Genes Controlled by DNA Methylation Are Involved in Wilms Tumor Progression
 ,
,  , ,
, , 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA and RNA Extraction
2.2. Infinium HumanMethylation450 BeadChip Arrays (Illumina) Procedures
2.3. Methylation Statistical Analysis
2.4. RNA Library Construction and Sequencing
2.5. Gene Expression Analysis
3. Results
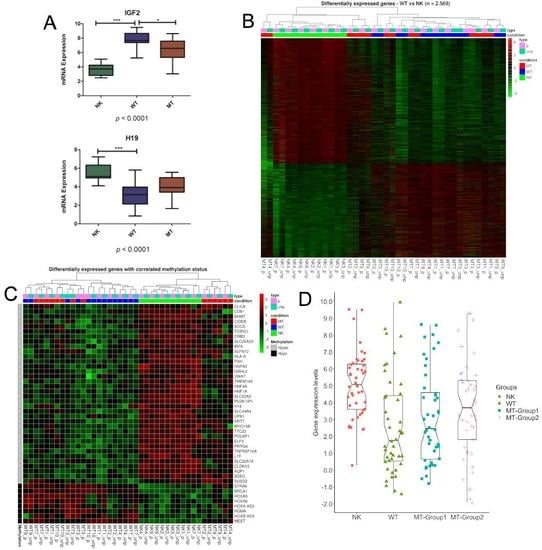
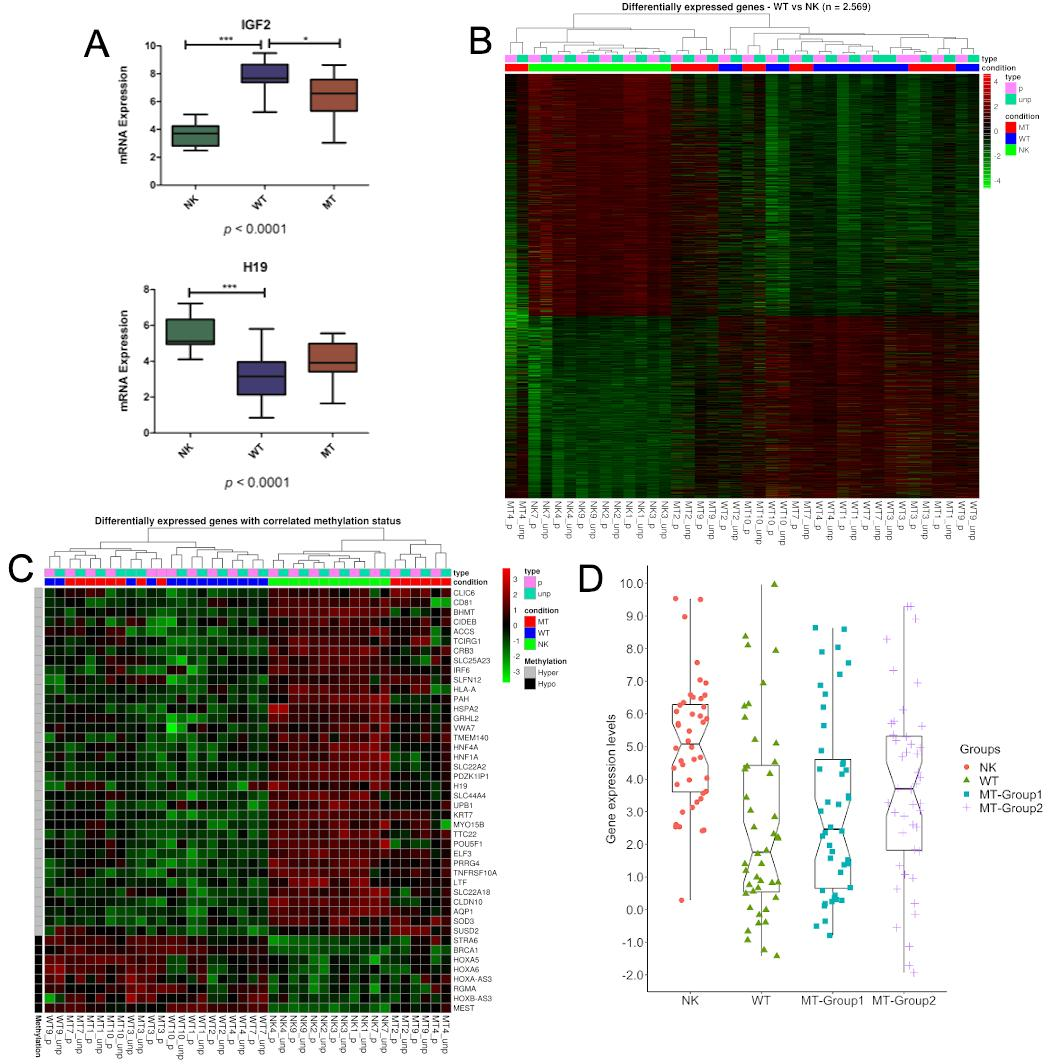
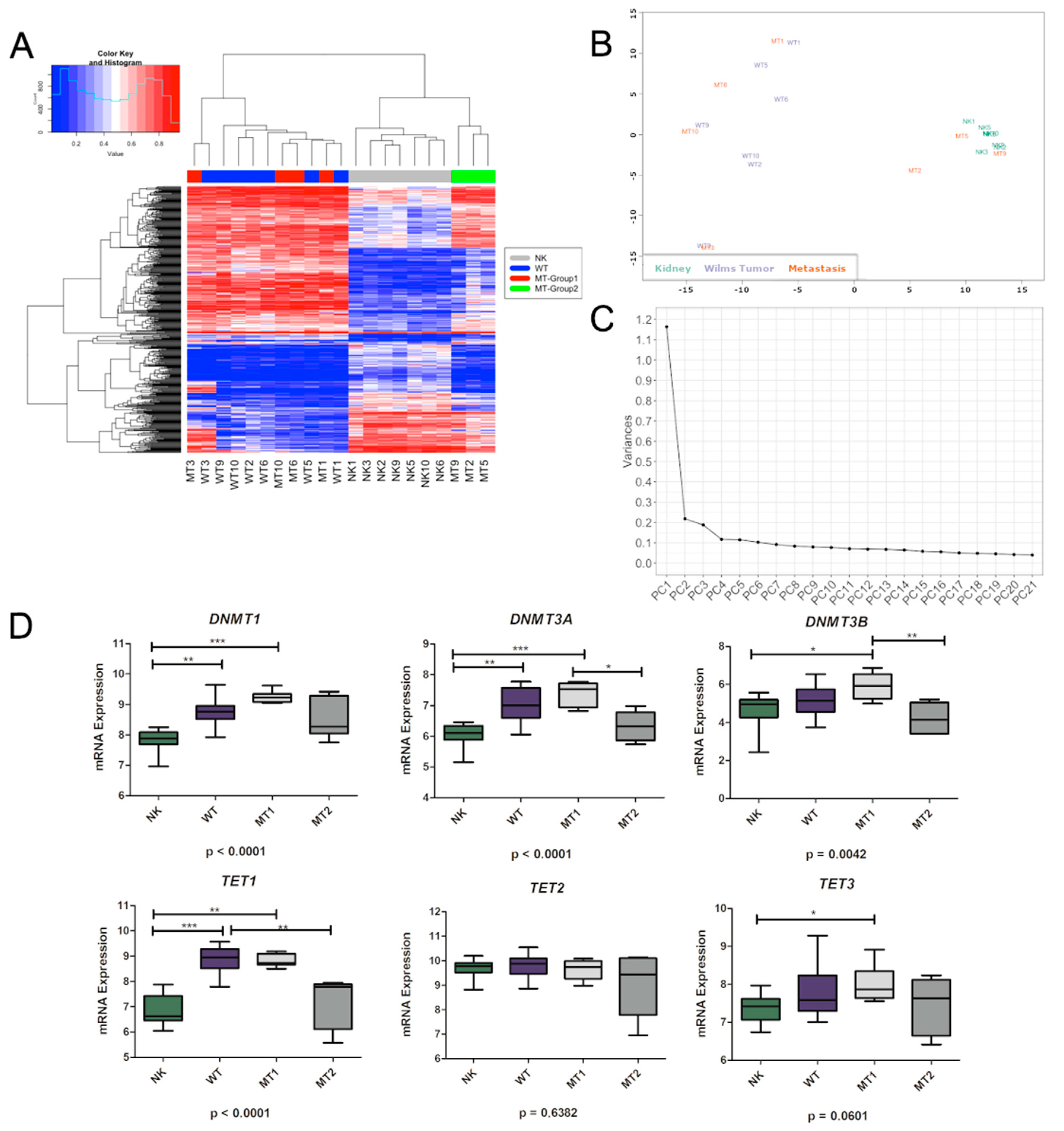
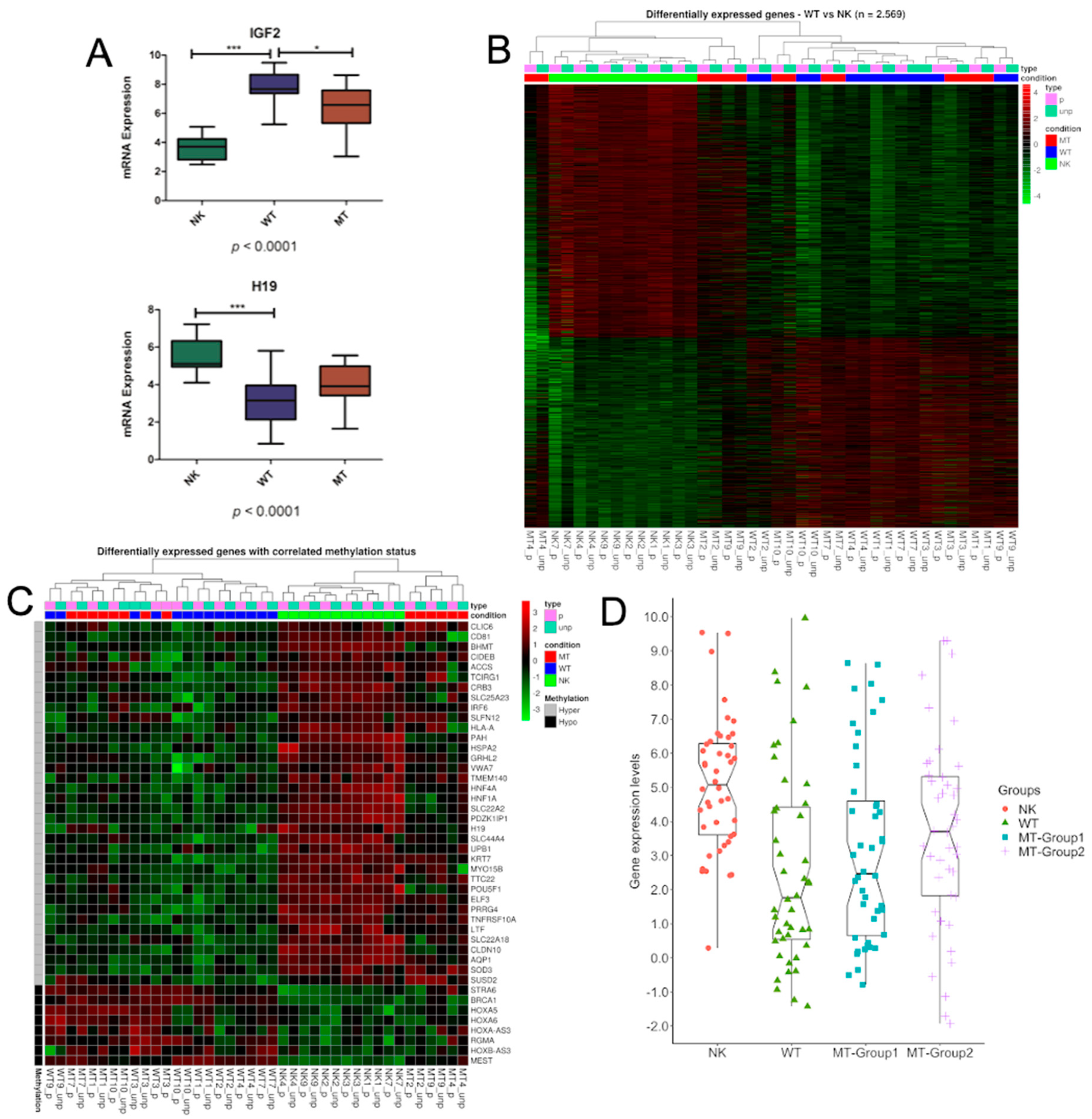
3.1. DNA Variability Suggests the Existence of Two Groups of Metastatic Tissues
3.2. Methylation Differences May Be Related to DNMTs and TETs Expression
3.3. Genes Controlled by DNA Methylation Confirm the Existence of Two Groups of Metastases
3.4. Characterization of the DMRs within the Metastatic Groups
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- de Camargo, B.; de Oliveira Ferreira, J.M.; de Souza Reis, R.; Ferman, S.; de Oliveira Santos, M.; Pombo-de-Oliveira, M.S. Socioeconomic status and the incidence of non-central nervous system childhood embryonic tumours in Brazil. BMC Cancer 2011, 11, 160. [Google Scholar] [CrossRef] [PubMed]
- Gadd, S.; Huff, V.; Huang, C.-C.; Ruteshouser, E.C.; Dome, J.S.; Grundy, P.E.; Breslow, N.; Jennings, L.; Green, D.M.; Beckwith, J.B.; et al. Clinically Relevant Subsets Identified by Gene Expression Patterns Support a Revised Ontogenic Model of Wilms Tumor: A Children’s Oncology Group Study. Neoplasia 2012, 14, 742–756. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.H.; Murray, A.; Baskcomb, L.; Turnbull, C.; Loveday, C.; Al-Saadi, R.; Williams, R.; Breatnach, F.; Gerrard, M.; Hale, J.; et al. Stratification of Wilms tumor by genetic and epigenetic analysis. Oncotarget 2012, 3, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Wegert, J.; Ishaque, N.; Vardapour, R.; Geörg, C.; Gu, Z.; Bieg, M.; Ziegler, B.; Bausenwein, S.; Nourkami, N.; Ludwig, N.; et al. Mutations in the SIX1/2 Pathway and the DROSHA/DGCR8 miRNA Microprocessor Complex Underlie High-Risk Blastemal Type Wilms Tumors. Cancer Cell 2015, 27, 298–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrezan, G.T.; Ferreira, E.N.; Nakahata, A.M.; Barros, B.D.F.; Castro, M.T.M.; Correa, B.R.; Krepischi, A.C.V.; Olivieri, E.H.R.; Cunha, I.W.; Tabori, U.; et al. Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour. Nat. Commun. 2014, 5, 4039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walz, A.L.; Ooms, A.; Gadd, S.; Gerhard, D.S.; Smith, M.A.; Guidry Auvil, J.M.; Meerzaman, D.; Chen, Q.-R.; Hsu, C.H.; Yan, C.; et al. Recurrent DGCR8, DROSHA, and SIX Homeodomain Mutations in Favorable Histology Wilms Tumors. Cancer Cell 2015, 27, 286–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadd, S.; Huff, V.; Walz, A.L.; Ooms, A.H.A.G.; Armstrong, A.E.; Gerhard, D.S.; Smith, M.A.; Guidry Auvil, J.M.; Meerzaman, D.; Chen, Q.R.; et al. A Children’s Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat. Genet. 2017, 49, 1487–1494. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.H.; Douglas, J.; Baskcomb, L.; Huxter, N.; Barker, K.; Hanks, S.; Craft, A.; Gerrard, M.; Kohler, J.A.; Levitt, G.A.; et al. Constitutional 11p15 abnormalities, including heritable imprinting center mutations, cause nonsyndromic Wilms tumor. Nat. Genet. 2008, 40, 1329–1334. [Google Scholar] [CrossRef]
- Maschietto, M.; Charlton, J.; Perotti, D.; Radice, P.; Geller, J.I.; Pritchard-Jones, K.; Weeks, M. The IGF signalling pathway in Wilms tumours—A report from the ENCCA Renal Tumours Biology-driven drug development workshop. Oncotarget 2014, 5, 8014–8026. [Google Scholar] [CrossRef]
- de Sá Pereira, B.M.; Montalvão-de-Azevedo, R.; Faria, P.A.; de Paula Silva, N.; Nicolau-Neto, P.; Maschietto, M.; de Camargo, B.; Soares Lima, S.C. Association between long interspersed nuclear element-1 methylation levels and relapse in Wilms tumors. Clin. Epigenet. 2017, 9. [Google Scholar] [CrossRef]
- Charlton, J.; Williams, R.D.; Sebire, N.J.; Popov, S.; Vujanic, G.; Chagtai, T.; Alcaide-German, M.; Morris, T.; Butcher, L.M.; Guilhamon, P.; et al. Comparative methylome analysis identifies new tumour subtypes and biomarkers for transformation of nephrogenic rests into Wilms tumour. Genome Med. 2015, 7, 11. [Google Scholar] [CrossRef]
- Dome, J.S.; Graf, N.; Geller, J.I.; Fernandez, C.V.; Mullen, E.A.; Spreafico, F.; Van Den Heuvel-Eibrink, M.; Pritchard-Jones, K. Advances in wilms tumor treatment and biology: Progress through international collaboration. J. Clin. Oncol. 2015, 33, 2999–3007. [Google Scholar] [CrossRef]
- Godzinski, J. The current status of treatment of Wilms′ tumor as per the SIOP trials. J. Indian Assoc. Pediatr. Surg. 2014, 20, 16. [Google Scholar] [CrossRef]
- Van Den Heuvel-Eibrink, M.M.; Hol, J.A.; Pritchard-Jones, K.; Van Tinteren, H.; Furtwängler, R.; Verschuur, A.C.; Vujanic, G.M.; Leuschner, I.; Brok, J.; Rübe, C.; et al. Position Paper: Rationale for the treatment of Wilms tumour in the UMBRELLA SIOP-RTSG 2016 protocol. Nat. Rev. Urol. 2017, 14, 743–752. [Google Scholar] [CrossRef]
- Szychot, E.; Apps, J.; Pritchard-Jones, K. Wilms’ tumor: Biology, diagnosis and treatment. Transl. Pediatr. 2014, 3, 12–24. [Google Scholar]
- Maschietto, M.; Williams, R.D.; Chagtai, T.; Popov, S.D.; Sebire, N.J.; Vujanic, G.; Perlman, E.; Anderson, J.R.; Grundy, P.; Dome, J.S.; et al. TP53 mutational status is a potential marker for risk stratification in Wilms tumour with diffuse anaplasia. PLoS ONE 2014, 9, e109924. [Google Scholar] [CrossRef] [PubMed]
- Ooms, A.H.A.G.; Gadd, S.; Gerhard, D.S.; Smith, M.A.; Guidry Auvil, J.M.; Meerzaman, D.; Chen, Q.R.; Hsu, C.H.; Yan, C.; Nguyen, C.; et al. Significance of TP53 mutation in Wilms tumors with diffuse anaplasia: A report from the Children’s Oncology Group. Clin. Cancer Res. 2016, 22, 5582–5591. [Google Scholar] [CrossRef]
- Wegert, J.; Vokuhl, C.; Ziegler, B.; Ernestus, K.; Leuschner, I.; Furtwängler, R.; Graf, N.; Gessler, M. TP53 alterations in Wilms tumour represent progression events with strong intratumour heterogeneity that are closely linked but not limited to anaplasia. J. Pathol. Clin. Res. 2017, 3, 234–248. [Google Scholar] [CrossRef] [Green Version]
- Chagtai, T.; Zill, C.; Dainese, L.; Wegert, J.; Savola, S.; Popov, S.; Mifsud, W.; Vujanić, G.; Sebire, N.; Le Bouc, Y.; et al. Gain of 1q as a prognostic biomarker in Wilms Tumors (WTs) treated with preoperative chemotherapy in the International Society of Paediatric Oncology (SIOP) WT 2001 trial: A SIOP renal tumours biology consortium study. J. Clin. Oncol. 2016, 34, 3195–3203. [Google Scholar] [CrossRef]
- Gratias, E.J.; Dome, J.S.; Jennings, L.J.; Chi, Y.Y.; Tian, J.; Anderson, J.; Grundy, P.; Mullen, E.A.; Geller, J.I.; Fernandez, C.V.; et al. Association of chromosome 1q gain with inferior survival in favorable-histology Wilms tumor: A report from the Children’s Oncology Group. J. Clin. Oncol. 2016, 34, 3189–3194. [Google Scholar] [CrossRef]
- Cone, E.B.; Dalton, S.S.; Van Noord, M.; Tracy, E.T.; Rice, H.E.; Routh, J.C. Biomarkers for Wilms Tumor: A Systematic Review. J. Urol. 2016, 196, 1530–1535. [Google Scholar] [CrossRef] [Green Version]
- Brok, J.; Pritchard-Jones, K.; Geller, J.I.; Spreafico, F. Review of phase I and II trials for Wilms’ tumour—Can we optimise the search for novel agents? Eur. J. Cancer 2017, 79, 205–213. [Google Scholar] [CrossRef]
- Wright, K.D.; Green, D.M.; Daw, N.C. Late effects of treatment for Wilms tumor. Pediatr. Hematol. Oncol. 2009, 26, 407–413. [Google Scholar] [CrossRef]
- Bal, A.S.K.; Yalcin, B.; Susam-Şen, H.; Aydin, B.; Varan, A.; Kutluk, T.; Akyüz, C. Renal late effects after the treatment of unilateral nonsyndromic wilms tumor. J. Pediatr. Hematol. Oncol. 2016, 38, e147–e150. [Google Scholar]
- Wong, K.F.; Reulen, R.C.; Winter, D.L.; Guha, J.; Fidler, M.M.; Kelly, J.; Lancashire, E.R.; Pritchard-Jones, K.; Jenkinson, H.C.; Sugden, E.; et al. Risk of adverse health and social outcomes up to 50 years after wilms tumor: The British childhood cancer survivor study. J. Clin. Oncol. 2016, 34, 1772–1779. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef]
- Nordlund, J.; Bäcklin, C.L.; Wahlberg, P.; Busche, S.; Berglund, E.C.; Eloranta, M.L.; Flaegstad, T.; Forestier, E.; Frost, B.M.; Harila-Saari, A.; et al. Genome-wide signatures of differential DNA methylation in pediatric acute lymphoblastic leukemia. Genome Biol. 2013, 14, r105. [Google Scholar] [CrossRef]
- Touleimat, N.; Tost, J. Complete pipeline for Infinium® Human Methylation 450K BeadChip data processing using subset quantile normalization for accurate DNA methylation estimation. Epigenomics 2012, 4, 325–341. [Google Scholar] [CrossRef]
- Leek, J.T.; Storey, J.D. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet. 2007, 3, 1724–1735. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3. [Google Scholar] [CrossRef]
- Peters, T.J.; Buckley, M.J.; Statham, A.L.; Pidsley, R.; Samaras, K.; Lord, R.V.; Clark, S.J.; Molloy, P.L. De novo identification of differentially methylated regions in the human genome. Epigenet. Chromatin 2015, 8, 6. [Google Scholar] [CrossRef]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Andrews, S. Babraham Bioinformatics FastQC: A Quality Control Tool for High Throughput Sequence Data; Manual; Babraham: Cambridge, UK, 2010. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Getz, G.; Guttman, M.; Mesirov, J.P.; Winckler, W.; Lander, E.S. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Vidal, E.; Sayols, S.; Moran, S.; Guillaumet-Adkins, A.; Schroeder, M.P.; Royo, R.; Orozco, M.; Gut, M.; Gut, I.; Lopez-Bigas, N.; et al. A DNA methylation map of human cancer at single base-pair resolution. Oncogene 2017, 36, 5648–5657. [Google Scholar] [CrossRef]
- Williams, L.A.; Mills, L.; Hooten, A.J.; Langer, E.; Roesler, M.; Frazier, A.L.; Krailo, M.; Nelson, H.H.; Bestrashniy, J.; Amatruda, J.F.; et al. Differences in DNA methylation profiles by histologic subtype of paediatric germ cell tumours: A report from the Children’s Oncology Group. Br. J. Cancer 2018, 119, 864–872. [Google Scholar] [CrossRef]
- Schultz, M.D.; He, Y.; Whitaker, J.W.; Hariharan, M.; Mukamel, E.A.; Leung, D.; Rajagopal, N.; Nery, J.R.; Urich, M.A.; Chen, H.; et al. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature 2015, 523, 212–216. [Google Scholar] [CrossRef]
- Charlton, J.; Williams, R.D.; Weeks, M.; Sebire, N.J.; Popov, S.; Vujanic, G.; Mifsud, W.; Alcaide-German, M.; Butcher, L.M.; Beck, S.; et al. Methylome analysis identifies a Wilms tumor epigenetic biomarker detectable in blood. Genome Biol. 2014, 15, 434. [Google Scholar] [CrossRef]
- Cui, H.; Hedborg, F.; He, L.; Nordenskjöld, A.; Sandstedt, B.; Pfeifer-Ohlsson, S.; Ohlsson, R. Inactivation of H19, an imprinted and putative tumor repressor gene, is a preneoplastic event during Wilms’ tumorigenesis. Cancer Res. 1997, 57, 4469–4473. [Google Scholar]
- Bjornsson, H.T.; Brown, L.J.; Fallin, M.D.; Rongione, M.A.; Bibikova, M.; Wickham, E.; Fan, J.B.; Feinberg, A.P. Epigenetic specificity of loss of imprinting of the IGF2 gene in wilms tumors. J. Natl. Cancer Inst. 2007, 99, 1270–1273. [Google Scholar] [CrossRef]
- Ludgate, J.L.; Le Mée, G.; Fukuzawa, R.; Rodger, E.J.; Weeks, R.J.; Reeve, A.E.; Morison, I.M. Global demethylation in loss of imprinting subtype of Wilms tumor. Genes. Chromosomes Cancer 2013, 52, 174–184. [Google Scholar] [CrossRef]
- Conway, E.; Healy, E.; Bracken, A.P. PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr. Opin. Cell Biol. 2015, 37, 42–48. [Google Scholar] [CrossRef]
- Shah, N.; Sukumar, S. The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer 2010, 10, 361–371. [Google Scholar] [CrossRef]
- Lau, H.H.; Ng, N.H.J.; Loo, L.S.W.; Jasmen, J.B.; Teo, A.K.K. The molecular functions of hepatocyte nuclear factors—In and beyond the liver. J. Hepatol. 2018, 68, 1033–1048. [Google Scholar] [CrossRef]
- Zhang, Q.; Lei, X.; Lu, H. Alterations of epigenetic signatures in hepatocyte nuclear factor 4α deficient mouse liver determined by improved ChIP-qPCR and (h)MeDIP-qPCR assays. PLoS ONE 2014, 9, e84925. [Google Scholar] [CrossRef]
- Walesky, C.; Apte, U. Role of hepatocyte nuclear factor 4α (HNF4α) in cell proliferation and cancer. Gene Expr. 2015, 16, 101–108. [Google Scholar] [CrossRef]
- Sadikovic, B.; Yoshimoto, M.; Chilton-MacNeill, S.; Thorner, P.; Squire, J.A.; Zielenska, M. Identification of interactive networks of gene expression associated with osteosarcoma oncogenesis by integrated molecular profiling. Hum. Mol. Genet. 2009, 18, 1962–1975. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.H.; Lim, S.W.; Kim, H.G.; Kim, D.Y.; Ryu, S.Y.; Joo, J.K.; Kim, J.C.; Lee, J.H. Lack of death receptor 4 (DR4) expression through gene promoter methylation in gastric carcinoma. Langenbeck Arch. Surg. 2009, 394, 661–670. [Google Scholar] [CrossRef]
- Martinez, R.; Setien, F.; Voelter, C.; Casado, S.; Quesada, M.P.; Schackert, G.; Esteller, M. CpG island promoter hypermethylation of the pro-apoptotic gene caspase-8 is a common hallmark of relapsed glioblastoma multiforme. Carcinogenesis 2007, 28, 1264–1268. [Google Scholar] [CrossRef] [Green Version]
- Martinez, R.; Martin-Subero, J.I.; Rohde, V.; Kirsch, M.; Alaminos, M.; Fernandez, A.F.; Ropero, S.; Schockert, G.; Esteller, M. A microarray-based DNA methylation study of glioblastoma multiforme. Epigenetics 2009, 4, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Wang, X.; Wang, P.; Li, T.; Hu, F.; Liu, Q.; Yang, F.; Wang, J.; Xu, T.; Han, W. SUSD2 is frequently downregulated and functions as a tumor suppressor in RCC and lung cancer. Tumor Biol. 2016, 37, 9919–9930. [Google Scholar] [CrossRef]
- Watson, A.P.; Evans, R.L.; Egland, K.A. Multiple Functions of Sushi Domain Containing 2 (SUSD2) in Breast Tumorigenesis. Mol. Cancer Res. 2012, 11, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Zeng, N.; Alowayed, N.; Singh, Y.; Cheng, A.; Lang, F.; Salker, M.S. Downregulation of endometrial mesenchymal marker SUSD2 causes cell senescence and cell death in endometrial carcinoma cells. PLoS ONE 2017, 12, e0183681. [Google Scholar] [CrossRef]
- Zhao, S.; Geybels, M.S.; Leonardson, A.; Rubicz, R.; Kolb, S.; Yan, Q.; Klotzle, B.; Bibikova, M.; Hurtado-Coll, A.; Troyer, D.; et al. Epigenome-wide tumor DNA methylation profiling identifies novel prognostic biomarkers of metastatic-lethal progression in men diagnosed with clinically localized prostate cancer. Clin. Cancer Res. 2017, 23, 311–319. [Google Scholar] [CrossRef]
- Saghafinia, S.; Mina, M.; Riggi, N.; Hanahan, D.; Ciriello, G. Pan-Cancer Landscape of Aberrant DNA Methylation across Human Tumors. Cell Rep. 2018, 25, 1066–1080. [Google Scholar] [CrossRef]
- Oostveen, R.M.; Pritchard-Jones, K. Pharmacotherapeutic Management of Wilms Tumor: An Update. Pediatr. Drugs 2019, 21, 1–13. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Sample (ID) | Age at Diagnosis (years) | Gender | Histology (Primary Tumor) | Patient Stage | Relapse Site | Histology (Metastasis) | Technique |
|---|---|---|---|---|---|---|---|
| 1 | 3 | F | Blastemal | III | Left lung | Mixed | RNA-Seq/450k |
| 2 | 7 | M | Regressive | II | Left lung | Epithelial | RNA-Seq/450k |
| 3 | 5 | M | Mixed | I | Left lung | Blastemal | RNA-Seq/450k |
| 4 | 3 | F | Mixed | II | Right lung | Blastemal | RNA-Seq |
| 5 | 3 | F | Mixed | II | Right and left lung | Epithelial | 450k |
| 6 | 9 | M | Mixed | I | Left lung | Mixed | 450k |
| 7 | 4 | M | Regressive | I | Right lung | Blastemal | RNA-Seq |
| 9 | 3 | M | Mixed | I | Right lung | Blastemal | RNA-Seq/450k |
| 10 | 6 | M | Mixed | I | Right lung | Mixed | RNA-Seq/450k |
| Gene | DMR Location | Number of CpGs | DMR Width (Bp) | Minimum p-value | Methylation Status (MaxBetaFC) | Methylation Status (MeanBetaFC) | Expression (Log2FC) |
|---|---|---|---|---|---|---|---|
| ACCS | chr11:44087396-44088257 | 12 | 862 | 5 × 10−19 | 0.5 | 0.3 | −1.4 |
| AQP1 | chr7:30951064-30951801 | 11 | 738 | 6 × 10−18 | 0.4 | 0.1 | −3.9 |
| BHMT | chr5:78407153-78407683 | 8 | 531 | 4 × 10−40 | 0.5 | 0.4 | −7.5 |
| BRCA1 | chr17:41277974-41279022 | 21 | 1049 | 2 × 10−25 | −0.5 | −0.2 | 2.2 |
| CD81 | chr11:2397255-2398336 | 21 | 1082 | 2 × 10−46 | 0.4 | 0.2 | −1.2 |
| CIDEB | chr14:24779793-24780926 | 13 | 1134 | 4 × 10−43 | 0.6 | 0.2 | −2.2 |
| CLDN10 | chr13:96204518-96204978 | 8 | 461 | 1 × 10−4 | 0.2 | 0.1 | −5.6 |
| CLIC6 | chr21:36041334-36041699 | 8 | 366 | 1 × 10−52 | 0.6 | 0.5 | −3.0 |
| CRB3 | chr19:6463949-6464275 | 9 | 327 | 1 × 10−23 | 0.4 | 0.3 | −5.3 |
| ELF3 | chr1:201979478-201979938 | 7 | 461 | 1 × 10−6 | 0.3 | 0.1 | −4.8 |
| GRHL2 | chr8:102504447-102504859 | 8 | 413 | 7 × 10−16 | 0.3 | 0.2 | −4.0 |
| H19 | chr11:2019452-2020560 | 29 | 1109 | 4 × 10−12 | 0.3 | 0.1 | −2.1 |
| HLA-A | chr6:29910411-29911095 | 8 | 685 | 4 × 10−18 | 0.4 | 0.2 | −2.2 |
| HNF1A | chr12:121416315-121416796 | 7 | 482 | 7 × 10−10 | 0.4 | 0.2 | −4.9 |
| HNF4A | chr20:42983920-42984878 | 12 | 959 | 1 × 10−17 | 0.4 | 0.2 | −5.4 |
| HOXA-AS3 | chr7:27183816-27185512 | 26 | 1697 | 5 × 10−16 | −0.3 | −0.2 | 2.6 |
| HOXA5 | chr7:27183816-27185512 | 26 | 1697 | 5 × 10−16 | −0.3 | −0.2 | 1.8 |
| HOXA6 | chr7:27183816-27185512 | 26 | 1697 | 5 × 10−16 | −0.3 | −0.2 | 2.3 |
| HOXB-AS3 | chr17:46669455-46670029 | 9 | 575 | 4 × 10−16 | −0.3 | −0.1 | 2.7 |
| HSPA2 | chr14:65006688-65007437 | 16 | 750 | 1 × 10−15 | 0.4 | 0.2 | −3.9 |
| IRF6 | chr1:209979111-209979779 | 9 | 669 | 3 × 10−22 | 0.5 | 0.3 | −2.8 |
| KRT7 | chr12:52626814-52627576 | 8 | 763 | 1 × 10−8 | 0.4 | 0.2 | −5.8 |
| LTF | chr3:46506104-46506554 | 9 | 451 | 3 × 10−5 | 0.3 | 0.2 | −7.0 |
| MEST | chr7:130130753-130131730 | 13 | 978 | 3 × 10−9 | −0.2 | −0.1 | 3.7 |
| MYO15B | chr17:73583839-73584360 | 9 | 522 | 1 × 10−7 | 0.3 | 0.2 | −1.6 |
| PAH | chr12:103310839-103311761 | 9 | 923 | 8 × 10−20 | 0.3 | 0.2 | −8.1 |
| PDZK1IP1 | chr1:47655599-47656423 | 7 | 825 | 7 × 10−7 | 0.3 | 0.2 | −7.3 |
| POU5F1 | chr6:31148404-31148748 | 7 | 345 | 1 × 10−6 | 0.2 | 0.1 | −4.7 |
| PRRG4 | chr11:32851087-32851531 | 9 | 445 | 1 × 10−4 | 0.2 | 0.1 | −4.2 |
| RGMA | chr15:93616894-93617168 | 11 | 275 | 2 × 10−9 | −0.3 | −0.2 | 2.7 |
| SLC22A18 | chr11:2925594-2925969 | 8 | 376 | 2 × 10−4 | 0.2 | 0.1 | −2.5 |
| SLC22A2 | chr6:160679391-160680162 | 10 | 772 | 3 × 10−13 | 0.3 | 0.2 | −8.0 |
| SLC25A23 | chr19:6463949-6464275 | 9 | 327 | 1 × 10−23 | 0.4 | 0.3 | −1.6 |
| SLC44A4 | chr6:31846769-31847028 | 8 | 260 | 1 × 10−7 | 0.2 | 0.2 | −4.1 |
| SLFN12 | chr17:33759512-33760527 | 11 | 1016 | 1 × 10−14 | 0.5 | 0.3 | −1.7 |
| SOD3 | chr4:24796689-24797176 | 7 | 488 | 1 × 10−3 | 0.2 | 0.1 | −2.3 |
| STRA6 | chr15:74494781-74495354 | 7 | 574 | 4 × 10−15 | −0.3 | −0.2 | 3.6 |
| SUSD2 | chr22:24577223-24577448 | 7 | 226 | 4 × 10−3 | 0.3 | 0.1 | −2.7 |
| TCIRG1 | chr11:67806118-67806668 | 7 | 551 | 3 × 10−30 | 0.6 | 0.4 | −2.2 |
| TMEM140 | chr7:134832544-134833299 | 7 | 756 | 9 × 10−10 | 0.4 | 0.2 | −3.1 |
| TNFRSF10A | chr8:23082634-23082961 | 7 | 328 | 2 × 10−4 | 0.3 | 0.2 | −3.1 |
| TTC22 | chr1:55266296-55267152 | 8 | 857 | 1 × 10−7 | 0.3 | 0.2 | −5.7 |
| UPB1 | chr22:24891141-24891666 | 8 | 526 | 2 × 10−10 | 0.3 | 0.2 | −3.1 |
| VWA7 | chr6:31740805-31741184 | 8 | 380 | 1 × 10−9 | 0.2 | 0.2 | −1.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerra, J.V.d.S.; Pereira, B.M.d.S.; Cruz, J.G.V.d.; Scherer, N.d.M.; Furtado, C.; Montalvão de Azevedo, R.; Oliveira, P.S.L.d.; Faria, P.; Boroni, M.; de Camargo, B.; et al. Genes Controlled by DNA Methylation Are Involved in Wilms Tumor Progression. Cells 2019, 8, 921. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8080921
Guerra JVdS, Pereira BMdS, Cruz JGVd, Scherer NdM, Furtado C, Montalvão de Azevedo R, Oliveira PSLd, Faria P, Boroni M, de Camargo B, et al. Genes Controlled by DNA Methylation Are Involved in Wilms Tumor Progression. Cells. 2019; 8(8):921. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8080921
Chicago/Turabian StyleGuerra, João Victor da Silva, Bruna Maria de Sá Pereira, Jéssica Gonçalves Vieira da Cruz, Nicole de Miranda Scherer, Carolina Furtado, Rafaela Montalvão de Azevedo, Paulo Sergio Lopes de Oliveira, Paulo Faria, Mariana Boroni, Beatriz de Camargo, and et al. 2019. "Genes Controlled by DNA Methylation Are Involved in Wilms Tumor Progression" Cells 8, no. 8: 921. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8080921