiao, S.; Song, Z. Curcumin induces autophagy, inhibits proliferation and invasion by downregulating AKT/mTOR signaling pathway in human melanoma cells. Oncol. Rep. 2016, 35, 1065–1074. [Google Scholar] [CrossRef]
- Rozzo, C.; Fanciulli, M.; Fraumene, C.; Corrias, A.; Cubeddu, T.; Sassu, I.; Cossu, S.; Nieddu, V.; Galleri, G.; Azara, E.; et al. Molecular changes induced by the curcumin analogue D6 in human melanoma cells. Mol. Cancer 2013, 12, 37. [Google Scholar] [CrossRef]
- Wang, M.; Yu, T.; Zhu, C.; Sun, H.; Qiu, Y.; Zhu, X.; Li, J. Resveratrol Triggers Protective Autophagy Through the Ceramide/Akt/mTOR Pathway in Melanoma B16 Cells. Nutr. Cancer 2014, 66, 435–440. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Darjatmoko, S.R.; Polans, A.S. Resveratrol modulates the malignant properties of cutaneous melanoma through changes in the activation and attenuation of the antiapoptotic protooncogenic protein Akt/PKB. Melanoma Res. 2011, 21, 180–187. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, B.B.; Bhardwaj, A.; Aggarwal, R.S.; Seeram, N.P.; Shishodia, S.; Takada, Y. Role of resveratrol in prevention and therapy of cancer: Preclinical and clinical studies. Anticancer Res. 2004, 24, 2783–2840. [Google Scholar]
- Kaushik, G.; Ramalingam, S.; Subramaniam, D.; Rangarajan, P.; Protti, P.; Rammamoorthy, P.; Anant, S.; Mammen, J.M.V. Honokiol induces cytotoxic and cytostatic effects in malignant melanoma cancer cells. Am. J. Surg. 2012, 204, 868–873. [Google Scholar] [CrossRef] [Green Version]
- Prieto, J.M.; Alqathama, A. Natural products with therapeutic potential in melanoma metastasis. Nat. Prod. Rep. 2015, 32, 1170–1182. [Google Scholar]
- Hambright, H.G.; Meng, P.; Kumar, A.P.; Ghosh, R. Inhibition of PI3K/AKT/mTOR axis disrupts oxidative stress-mediated survival of melanoma cells. Oncotarget 2015, 6, 7195–7208. [Google Scholar] [CrossRef]
- Gong, J.; Munoz, A.R.; Chan, D.; Ghosh, R.; Kumar, A.P. STAT3 down regulates LC3 to inhibit autophagy and pancreatic cancer cell growth. Oncotarget 2014, 5, 2529–2541. [Google Scholar] [CrossRef]
- Huang, K.-J.; Kuo, C.-H.; Chen, S.-H.; Lin, C.-Y.; Lee, Y.-R. Honokiol inhibits in vitro and in vivo growth of oral squamous cell carcinoma through induction of apoptosis, cell cycle arrest and autophagy. J. Cell. Mol. Med. 2018, 22, 1894–1908. [Google Scholar] [CrossRef] [Green Version]
- Surdu, S. Non-melanoma skin cancer: Occupational risk from UV light and arsenic exposure. Rev. Environ. Health 2014, 29, 255–264. [Google Scholar] [CrossRef]
- Burton, K.A.; Ashack, K.A.; Khachemoune, A. Cutaneous Squamous Cell Carcinoma: A Review of High-Risk and Metastatic Disease. Am. J. Clin. Dermatol. 2016, 17, 491–508. [Google Scholar] [CrossRef]
- Cohen, B.J.; Cohen, E.S.; Cohen, P.R. Basal Cell Carcinoma: A Patient and Physician’s Experience. Dermatol. Ther. 2018, 8, 329–337. [Google Scholar] [CrossRef]
- Wong, C.S.M.; Strange, R.C.; Lear, J.T. Basal cell carcinoma. BMJ 2003, 327, 794–798. [Google Scholar] [CrossRef]
- Marzuka, A.G.; Book, S.E. Basal Cell Carcinoma: Pathogenesis, Epidemiology, Clinical Features, Diagnosis, Histopathology, and Management. Yale J. Boil. Med. 2015, 88, 167–179. [Google Scholar]
- Rogers, H.W.; Weinstock, M.A.; Feldman, S.R.; Coldiron, B.M. Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the U.S. Population, 2012. JAMA Dermatol. 2015, 151, 1081–1086. [Google Scholar] [CrossRef]
- Eisemann, N.; Waldmann, A.; Geller, A.C.; Weinstock, M.A.; Volkmer, B.; Greinert, R.; Breitbart, E.W.; Katalinic, A. Non-Melanoma Skin Cancer Incidence and Impact of Skin Cancer Screening on Incidence. J. Investig. Dermatol. 2014, 134, 43–50. [Google Scholar] [CrossRef] [Green Version]
- Guy, G.P.; Thomas, C.C.; Thompson, T.; Watson, M.; Massetti, G.M.; Richardson, L.C. Vital Signs: Melanoma Incidence and Mortality Trends and Projections—United States, 1982–2030. MMWR. Morb. Mortal. Wkly. Rep. 2015, 64, 591–596. [Google Scholar]
- Rigel, D.S. Cutaneous ultraviolet exposure and its relationship to the development of skin cancer. J. Am. Acad. Dermatol. 2008, 58, S129–S132. [Google Scholar] [CrossRef]
- Tarallo, M.; Cigna, E.; Frati, R.; Delfino, S.; Innocenzi, D.; Fama, U.; Corbianco, A.; Scuderi, N. Metatypical basal cell carcinoma: A clinical review. J. Exp. Clin. Cancer Res. 2008, 27, 65. [Google Scholar] [CrossRef]
- Athar, M.; Li, C.; Kim, A.L.; Spiegelman, V.S.; Bickers, D.R. Sonic Hedgehog Signaling in Basal Cell Nevus Syndrome. Cancer Res. 2014, 74, 4967–4975. [Google Scholar] [CrossRef] [Green Version]
- Noubissi, F.K.; Kim, T.; Kawahara, T.N.; Aughenbaugh, W.D.; Berg, E.; Longley, B.J.; Athar, M.; Spiegelman, V.S. Role of CRD-BP in the growth of human Basal Cell Carcinoma Cells. J. Investig. Dermatol. 2014, 134, 1718–1724. [Google Scholar] [CrossRef]
- Chamcheu, J.C.; Rady, I.; Chamcheu, R.-C.N.; Siddique, A.B.; Bloch, M.B.; Mbeumi, S.B.; Babatunde, A.S.; Uddin, M.B.; Noubissi, F.K.; Jurutka, P.W.; et al. Graviola (Annona muricata) Exerts Anti-Proliferative, Anti-Clonogenic and Pro-Apoptotic Effects in Human Non-Melanoma Skin Cancer UW-BCC1 and A431 Cells In Vitro: Involvement of Hedgehog Signaling. Int. J. Mol. Sci. 2018, 19, 1791. [Google Scholar] [CrossRef]
- Kasper, M.; Jaks, V.; Hohl, D.; Toftgård, R. Basal cell carcinoma—Molecular biology and potential new therapies. J. Clin. Investig. 2012, 122, 455–463. [Google Scholar] [CrossRef]
- Bakshi, A.; Chaudhary, S.C.; Rana, M.; Elmets, C.A.; Athar, M. Basal cell carcinoma pathogenesis and therapy involving hedgehog signaling and beyond. Mol. Carcinog. 2017, 56, 2543–2557. [Google Scholar] [CrossRef]
- Alter, M.; Hillen, U.; Leiter, U.; Sachse, M.; Gutzmer, R. Current diagnosis and treatment of basal cell carcinoma. J. Dtsch. Dermatol. Ges. 2015, 13, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Tsubamoto, H.; Ueda, T.; Inoue, K.; Sakata, K.; Shibahara, H.; Sonoda, T. Repurposing itraconazole as an anticancer agent. Oncol. Lett. 2017, 14, 1240–1246. [Google Scholar] [CrossRef]
- Alam, M.; Ratner, D. Cutaneous squamous-cell carcinoma. N. Engl. J. Med. 2001, 344, 975–983. [Google Scholar] [CrossRef]
- Lansbury, L.; Bath-Hextall, F.; Perkins, W.; Stanton, W.; Leonardi-Bee, J. Interventions for non-metastatic squamous cell carcinoma of the skin: Systematic review and pooled analysis of observational studies. BMJ 2013, 347, f6153. [Google Scholar] [CrossRef]
- Agamia, N.F.; Abdallah, D.M.; Sorour, O.; Mourad, B.; Younan, D.N. Skin expression of mammalian target of rapamycin and forkhead box transcription factor O1, and serum insulin-like growth factor-1 in patients with acne vulgaris and their relationship with diet. Br. J. Dermatol. 2016, 174, 1299–1307. [Google Scholar] [CrossRef]
- Gurney, B.; Newlands, C. Management of regional metastatic disease in head and neck cutaneous malignancy.1. Cutaneous squamous cell carcinoma. Br. J. Oral Maxillofac. Surg. 2014, 52, 294–300. [Google Scholar] [CrossRef]
- Azimi, A.; Kaufman, K.L.; Ali, M.; Arthur, J.; Kossard, S.; Fernandez-Penas, P. Differential proteomic analysis of actinic keratosis, Bowen’s disease and cutaneous squamous cell carcinoma by label-free LC–MS/MS. J. Dermatol. Sci. 2018, 91, 69–78. [Google Scholar] [CrossRef]
- Voß, M.; Plasmeijer, E.I.; Van Bemmel, B.C.; Van Der Bij, W.; Klaver, N.S.; Erasmus, M.E.; De Bock, G.H.; Verschuuren, E.A.; Rácz, E. Azathioprine to mycophenolate mofetil transition and risk of squamous cell carcinoma after lung transplantation. J. Hear. Lung Transplant. 2018, 37, 853–859. [Google Scholar] [CrossRef] [Green Version]
- Lewis, C.M.; Glisson, B.S.; Feng, L.; Wan, F.; Tang, X.; Wistuba, I.I.; El-Naggar, A.K.; Rosenthal, D.I.; Chambers, M.S.; Lustig, R.A.; et al. A Phase II Study of Gefitinib for Aggressive Cutaneous Squamous Cell Carcinoma of the Head and Neck. Clin. Cancer Res. 2012, 18, 1435–1446. [Google Scholar] [CrossRef] [Green Version]
- Karayannopoulou, G.; Euvrard, S.; Kanitakis, J. Differential expression of p-mTOR in cutaneous basal and squamous cell carcinomas likely explains their different response to mTOR inhibitors in organ-transplant recipients. Anticancer Res. 2013, 33, 3711–3714. [Google Scholar]
- Wu, N.; Du, Z.; Zhu, Y.; Song, Y.; Pang, L.; Chen, Z. The Expression and Prognostic Impact of the PI3K/AKT/mTOR Signaling Pathway in Advanced Esophageal Squamous Cell Carcinoma. Technol. Cancer Res. Treat. 2018, 17, 1533033818758772. [Google Scholar] [CrossRef]
- Euvrard, S.; Claudy, A.; Kanitakis, J. Skin Cancers after Organ Transplantation. N. Engl. J. Med. 2003, 348, 1681–1691. [Google Scholar] [CrossRef]
- Chen, S.-J.; Nakahara, T.; Takahara, M.; Kido, M.; Dugu, L.; Uchi, H.; Takeuchi, S.; Tu, Y.-T.; Moroi, Y.; Furue, M. Activation of the mammalian target of rapamycin signalling pathway in epidermal tumours and its correlation with cyclin-dependent kinase 2. Br. J. Dermatol. 2009, 160, 442–445. [Google Scholar] [CrossRef]
- Monaco, A.P. The Role of mTOR Inhibitors in the Management of Posttransplant Malignancy. Transplant. 2009, 87, 157–163. [Google Scholar] [CrossRef]
- Einspahr, J.G.; Calvert, V.; Alberts, D.S.; Curiel-Lewandrowski, C.; Warneke, J.; Krouse, R.; Stratton, S.P.; Liotta, L.; Longo, C.; Pellicani, G.; et al. Functional protein pathway activation mapping of the progression of normal skin to squamous cell carcinoma. Cancer Prev. Res. 2012, 5, 403–413. [Google Scholar] [CrossRef]
- Massarelli, E.; Lin, H.; Ginsberg, L.E.; Tran, H.T.; Lee, J.J.; Canales, J.R.; Williams, M.D.; Blumenschein, G.R.; Lu, C.; Heymach, J.V.; et al. Phase II trial of everolimus and erlotinib in patients with platinum-resistant recurrent and/or metastatic head and neck squamous cell carcinoma. Ann. Oncol. 2015, 26, 1476–1480. [Google Scholar] [CrossRef]
- Schrama, D.; Ugurel, S.; Becker, J.C. Merkel cell carcinoma: Recent insights and new treatment options. Curr. Opin. Oncol. 2012, 24, 141–149. [Google Scholar] [CrossRef]
- Shuda, M.; Kwun, H.J.; Feng, H.; Chang, Y.; Moore, P.S. Human Merkel cell polyomavirus small T antigen is an oncoprotein targeting the 4E-BP1 translation regulator. J. Clin. Investig. 2011, 121, 3623–3634. [Google Scholar] [CrossRef]
- Fahmy, M.D.; Gupta, A.; Padilla, R.J.; Segura, A.; Brookes, C.D. Desmoplastic fibroma associated with tuberous sclerosis: Case report and literature review. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 2019, 128, e92–e99. [Google Scholar] [CrossRef]
- Ekure, E.N.; Addissie, Y.A.; Sokunbi, O.J.; Kruszka, P.; Muenke, M.; Adeyemo, A.A. Tuberous sclerosis in a patient from Nigeria. Am. J. Med. Genet. Part A 2019, 179, 1423–1425. [Google Scholar] [CrossRef]
- Curatolo, P.; Bombardieri, R.; Jozwiak, S. Tuberous sclerosis. Lancet 2008, 372, 657–668. [Google Scholar] [CrossRef]
- Islam, M.P.; Roach, E.S. Tuberous sclerosis complex. In Handbook of Clinical Neurology; Oxford University Press: Oxford, UK, 2015; Volume 132, pp. 97–109. [Google Scholar]
- Northrup, H.; Koenig, M.K.; Pearson, D.A.; Au, K.S. Tuberous Sclerosis Complex; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Eds.; Oxford University Press: Seattle, WA, USA, 1993. [Google Scholar]
- Wei, C.-C.; Sheu, J.-N.; Liu, J.-T.; Yang, S.-H.; Chou, I.-C.; Tsai, J.-D. Trend of seizure remission in patients with tuberous sclerosis complex: A retrospective medical review. J. Chin. Med. Assoc. 2018, 81, 724–728. [Google Scholar] [CrossRef]
- Volpi, A.; Sala, G.; Lesma, E.; Labriola, F.; Righetti, M.; Alfano, R.M.; Cozzolino, M. Tuberous sclerosis complex: New insights into clinical and therapeutic approach. J. Nephrol. 2019, 32, 355–363. [Google Scholar] [CrossRef]
- Crino, P.B.; Nathanson, K.L.; Henske, E.P. The tuberous sclerosis complex. N. Engl. J. Med. 2006, 355, 1345–1356. [Google Scholar] [CrossRef]
- Curatolo, P.; Moavero, R. mTOR Inhibitors in Tuberous Sclerosis Complex. Curr. Neuropharmacol. 2012, 10, 404–415. [Google Scholar] [CrossRef]
- Adil, A.; Singh, A.K. Neurofibromatosis Type 1 (Von Recklinghausen); StatPearls Publishing: Treasure Island, FL, USA, 2019. [Google Scholar]
- Treichel, A.M.; Hamieh, L.; Nathan, N.R.; Tyburczy, M.E.; Wang, J.-A.; Oyerinde, O.; Raiciulescu, S.; Julien-Williams, P.; Jones, A.M.; Gopalakrishnan, V.; et al. Phenotypic distinctions between mosaic forms of tuberous sclerosis complex. Genet. Med. 2019, 1. [Google Scholar] [CrossRef]
- Giannikou, K.; Lasseter, K.D.; Grevelink, J.M.; Tyburczy, M.E.; Dies, K.A.; Zhu, Z.; Hamieh, L.; Wollison, B.M.; Thorner, A.R.; Ruoss, S.J.; et al. Low-level mosaicism in tuberous sclerosis complex: Prevalence, clinical features, and risk of disease transmission. Genet. Med. 2019, 1. [Google Scholar] [CrossRef]
- Leducq, S.; Giraudeau, B.; Tavernier, E.; Maruani, A. Topical use of mammalian target of rapamycin inhibitors in dermatology: A systematic review with meta-analysis. J. Am. Acad. Dermatol. 2019, 80, 735–742. [Google Scholar] [CrossRef]
- Combes, F.P.; Baneyx, G.; Coello, N.; Zhu, P.; Sallas, W.; Yin, H.; Nedelman, J. Population pharmacokinetics–pharmacodynamics of oral everolimus in patients with seizures associated with tuberous sclerosis complex. J. Pharmacokinet. Pharmacodyn. 2018, 45, 707–719. [Google Scholar] [CrossRef]
- Weidinger, S.; Novak, N. Atopic dermatitis. Lancet 2016, 387, 1109–1122. [Google Scholar] [CrossRef]
- Boehncke, W.-H.; Schon, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Desmet, S.J.; De Bosscher, K. Glucocorticoid receptors: Finding the middle ground. J. Clin. Investig. 2017, 127, 1136–1145. [Google Scholar] [CrossRef]
- Das, L.; Bhaumik, E.; Raychaudhuri, U.; Chakraborty, R. Role of nutraceuticals in human health. J. Food Sci. Technol. 2012, 49, 173–183. [Google Scholar] [CrossRef]
- Proksch, E.; Schunck, M.; Zague, V.; Segger, D.; Degwert, J.; Oesser, S. Oral intake of specific bioactive collagen peptides reduces skin wrinkles and increases dermal matrix synthesis. Skin Pharmacol. Physiol. 2014, 27, 113–119. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lead Compound(s) | Protein Target(s) | Skin Cancer Type | Compound Structure | References |
|---|---|---|---|---|
| Everolimus (RAD-001) | mTOR | Melanoma, Basal Cell Carcinoma |  | [92,93] |
| Erufosine | mTOR | Oral Squamous Cell Carcinoma | [94] | |
| GDC-0084 | PI3K, mTOR, Akt | cutaneous Squamous Cell Carcinoma | [95] | |
| Isoselenocyanate-4 | Akt | Melanoma |  | [96] |
| MLN0128 (Sapanisertib) | mTOR | Melanoma Merkel Cell Carcinoma |  | [97,98,99] |
| NVP-BEZ235 | PI3K, Akt, mTOR | Melanoma Merkel Cell Carcinoma |  | [100,101] |
| NC1 domain of collagen Type XIX [NC1(XIX)] | PI3K, Akt, mTOR, FAK | Melanoma | [89,90,91] | |
| PBISe | Akt3 | Invasive metastatic Melanoma |  | [102] |
| Rapamycin | PI3k, Akt, mTOR | Melanoma, Esophageal squamous cell carcinoma |  | [77,78,103] |
| SKLB-M8 | Akt, mTOR | Melanoma | [87,88] | |
| PI-103 | PI3K, mTOR | Melanoma | [104] | |
| Perifosine | Akt | Metastatic Melanoma | [105] | |
| Tazarotene | IGFR, PI3K, Akt, mTOR | Basal cell carcinoma | [106,107] | |
| Temsirolimus | mTOR | Metastatic melanoma |  | [108] |
| WYE-354 | mTOR | Merkel cell carcinoma | [109] | |
| VS-5584 | PI3K and mTOR | Melanoma | [67,84,85,86] | |
| Itraconazole | PI3K and mTOR | Melanoma Basal Cell Carcinoma | [110,111] | |
| LY3023414 | PI3K/mTOR | Cutaneous Basal Cell Carcinoma, cutaneous Squamous Cell Carcinoma | [112] | |
| Ku-0063794 | mTORC1 and mTORC2 | BRAF-Mutant Melanoma in combination with MEK inhibitory agents Merkel cell carcinoma |  | [113] |
| Lead Compound (s) | Protein Targets | Skin Cancer Type | Compound Structure | References |
|---|---|---|---|---|
| Acacetin | PI3K, Akt, mTOR | Malignant Melanoma |  | [124] |
| Bee Venom Melittin | PI3K, Akt, mTOR | Melanoma | [129] | |
| Capsaicin | PI3K, Akt, Rac1 | Melanoma |  | [126] |
| Curcumin | PI3K, Akt, mTOR | Melanoma |  | [142,143] |
| Epigallocatechin-3 (EGCG) | mTOR | Melanoma |  | [148] |
| Evodiamine | PI3K, Akt | Melanoma |  | [127] |
| Fisetin | PI3K, Akt, mTOR | Melanoma |  | [68,139,140,141] |
| Isoliquiritigenin | mTORC2, Akt, GSK-3β | Melanoma cachexia |  | [128] |
| Harmine | Akt, mTOR and ERK1/2 | Melanoma |  | [131] |
| Obatoclax | Akt, mTOR | Melanoma | [133] | |
| Panduratin A | mTOR | Melanoma |  | [130] |
| Prodigiosin | Akt, mTOR | Melanoma | [133] | |
| Resveratrol | Akt, mTOR | Melanoma |  | [46,144,145,146] |
| Sinomenine | PI3K, Akt, mTOR | Melanoma |  | [132] |
| Honokiol | mTOR | Melanoma, Oral squamous cell carcinoma |  | [63,147,151] |
| NexrutineR | PI3K/Akt/mTOR | Melanoma | [149,150] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chamcheu, J.C.; Roy, T.; Uddin, M.B.; Banang-Mbeumi, S.; Chamcheu, R.-C.N.; Walker, A.L.; Liu, Y.-Y.; Huang, S. Role and Therapeutic Targeting of the PI3K/Akt/mTOR Signaling Pathway in Skin Cancer: A Review of Current Status and Future Trends on Natural and Synthetic Agents Therapy. Cells 2019, 8, 803. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8080803
Chamcheu JC, Roy T, Uddin MB, Banang-Mbeumi S, Chamcheu R-CN, Walker AL, Liu Y-Y, Huang S. Role and Therapeutic Targeting of the PI3K/Akt/mTOR Signaling Pathway in Skin Cancer: A Review of Current Status and Future Trends on Natural and Synthetic Agents Therapy. Cells. 2019; 8(8):803. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8080803
Chicago/Turabian StyleChamcheu, Jean Christopher, Tithi Roy, Mohammad Burhan Uddin, Sergette Banang-Mbeumi, Roxane-Cherille N. Chamcheu, Anthony L. Walker, Yong-Yu Liu, and Shile Huang. 2019. "Role and Therapeutic Targeting of the PI3K/Akt/mTOR Signaling Pathway in Skin Cancer: A Review of Current Status and Future Trends on Natural and Synthetic Agents Therapy" Cells 8, no. 8: 803. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8080803