The Intricate Interplay between Epigenetic Events, Alternative Splicing and Noncoding RNA Deregulation in Colorectal Cancer

 , , and
, , and
Abstract
:1. Introduction
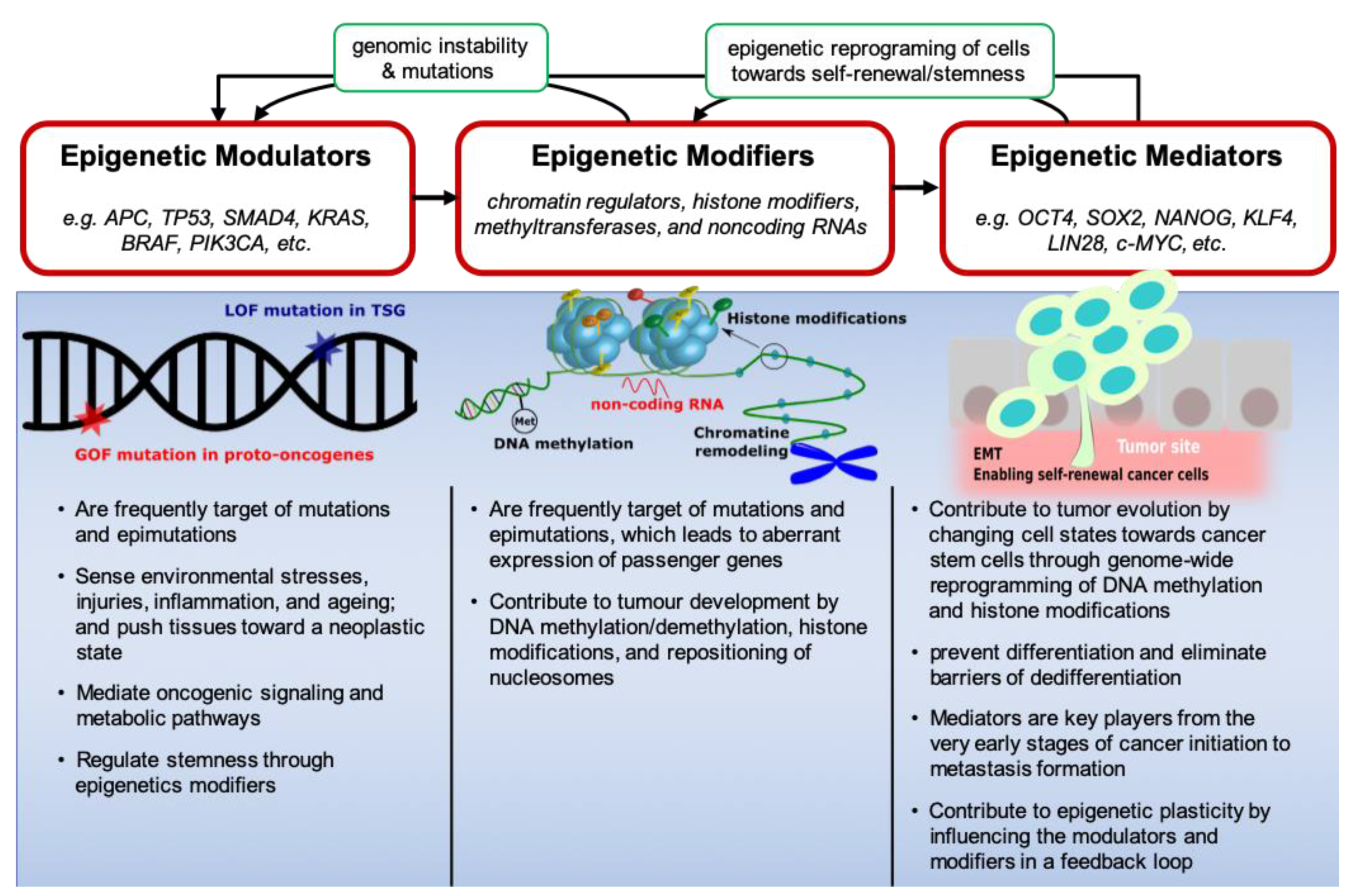
2. Epigenetic Modulators, Modifiers and Mediators in CRC
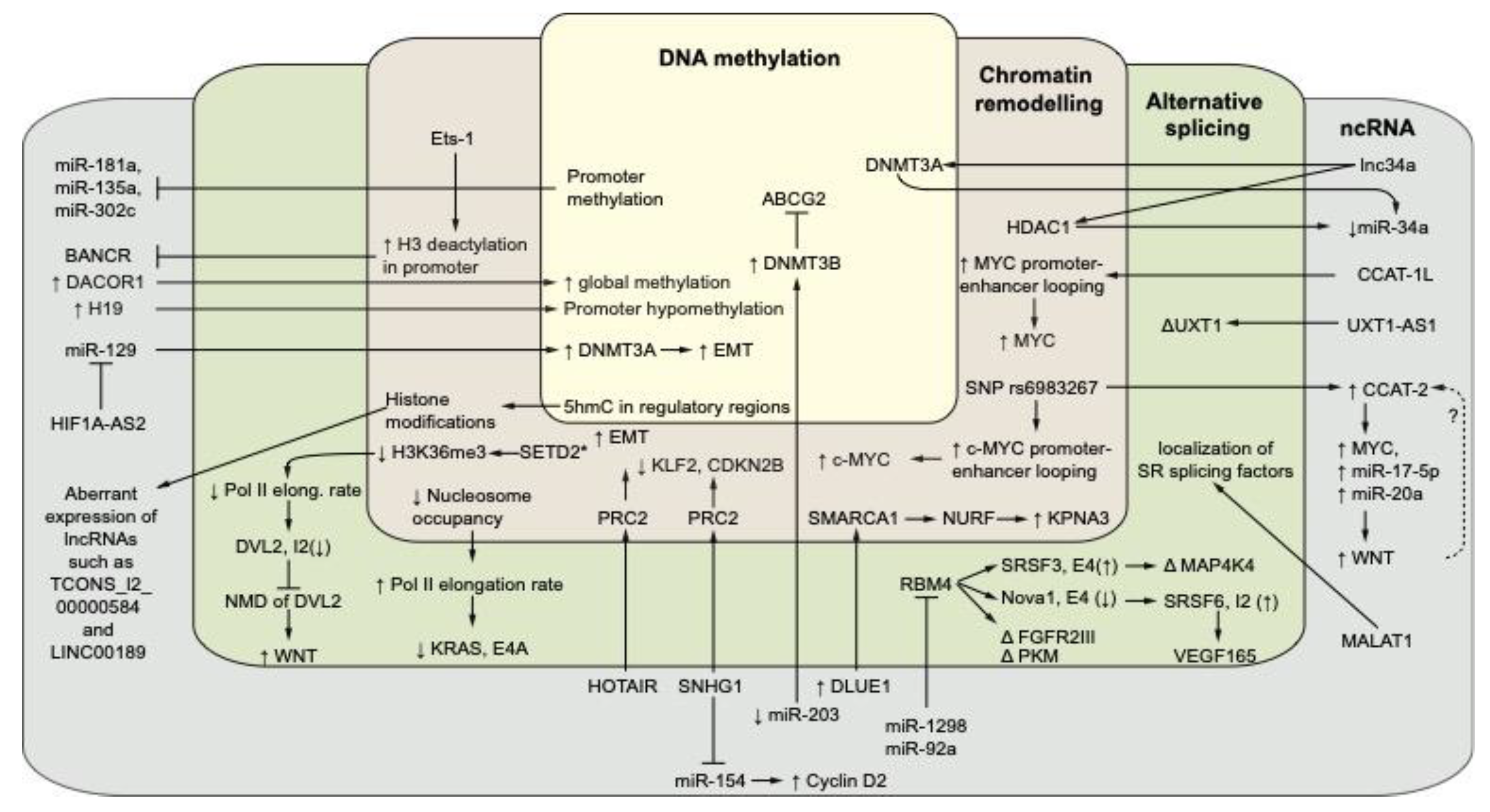
3. The Interplay between Non-Coding RNAs and Epigenetics in CRC
3.1. Noncoding RNAs and DNA Methylation in CRC
3.2. Noncoding RNAs, Chromatin Remodeling, and Histone Modifications in CRC
4. Regulation of Alternative Splicing in CRC
4.1. Epigenetic Regulators of Alternative Splicing in CRC
4.1.1. Modulation of RNA Pol II Elongation Rate
4.1.2. Splicing Factor Recruitment or Sequestration
4.2. Non-Coding RNAs and the Regulation of Alternative Splicing in CRC
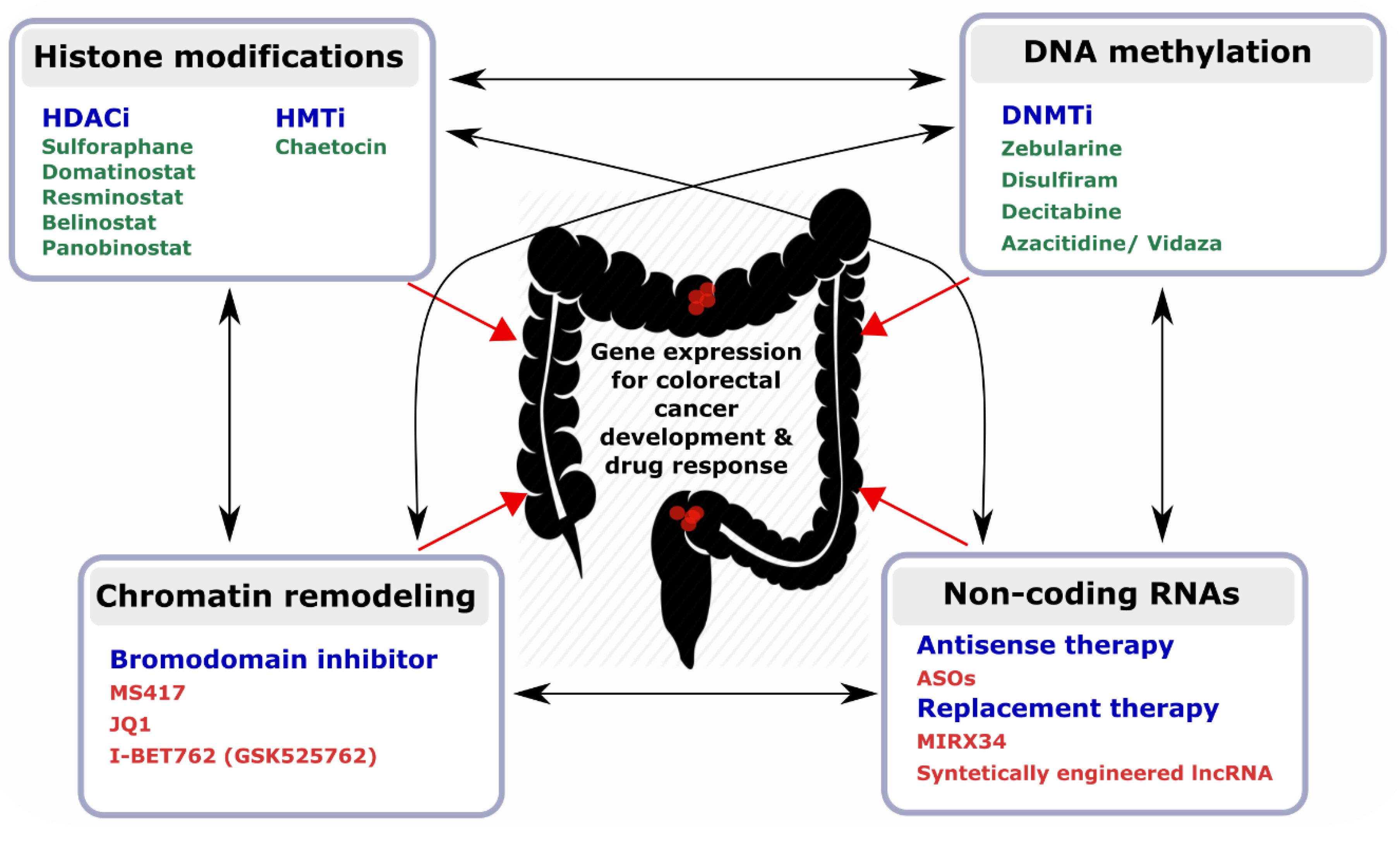
5. Epi-Biomarkers and Promising Targets for the Design of epi-Drugs
6. Future Directions
Supplementary Materials
Funding
Conflicts of Interest
References
- Shen, H.; Laird, P.W. Interplay between the cancer genome and epigenome. Cell 2013, 153, 38–55. [Google Scholar] [CrossRef] [PubMed]
- Assenov, Y.; Brocks, D.; Gerhauser, C. Intratumor heterogeneity in epigenetic patterns. Semin. Cancer Biol. 2018, 51, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Vaiopoulos, A.G.; Athanasoula, K.; Papavassiliou, A.G. Epigenetic modifications in colorectal cancer: Molecular insights and therapeutic challenges. Biochim. Biophys. Acta 2014, 1842, 971–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migliore, L.; Migheli, F.; Spisni, R.; Coppede, F. Genetics, cytogenetics, and epigenetics of colorectal cancer. J. Biomed. Biotechnol. 2011, 2011, 792362. [Google Scholar] [CrossRef] [PubMed]
- Guinney, J.; Dienstmann, R.; Wang, X.; de Reynies, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Duong, H.Q. The molecular characteristics of colorectal cancer: Implications for diagnosis and therapy. Oncol. Lett. 2018, 16, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.N. Genetic and epigenetic alterations of colorectal cancer. Intest. Res. 2018, 16, 327–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, K.; Chen, X.; Hu, X.; Liu, X.; Xu, T.; Sun, H.; Pan, Y.; He, B.; Wang, S. Lactb, a novel epigenetic silenced tumor suppressor, inhibits colorectal cancer progression by attenuating MDM2-mediated p53 ubiquitination and degradation. Oncogene 2018, 37, 5534–5551. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Koldobskiy, M.A.; Gondor, A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genet. 2016, 17, 284–299. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cardus, A.; Moran, S.; Musulen, E.; Moutinho, C.; Manzano, J.L.; Martinez-Balibrea, E.; Tierno, M.; Elez, E.; Landolfi, S.; Lorden, P.; et al. Epigenetic homogeneity within colorectal tumors predicts shorter relapse-free and overall survival times for patients with locoregional cancer. Gastroenterology 2016, 151, 961–972. [Google Scholar] [CrossRef]
- Widschwendter, M.; Fiegl, H.; Egle, D.; Mueller-Holzner, E.; Spizzo, G.; Marth, C.; Weisenberger, D.J.; Campan, M.; Young, J.; Jacobs, I.; et al. Epigenetic stem cell signature in cancer. Nat. Genet. 2007, 39, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Gotea, V.; Margolin, G.; Elnitski, L. Significant associations between driver gene mutations and DNA methylation alterations across many cancer types. PLoS Comput. Biol. 2017, 13, e1005840. [Google Scholar] [CrossRef] [PubMed]
- Hinoue, T.; Weisenberger, D.J.; Pan, F.; Campan, M.; Kim, M.; Young, J.; Whitehall, V.L.; Leggett, B.A.; Laird, P.W. Analysis of the association between CIMP and BRAF in colorectal cancer by DNA methylation profiling. PLoS ONE 2009, 4, e8357. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, E.; Suzuki, H.; Yamano, H.O.; Maruyama, R.; Nojima, M.; Kamimae, S.; Sawada, T.; Ashida, M.; Yoshikawa, K.; Kimura, T.; et al. Molecular dissection of premalignant colorectal lesions reveals early onset of the CPG island methylator phenotype. Am. J. Pathol. 2012, 181, 1847–1861. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.Y.; Chen, J.S.; Chang, S.C.; Wang, M.C.; Chang, N.C.; Wen, Y.H.; Tsai, W.S.; Liu, W.H.; Liu, H.L.; Lu, J.J. Acquired somatic TP53 or PIK3CA mutations are potential predictors of when polyps evolve into colorectal cancer. Oncotarget 2017, 8, 72352–72362. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Jones, P.A. Epigenetic determinants of cancer. Cold Spring Harb. Perspect. Biol. 2016, 8, a019505. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.H.; Li, Y.; Liu, X.H.; Sui, H.M.; Liu, Y.X.; Xiao, Z.Q.; Zheng, P.; Chen, L.; Yao, S.; Xing, C.; et al. A signature for induced pluripotent stem cell-associated genes in colorectal cancer. Med. Oncol. 2013, 30, 426. [Google Scholar] [CrossRef]
- Mathonnet, M.; Perraud, A.; Christou, N.; Akil, H.; Melin, C.; Battu, S.; Jauberteau, M.O.; Denizot, Y. Hallmarks in colorectal cancer: Angiogenesis and cancer stem-like cells. World J. Gastroenterol. 2014, 20, 4189–4196. [Google Scholar] [CrossRef]
- Matano, M.; Date, S.; Shimokawa, M.; Takano, A.; Fujii, M.; Ohta, Y.; Watanabe, T.; Kanai, T.; Sato, T. Modeling colorectal cancer using crispr-cas9-mediated engineering of human intestinal organoids. Nat. Med. 2015, 21, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; van Jaarsveld, R.H.; Ponsioen, B.; Zimberlin, C.; van Boxtel, R.; Buijs, A.; Sachs, N.; Overmeer, R.M.; Offerhaus, G.J.; Begthel, H.; et al. Sequential cancer mutations in cultured human intestinal stem cells. Nature 2015, 521, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Fumagalli, A.; Drost, J.; Suijkerbuijk, S.J.; van Boxtel, R.; de Ligt, J.; Offerhaus, G.J.; Begthel, H.; Beerling, E.; Tan, E.H.; Sansom, O.J.; et al. Genetic dissection of colorectal cancer progression by orthotopic transplantation of engineered cancer organoids. PNAS 2017, 114, E2357–E2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lannagan, T.R.M.; Lee, Y.K.; Wang, T.; Roper, J.; Bettington, M.L.; Fennell, L.; Vrbanac, L.; Jonavicius, L.; Somashekar, R.; Gieniec, K.; et al. Genetic editing of colonic organoids provides a molecularly distinct and orthotopic preclinical model of serrated carcinogenesis. Gut 2019, 68, 684–692. [Google Scholar] [CrossRef] [PubMed]
- Fang, M.; Ou, J.; Hutchinson, L.; Green, M.R. The braf oncoprotein functions through the transcriptional repressor MAFG to mediate the CPG island methylator phenotype. Mol. Cell 2014, 55, 904–915. [Google Scholar] [CrossRef] [PubMed]
- Fennell, L.; Dumenil, T.; Wockner, L.; Hartel, G.; Nones, K.; Bond, C.; Borowsky, J.; Liu, C.; McKeone, D.; Bowdler, L.; et al. Integrative genome-scale DNA methylation analysis of a large and unselected cohort reveals five distinct subtypes of colorectal adenocarcinomas. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Serra, R.W.; Fang, M.; Park, S.M.; Hutchinson, L.; Green, M.R. A kras-directed transcriptional silencing pathway that mediates the CPG island methylator phenotype. Elife 2014, 3, e02313. [Google Scholar] [CrossRef] [PubMed]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Nebbioso, A.; Tambaro, F.P.; Dell’Aversana, C.; Altucci, L. Cancer epigenetics: Moving forward. PLoS Genet. 2018, 14, e1007362. [Google Scholar] [CrossRef]
- Plass, C.; Pfister, S.M.; Lindroth, A.M.; Bogatyrova, O.; Claus, R.; Lichter, P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat. Rev. Genet. 2013, 14, 765–780. [Google Scholar] [CrossRef]
- Pfister, S.X.; Ashworth, A. Marked for death: Targeting epigenetic changes in cancer. Nat. Rev. Drug Discov. 2017, 16, 241–263. [Google Scholar] [CrossRef]
- Lizarbe, M.A.; Calle-Espinosa, J.; Fernandez-Lizarbe, E.; Fernandez-Lizarbe, S.; Robles, M.A.; Olmo, N.; Turnay, J. Colorectal cancer: From the genetic model to posttranscriptional regulation by noncoding RNAs. Biomed. Res. Int. 2017, 2017, 7354260. [Google Scholar] [CrossRef]
- Han, D.; Wang, M.; Ma, N.; Xu, Y.; Jiang, Y.; Gao, X. Long noncoding RNAs: Novel players in colorectal cancer. Cancer Lett. 2015, 361, 13–21. [Google Scholar] [CrossRef]
- Kim, T.; Croce, C.M. Long noncoding RNAs: Undeciphered cellular codes encrypting keys of colorectal cancer pathogenesis. Cancer Lett. 2018, 417, 89–95. [Google Scholar] [CrossRef]
- Li, H.; Ma, S.Q.; Huang, J.; Chen, X.P.; Zhou, H.H. Roles of long noncoding RNAs in colorectal cancer metastasis. Oncotarget 2017, 8, 39859–39876. [Google Scholar] [CrossRef]
- Schmitz, U.; Naderi-Meshkin, H.; Gupta, S.K.; Wolkenhauer, O.; Vera, J. The RNA world in the 21st century-a systems approach to finding non-coding keys to clinical questions. Brief. Bioinform. 2016, 17, 380–392. [Google Scholar] [CrossRef]
- Wu, L.; Murat, P.; Matak-Vinkovic, D.; Murrell, A.; Balasubramanian, S. Binding interactions between long noncoding RNA HOTAIR and PRC2 proteins. Biochemistry 2013, 52, 9519–9527. [Google Scholar] [CrossRef]
- Wang, K.; Jin, W.; Song, Y.; Fei, X. Lncrna rp11-436h11.5, functioning as a competitive endogenous Rna, upregulates bcl-w expression by sponging mir-335-5p and promotes proliferation and invasion in renal cell carcinoma. Mol. Cancer 2017, 16, 166. [Google Scholar] [CrossRef]
- Neve, B.; Jonckheere, N.; Vincent, A.; Van Seuningen, I. Epigenetic regulation by lncRNAs: An overview focused on uca1 in colorectal cancer. Cancers 2018, 10, 440. [Google Scholar] [CrossRef]
- Liu, T.; Han, Z.; Li, H.; Zhu, Y.; Sun, Z.; Zhu, A. Lncrna dleu1 contributes to colorectal cancer progression via activation of KPNA3. Mol. Cancer 2018, 17, 118. [Google Scholar] [CrossRef]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 79–92. [Google Scholar] [CrossRef]
- Liu, Y.; Sethi, N.S.; Hinoue, T.; Schneider, B.G.; Cherniack, A.D.; Sanchez-Vega, F.; Seoane, J.A.; Farshidfar, F.; Bowlby, R.; Islam, M.; et al. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell 2018, 33, 721–735.e8. [Google Scholar] [CrossRef]
- Matsuzaki, K.; Deng, G.; Tanaka, H.; Kakar, S.; Miura, S.; Kim, Y.S. The relationship between global methylation level, loss of heterozygosity, and microsatellite instability in sporadic colorectal cancer. Clin. Cancer Res. 2005, 11, 8564–8569. [Google Scholar] [CrossRef]
- Deng, G.; Nguyen, A.; Tanaka, H.; Matsuzaki, K.; Bell, I.; Mehta, K.R.; Terdiman, J.P.; Waldman, F.M.; Kakar, S.; Gum, J.; et al. Regional hypermethylation and global hypomethylation are associated with altered chromatin conformation and histone acetylation in colorectal cancer. Int. J. Cancer 2006, 118, 2999–3005. [Google Scholar] [CrossRef]
- Nazemalhosseini Mojarad, E.; Kuppen, P.J.; Aghdaei, H.A.; Zali, M.R. The cpg island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol Hepatol. Bed. Bench. 2013, 6, 120–128. [Google Scholar]
- Rokavec, M.; Horst, D.; Hermeking, H. Cellular model of colon cancer progression reveals signatures of mRNAs, miRNA, lncRNAs, and epigenetic modifications associated with metastasis. Cancer Res. 2017, 77, 1854–1867. [Google Scholar] [CrossRef]
- McCleland, M.L.; Mesh, K.; Lorenzana, E.; Chopra, V.S.; Segal, E.; Watanabe, C.; Haley, B.; Mayba, O.; Yaylaoglu, M.; Gnad, F.; et al. CCAT1 is an enhancer-templated RNA that predicts BET sensitivity in colorectal cancer. J. Clin. Investig. 2016, 126, 639–652. [Google Scholar] [CrossRef]
- Mio, C.; Bulotta, S.; Russo, D.; Damante, G. Reading cancer: Chromatin readers as druggable targets for cancer treatment. Cancers 2019, 11, 61. [Google Scholar] [CrossRef]
- Merry, C.R.; Forrest, M.E.; Sabers, J.N.; Beard, L.; Gao, X.H.; Hatzoglou, M.; Jackson, M.W.; Wang, Z.; Markowitz, S.D.; Khalil, A.M. DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum. Mol. Genet. 2015, 24, 6240–6253. [Google Scholar] [CrossRef]
- Somasundaram, S.; Forrest, M.E.; Moinova, H.; Cohen, A.; Varadan, V.; LaFramboise, T.; Markowitz, S.; Khalil, A.M. The dnmt1-associated lincrna dacor1 reprograms genome-wide DNA methylation in colon cancer. Clin. Epigenet. 2018, 10, 127. [Google Scholar] [CrossRef]
- Uribe-Lewis, S.; Stark, R.; Carroll, T.; Dunning, M.J.; Bachman, M.; Ito, Y.; Stojic, L.; Halim, S.; Vowler, S.L.; Lynch, A.G.; et al. 5-hydroxymethylcytosine marks promoters in colon that resist DNA hypermethylation in cancer. Genome Biol. 2015, 16, 69. [Google Scholar] [CrossRef] [Green Version]
- Tian, Y.; Pan, F.; Sun, X.; Gan, M.; Lin, A.; Zhang, D.; Zhu, Y.; Lai, M. Association of tet1 expression with colorectal cancer progression. Scand. J. Gastroenterol. 2017, 52, 312–320. [Google Scholar] [CrossRef]
- Hu, H.; Shu, M.; He, L.; Yu, X.; Liu, X.; Lu, Y.; Chen, Y.; Miao, X.; Chen, X. Epigenomic landscape of 5-hydroxymethylcytosine reveals its transcriptional regulation of LncRNAs in colorectal cancer. Br. J. Cancer 2017, 116, 658–668. [Google Scholar] [CrossRef]
- Puig, I.; Tenbaum, S.P.; Chicote, I.; Arques, O.; Martinez-Quintanilla, J.; Cuesta-Borras, E.; Ramirez, L.; Gonzalo, P.; Soto, A.; Aguilar, S.; et al. TET2 controls chemoresistant slow-cycling cancer cell survival and tumor recurrence. J. Clin. Investig. 2018, 128, 3887–3905. [Google Scholar] [CrossRef]
- Mitrea, C.; Wijesinghe, P.; Dyson, G.; Kruger, A.; Ruden, D.M.; Draghici, S.; Bollig-Fischer, A. Integrating 5hmc and gene expression data to infer regulatory mechanisms. Bioinformatics 2018, 34, 1441–1447. [Google Scholar] [CrossRef]
- Lin, J.; Shi, Z.; Yu, Z.; He, Z. LncRNA HIF1A-AS2 positively affects the progression and EMT formation of colorectal cancer through regulating miR-129-5p and DNMT3A. Biomed. Pharmacother. 2018, 98, 433–439. [Google Scholar] [CrossRef]
- Han, L.; Witmer, P.D.; Casey, E.; Valle, D.; Sukumar, S. DNA methylation regulates microRNA expression. Cancer Biol. Ther. 2007, 6, 1284–1288. [Google Scholar] [CrossRef]
- Shi, L.; Li, X.; Wu, Z.; Nie, J.; Guo, M.; Mei, Q.; Han, W. DNA methylation-mediated repression of miR-181a/135a/302c expression promotes the microsatellite-unstable colorectal cancer development and 5-FU resistance via targeting PLAG1. J. Genet. Genom. 2018, 45, 205–214. [Google Scholar] [CrossRef]
- Qin, J.; Ke, J.; Xu, J.; Wang, F.; Zhou, Y.; Jiang, Y.; Wang, Z. Downregulation of microRNA-132 by DNA hypermethylation is associated with cell invasion in colorectal cancer. Oncotargets Ther. 2015, 8, 3639–3648. [Google Scholar] [Green Version]
- Lv, L.V.; Zhou, J.; Lin, C.; Hu, G.; Yi, L.U.; Du, J.; Gao, K.; Li, X. DNA methylation is involved in the aberrant expression of miR-133b in colorectal cancer cells. Oncol. Lett. 2015, 10, 907–912. [Google Scholar] [CrossRef] [Green Version]
- To, K.K.; Leung, W.W.; Ng, S.S. A novel miR-203-DNMT3b-ABCG2 regulatory pathway predisposing colorectal cancer development. Mol. Carcinog. 2017, 56, 464–477. [Google Scholar] [CrossRef]
- Tian, F.; Tang, Z.; Song, G.; Pan, Y.; He, B.; Bao, Q.; Wang, S. Loss of imprinting of IGF2 correlates with hypomethylation of the H19 differentially methylated region in the tumor tissue of colorectal cancer patients. Mol. Med. Rep. 2012, 5, 1536–1540. [Google Scholar]
- Cui, H.; Onyango, P.; Brandenburg, S.; Wu, Y.; Hsieh, C.L.; Feinberg, A.P. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. 2002, 62, 6442–6446. [Google Scholar]
- Cheng, Y.W.; Idrees, K.; Shattock, R.; Khan, S.A.; Zeng, Z.; Brennan, C.W.; Paty, P.; Barany, F. Loss of imprinting and marked gene elevation are 2 forms of aberrant IGF2 expression in colorectal cancer. Int. J. Cancer 2010, 127, 568–577. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Bu, J.; Liu, X.; Wang, W.; Mai, W.; Lv, B.; Zou, J.; Mo, X.; Li, X.; Wang, J.; et al. miR-133b suppresses metastasis by targeting HOXA9 in human colorectal cancer. Oncotarget 2017, 8, 63935–63948. [Google Scholar] [CrossRef]
- Ren, S.; Wang, F.; Shen, J.; Sun, Y.; Xu, W.; Lu, J.; Wei, M.; Xu, C.; Wu, C.; Zhang, Z.; et al. Long non-coding rna metastasis associated in lung adenocarcinoma transcript 1 derived miniRNA as a novel plasma-based biomarker for diagnosing prostate cancer. Eur. J. Cancer 2013, 49, 2949–2959. [Google Scholar] [CrossRef]
- Tang, J.T.; Wang, J.L.; Du, W.; Hong, J.; Zhao, S.L.; Wang, Y.C.; Xiong, H.; Chen, H.M.; Fang, J.Y. Microrna 345, a methylation-sensitive microRNA is involved in cell proliferation and invasion in human colorectal cancer. Carcinogenesis 2011, 32, 1207–1215. [Google Scholar] [CrossRef]
- Ling, H.; Spizzo, R.; Atlasi, Y.; Nicoloso, M.; Shimizu, M.; Redis, R.S.; Nishida, N.; Gafa, R.; Song, J.; Guo, Z.; et al. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome Res. 2013, 23, 1446–1461. [Google Scholar] [CrossRef]
- Kasagi, Y.; Oki, E.; Ando, K.; Ito, S.; Iguchi, T.; Sugiyama, M.; Nakashima, Y.; Ohgaki, K.; Saeki, H.; Mimori, K.; et al. The expression of CCAT2, a novel long noncoding RNA transcript, and rs6983267 single-nucleotide polymorphism genotypes in colorectal cancers. Oncology 2017, 92, 48–54. [Google Scholar] [CrossRef]
- Xiang, J.F.; Yin, Q.F.; Chen, T.; Zhang, Y.; Zhang, X.O.; Wu, Z.; Zhang, S.; Wang, H.B.; Ge, J.; Lu, X.; et al. Human colorectal cancer-specific CCAT1-l LncRNA regulates long-range chromatin interactions at the MYC locus. Cell Res. 2014, 24, 513–531. [Google Scholar] [CrossRef]
- Ozawa, T.; Matsuyama, T.; Toiyama, Y.; Takahashi, N.; Ishikawa, T.; Uetake, H.; Yamada, Y.; Kusunoki, M.; Calin, G.; Goel, A. CCAT1 and CCAT2 long noncoding rNAS, located within the 8q.24.21 ‘gene desert’, serve as important prognostic biomarkers in colorectal cancer. Ann. Oncol. 2017, 28, 1882–1888. [Google Scholar] [CrossRef]
- Kogo, R.; Shimamura, T.; Mimori, K.; Kawahara, K.; Imoto, S.; Sudo, T.; Tanaka, F.; Shibata, K.; Suzuki, A.; Komune, S.; et al. Long noncoding rna hotair regulates polycomb-dependent chromatin modification and is associated with poor prognosis in colorectal cancers. Cancer Res. 2011, 71, 6320–6326. [Google Scholar] [CrossRef]
- Chen, X.; Liu, B.; Yang, R.; Guo, Y.; Li, F.; Wang, L.; Hu, H. Integrated analysis of long non-coding rNAS in human colorectal cancer. Oncotarget 2016, 7, 23897–23908. [Google Scholar] [CrossRef]
- Xu, M.; Chen, X.; Lin, K.; Zeng, K.; Liu, X.; Pan, B.; Xu, X.; Xu, T.; Hu, X.; Sun, L.; et al. The long noncoding rna snhg1 regulates colorectal cancer cell growth through interactions with EZH2 and miR-154-5p. Mol. Cancer 2018, 17, 141. [Google Scholar] [CrossRef]
- Ding, J.; Li, J.; Wang, H.; Tian, Y.; Xie, M.; He, X.; Ji, H.; Ma, Z.; Hui, B.; Wang, K.; et al. Long noncoding RNA crnde promotes colorectal cancer cell proliferation via epigenetically silencing DUSP5/CDKN1a expression. Cell Death Dis. 2017, 8, e2997. [Google Scholar] [CrossRef]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.P.; Goh, B.C. Exosome-mediated metastasis: From epithelial-mesenchymal transition to escape from immunosurveillance. Trends Pharmacol. Sci. 2016, 37, 606–617. [Google Scholar] [CrossRef]
- Wang, L.; Bu, P.; Ai, Y.; Srinivasan, T.; Chen, H.J.; Xiang, K.; Lipkin, S.M.; Shen, X. A long non-coding RNA targets microrna miR-34a to regulate colon cancer stem cell asymmetric division. Elife 2016, 5, e14620. [Google Scholar] [CrossRef]
- Li, A.X.; Xin, W.Q.; Ma, C.G. Fentanyl inhibits the invasion and migration of colorectal cancer cells via inhibiting the negative regulation of ETS-1 on bancr. Biochem. Biophys. Res. Commun. 2015, 465, 594–600. [Google Scholar] [CrossRef]
- Alaiyan, B.; Ilyayev, N.; Stojadinovic, A.; Izadjoo, M.; Roistacher, M.; Pavlov, V.; Tzivin, V.; Halle, D.; Pan, H.; Trink, B.; et al. Differential expression of colon cancer associated transcript1 (CCAT1) along the colonic adenoma-carcinoma sequence. BMC Cancer 2013, 13, 196. [Google Scholar] [CrossRef]
- Li, P.; Zhang, X.; Wang, H.; Wang, L.; Liu, T.; Du, L.; Yang, Y.; Wang, C. MALAT1 is associated with poor response to oxaliplatin-based chemotherapy in colorectal cancer patients and promotes chemoresistance through EZH2. Mol. Cancer Ther. 2017, 16, 739–751. [Google Scholar] [CrossRef]
- Yang, X.J.; Huang, C.Q.; Peng, C.W.; Hou, J.X.; Liu, J.Y. Long noncoding RNA HULC promotes colorectal carcinoma progression through epigenetically repressing NKD2 expression. Gene 2016, 592, 172–178. [Google Scholar] [CrossRef]
- Ma, Z.; Gu, S.; Song, M.; Yan, C.; Hui, B.; Ji, H.; Wang, J.; Zhang, J.; Wang, K.; Zhao, Q. Long non-coding RNA SNHG17 is an unfavourable prognostic factor and promotes cell proliferation by epigenetically silencing p57 in colorectal cancer. Mol. Biosyst. 2017, 13, 2350–2361. [Google Scholar] [CrossRef]
- Ma, Z.; Peng, P.; Zhou, J.; Hui, B.; Ji, H.; Wang, J.; Wang, K. Long non-coding RNA SH3PXD2A-AS1 promotes cell progression partly through epigenetic silencing p57 and KLF2 in colorectal cancer. Cell. Physiol. Biochem. 2018, 46, 2197–2214. [Google Scholar] [CrossRef]
- Li, Z.; Qiu, R.; Qiu, X.; Tian, T. SNHG6 promotes tumor growth via repression of p21 in colorectal cancer. Cell. Physiol. Biochem. 2018, 49, 463–478. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, P.; Gao, Y.; Ta, N.; Zhang, Y.; Cai, J.; Zhao, Y.; Liu, S.; Zheng, J. MEG3 activated by vitamin d inhibits colorectal cancer cells proliferation and migration via regulating clusterin. EBioMedicine 2018, 30, 148–157. [Google Scholar] [CrossRef]
- Marin-Bejar, O.; Marchese, F.P.; Athie, A.; Sanchez, Y.; Gonzalez, J.; Segura, V.; Huang, L.; Moreno, I.; Navarro, A.; Monzo, M.; et al. Pint LincRNA connects the p53 pathway with epigenetic silencing by the polycomb repressive complex 2. Genome Biol. 2013, 14, R104. [Google Scholar] [CrossRef]
- Marin-Bejar, O.; Mas, A.M.; Gonzalez, J.; Martinez, D.; Athie, A.; Morales, X.; Galduroz, M.; Raimondi, I.; Grossi, E.; Guo, S.; et al. The human lncrna linc-pint inhibits tumor cell invasion through a highly conserved sequence element. Genome Biol. 2017, 18, 202. [Google Scholar] [CrossRef]
- Huang, W.; Su, G.; Huang, X.; Zou, A.; Wu, J.; Yang, Y.; Zhu, Y.; Liang, S.; Li, D.; Ma, F.; et al. Long noncoding rna pcat6 inhibits colon cancer cell apoptosis by regulating anti-apoptotic protein arc expression via EZH2. Cell Cycle 2019, 18, 69–83. [Google Scholar] [CrossRef]
- Li, T.; Xu, C.; Cai, B.; Zhang, M.; Gao, F.; Gan, J. Expression and clinicopathological significance of the LncRNA HOXA11-as in colorectal cancer. Oncol. Lett. 2016, 12, 4155–4160. [Google Scholar] [CrossRef]
- Sun, M.; Nie, F.; Wang, Y.; Zhang, Z.; Hou, J.; He, D.; Xie, M.; Xu, L.; De, W.; Wang, Z.; et al. LncRNA HOXA11-as promotes proliferation and invasion of gastric cancer by scaffolding the chromatin modification factors PRC2, LSD1, and DNMT1. Cancer Res. 2016, 76, 6299–6310. [Google Scholar] [CrossRef]
- Bisognin, A.; Pizzini, S.; Perilli, L.; Esposito, G.; Mocellin, S.; Nitti, D.; Zanovello, P.; Bortoluzzi, S.; Mandruzzato, S. An integrative framework identifies alternative splicing events in colorectal cancer development. Mol. Oncol. 2014, 8, 129–141. [Google Scholar] [CrossRef]
- Zong, Z.; Li, H.; Yi, C.; Ying, H.; Zhu, Z.; Wang, H. Genome-wide profiling of prognostic alternative splicing signature in colorectal cancer. Front. Oncol. 2018, 8, 537. [Google Scholar] [CrossRef]
- Xiong, Y.; Deng, Y.; Wang, K.; Zhou, H.; Zheng, X.; Si, L.; Fu, Z. Profiles of alternative splicing in colorectal cancer and their clinical significance: A study based on large-scale sequencing data. EBioMedicine 2018, 36, 183–195. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, H.; Shen, S.; Sun, L.; Yuan, Y.; Xing, C. Alternative splicing events implicated in carcinogenesis and prognosis of colorectal cancer. J. Cancer 2018, 9, 1754–1764. [Google Scholar] [CrossRef] [Green Version]
- Kahles, A.; Lehmann, K.V.; Toussaint, N.C.; Huser, M.; Stark, S.G.; Sachsenberg, T.; Stegle, O.; Kohlbacher, O.; Sander, C.; Ratsch, G. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell 2018, 34, 211–224.e6. [Google Scholar] [CrossRef]
- Eilertsen, I.A.; Sveen, A.; Stromme, J.M.; Skotheim, R.I.; Nesbakken, A.; Lothe, R.A. Alternative splicing expands the prognostic impact of KRAS in microsatellite stable primary colorectal cancer. Int. J. Cancer 2019, 144, 841–847. [Google Scholar] [CrossRef]
- Sebestyen, E.; Singh, B.; Minana, B.; Pages, A.; Mateo, F.; Pujana, M.A.; Valcarcel, J.; Eyras, E. Large-scale analysis of genome and transcriptome alterations in multiple tumors unveils novel cancer-relevant splicing networks. Genome Res. 2016, 26, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Ghigna, C.; Giordano, S.; Shen, H.; Benvenuto, F.; Castiglioni, F.; Comoglio, P.M.; Green, M.R.; Riva, S.; Biamonti, G. Cell motility is controlled by SF2/ASF through alternative splicing of the RON protooncogene. Mol. Cell 2005, 20, 881–890. [Google Scholar] [CrossRef]
- Thorsen, K.; Mansilla, F.; Schepeler, T.; Oster, B.; Rasmussen, M.H.; Dyrskjot, L.; Karni, R.; Akerman, M.; Krainer, A.R.; Laurberg, S.; et al. Alternative splicing of SLC39A14 in colorectal cancer is regulated by the wnt pathway. Mol. Cell. Proteom. 2011, 10, M110-002998. [Google Scholar] [CrossRef]
- Goncalves, V.; Henriques, A.F.; Pereira, J.F.; Neves Costa, A.; Moyer, M.P.; Moita, L.F.; Gama-Carvalho, M.; Matos, P.; Jordan, P. Phosphorylation of SRSF1 by SRPK1 regulates alternative splicing of tumor-related rac1b in colorectal cells. RNA 2014, 20, 474–482. [Google Scholar] [CrossRef]
- Cohen-Eliav, M.; Golan-Gerstl, R.; Siegfried, Z.; Andersen, C.L.; Thorsen, K.; Orntoft, T.F.; Mu, D.; Karni, R. The splicing factor SRSF6 is amplified and is an oncoprotein in lung and colon cancers. J. Pathol. 2013, 229, 630–639. [Google Scholar] [CrossRef]
- Neklason, D.W.; Solomon, C.H.; Dalton, A.L.; Kuwada, S.K.; Burt, R.W. Intron 4 mutation in APC gene results in splice defect and attenuated FAP phenotype. Fam. Cancer 2004, 3, 35–40. [Google Scholar] [CrossRef]
- McVety, S.; Li, L.; Gordon, P.H.; Chong, G.; Foulkes, W.D. Disruption of an exon splicing enhancer in exon 3 of MLH1 is the cause of HNPCC in a quebec family. J. Med Genet. 2006, 43, 153–156. [Google Scholar] [CrossRef]
- Goncalves, V.; Theisen, P.; Antunes, O.; Medeira, A.; Ramos, J.S.; Jordan, P.; Isidro, G. A missense mutation in the apc tumor suppressor gene disrupts an ASF/SF2 splicing enhancer motif and causes pathogenic skipping of exon 14. Mutat. Res. 2009, 662, 33–36. [Google Scholar] [CrossRef]
- Urbanski, L.M.; Leclair, N.; Anczukow, O. Alternative-splicing defects in cancer: Splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip. Rev. RNA 2018, 9, e1476. [Google Scholar] [CrossRef]
- Lin, J.C.; Lee, Y.C.; Liang, Y.C.; Fann, Y.C.; Johnson, K.R.; Lin, Y.J. The impact of the rbm4-initiated splicing cascade on modulating the carcinogenic signature of colorectal cancer cells. Sci. Rep. 2017, 7, 44204. [Google Scholar] [CrossRef]
- Lin, J.C.; Lee, Y.C.; Tan, T.H.; Liang, Y.C.; Chuang, H.C.; Fann, Y.C.; Johnson, K.R.; Lin, Y.J. RBM4-SRSF3-MAP4K4 splicing cascade modulates the metastatic signature of colorectal cancer cell. Biochim. Biophys. Acta. Mol. Cell Res. 2018, 1865, 259–272. [Google Scholar] [CrossRef]
- Wiencke, J.K.; Zheng, S.; Morrison, Z.; Yeh, R.F. Differentially expressed genes are marked by histone 3 lysine 9 trimethylation in human cancer cells. Oncogene 2008, 27, 2412–2421. [Google Scholar] [CrossRef]
- Riffo-Campos, A.L.; Gimeno-Valiente, F.; Rodriguez, F.M.; Cervantes, A.; Lopez-Rodas, G.; Franco, L.; Castillo, J. Role of epigenetic factors in the selection of the alternative splicing isoforms of human KRAS in colorectal cancer cell lines. Oncotarget 2018, 9, 20578–20589. [Google Scholar] [CrossRef] [Green Version]
- Matveeva, E.A.; Al-Tinawi, Q.M.H.; Rouchka, E.C.; Fondufe-Mittendorf, Y.N. Coupling of PARP1-mediated chromatin structural changes to transcriptional RNA polymerase ii elongation and cotranscriptional splicing. Epigenetics Chromatin 2019, 12, 15. [Google Scholar] [CrossRef]
- Yuan, H.; Li, N.; Fu, D.; Ren, J.; Hui, J.; Peng, J.; Liu, Y.; Qiu, T.; Jiang, M.; Pan, Q.; et al. Histone methyltransferase SETD2 modulates alternative splicing to inhibit intestinal tumorigenesis. J. Clin. Invest. 2017, 127, 3375–3391. [Google Scholar] [CrossRef]
- Sureau, A.; Gattoni, R.; Dooghe, Y.; Stevenin, J.; Soret, J. SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J. 2001, 20, 1785–1796. [Google Scholar] [CrossRef] [Green Version]
- Tilgner, H.; Nikolaou, C.; Althammer, S.; Sammeth, M.; Beato, M.; Valcarcel, J.; Guigo, R. Nucleosome positioning as a determinant of exon recognition. Nat. Struct. Mol. Biol. 2009, 16, 996–1001. [Google Scholar] [CrossRef]
- Schwartz, S.; Meshorer, E.; Ast, G. Chromatin organization marks exon-intron structure. Nat. Struct. Mol. Biol. 2009, 16, 990–995. [Google Scholar] [CrossRef]
- Shukla, S.; Kavak, E.; Gregory, M.; Imashimizu, M.; Shutinoski, B.; Kashlev, M.; Oberdoerffer, P.; Sandberg, R.; Oberdoerffer, S. CTCF-promoted RNA polymerase ii pausing links DNA methylation to splicing. Nature 2011, 479, 74–79. [Google Scholar] [CrossRef]
- Iannone, C.; Pohl, A.; Papasaikas, P.; Soronellas, D.; Vicent, G.P.; Beato, M.; ValcaRcel, J. Relationship between nucleosome positioning and progesterone-induced alternative splicing in breast cancer cells. RNA 2015, 21, 360–374. [Google Scholar] [CrossRef] [Green Version]
- Matveeva, E.; Maiorano, J.; Zhang, Q.; Eteleeb, A.M.; Convertini, P.; Chen, J.; Infantino, V.; Stamm, S.; Wang, J.; Rouchka, E.C.; et al. Involvement of PARP1 in the regulation of alternative splicing. Cell Discov. 2016, 2, 15046. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.E.; Park, C.; Kim, K.E.; Kim, K.K. Histone and rna-binding protein interaction creates crosstalk network for regulation of alternative splicing. Biochem. Biophys. Res. Commun. 2018, 499, 30–36. [Google Scholar] [CrossRef]
- Kfir, N.; Lev-Maor, G.; Glaich, O.; Alajem, A.; Datta, A.; Sze, S.K.; Meshorer, E.; Ast, G. SF3B1 association with chromatin determines splicing outcomes. Cell Rep. 2015, 11, 618–629. [Google Scholar] [CrossRef]
- Maunakea, A.K.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MECP2 to promote exon recognition. Cell Res. 2013, 23, 1256–1269. [Google Scholar] [CrossRef]
- Gu, H.; Bock, C.; Mikkelsen, T.S.; Jager, N.; Smith, Z.D.; Tomazou, E.; Gnirke, A.; Lander, E.S.; Meissner, A. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat. Methods 2010, 7, 133–136. [Google Scholar] [CrossRef] [Green Version]
- Gelfman, S.; Cohen, N.; Yearim, A.; Ast, G. DNA-methylation effect on cotranscriptional splicing is dependent on GC architecture of the exon-intron structure. Genome Res. 2013, 23, 789–799. [Google Scholar] [CrossRef]
- Yearim, A.; Gelfman, S.; Shayevitch, R.; Melcer, S.; Glaich, O.; Mallm, J.P.; Nissim-Rafinia, M.; Cohen, A.H.; Rippe, K.; Meshorer, E.; et al. HP1 is involved in regulating the global impact of DNA methylation on alternative splicing. Cell Rep. 2015, 10, 1122–1134. [Google Scholar] [CrossRef]
- Fan, Y.; Li, H.; Liang, X.; Xiang, Z. Cbx3 promotes colon cancer cell proliferation by CDK6 kinase-independent function during cell cycle. Oncotarget 2017, 8, 19934–19946. [Google Scholar] [CrossRef]
- Liu, M.; Huang, F.; Zhang, D.; Ju, J.; Wu, X.B.; Wang, Y.; Wu, Y.; Nie, M.; Li, Z.; Ma, C.; et al. Heterochromatin protein hp1gamma promotes colorectal cancer progression and is regulated by miR-30a. Cancer Res. 2015, 75, 4593–4604. [Google Scholar] [CrossRef]
- Davie, J.R.; Xu, W.; Delcuve, G.P. Histone H3K4 trimethylation: Dynamic interplay with pre-mRNA splicing. Biochem. Cell Biol. 2016, 94, 1–11. [Google Scholar] [CrossRef]
- Dhami, P.; Saffrey, P.; Bruce, A.W.; Dillon, S.C.; Chiang, K.; Bonhoure, N.; Koch, C.M.; Bye, J.; James, K.; Foad, N.S.; et al. Complex exon-intron marking by histone modifications is not determined solely by nucleosome distribution. PLoS ONE 2010, 5, e12339. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Lev-Maor, G.; Ast, G. Pre-mrna splicing is a determinant of nucleosome organization. PLoS ONE 2013, 8, e53506. [Google Scholar] [CrossRef]
- Roda, D.; Castillo, J.; Telechea-Fernandez, M.; Gil, A.; Lopez-Rodas, G.; Franco, L.; Gonzalez-Rodriguez, P.; Rosello, S.; Perez-Fidalgo, J.A.; Garcia-Trevijano, E.R.; et al. EGF-induced acetylation of heterogeneous nuclear ribonucleoproteins is dependent on KRAS mutational status in colorectal cancer cells. PLoS ONE 2015, 10, e0130543. [Google Scholar] [CrossRef]
- Riffo-Campos, A.L.; Castillo, J.; Vallet-Sanchez, A.; Ayala, G.; Cervantes, A.; Lopez-Rodas, G.; Franco, L. In silico RNA-seq and experimental analyses reveal the differential expression and splicing of EPDR1 and ZNF518B genes in relation to KRAS mutations in colorectal cancer cells. Oncol. Rep. 2016, 36, 3627–3634. [Google Scholar] [CrossRef] [Green Version]
- Amirkhah, R.; Schmitz, U.; Linnebacher, M.; Wolkenhauer, O.; Farazmand, A. Microrna-mRNA interactions in colorectal cancer and their role in tumor progression. Genes Chromosomes Cancer 2015, 54, 129–141. [Google Scholar] [CrossRef]
- Tao, Y.; Ma, C.; Fan, Q.; Wang, Y.; Han, T.; Sun, C. Microrna-1296 facilitates proliferation, migration and invasion of colorectal cancer cells by targeting SFPQ. J. Cancer 2018, 9, 2317–2326. [Google Scholar] [CrossRef]
- Liang, Y.C.; Lin, W.C.; Lin, Y.J.; Lin, J.C. The impact of RNA binding motif protein 4-regulated splicing cascade on the progression and metabolism of colorectal cancer cells. Oncotarget 2015, 6, 38046–38060. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, I.; Munita, R.; Agirre, E.; Dittmer, T.A.; Gysling, K.; Misteli, T.; Luco, R.F. A LncRNA regulates alternative splicing via establishment of a splicing-specific chromatin signature. Nat. Struct. Mol. Biol. 2015, 22, 370–376. [Google Scholar] [CrossRef]
- Luco, R.F.; Pan, Q.; Tominaga, K.; Blencowe, B.J.; Pereira-Smith, O.M.; Misteli, T. Regulation of alternative splicing by histone modifications. Science 2010, 327, 996–1000. [Google Scholar] [CrossRef]
- Taniguchi, K.; Sakai, M.; Sugito, N.; Kumazaki, M.; Shinohara, H.; Yamada, N.; Nakayama, T.; Ueda, H.; Nakagawa, Y.; Ito, Y.; et al. PTBP1-associated microRNA-1 and -133b suppress the warburg effect in colorectal tumors. Oncotarget 2016, 7, 18940–18952. [Google Scholar] [CrossRef]
- Romero-Barrios, N.; Legascue, M.F.; Benhamed, M.; Ariel, F.; Crespi, M. Splicing regulation by long noncoding RNAs. Nucleic Acids Res. 2018, 46, 2169–2184. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.Y.; Zhu, Y.R.; Dai, D.J.; Wang, X.; Jin, H.C. Epigenetic regulation of alternative splicing. Am. J. Cancer Res. 2018, 8, 2346–2358. [Google Scholar]
- Song, X.; Zeng, Z.; Wei, H.; Wang, Z. Alternative splicing in cancers: From aberrant regulation to new therapeutics. Semin. Cell Dev. Biol. 2018, 75, 13–22. [Google Scholar] [CrossRef]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar] [CrossRef]
- Kong, J.; Sun, W.; Li, C.; Wan, L.; Wang, S.; Wu, Y.; Xu, E.; Zhang, H.; Lai, M. Long non-coding RNA LINC01133 inhibits epithelial-mesenchymal transition and metastasis in colorectal cancer by interacting with SRSF6. Cancer Lett. 2016, 380, 476–484. [Google Scholar] [CrossRef]
- Wan, L.; Yu, W.; Shen, E.; Sun, W.; Liu, Y.; Kong, J.; Wu, Y.; Han, F.; Zhang, L.; Yu, T.; et al. SRSF6-regulated alternative splicing that promotes tumour progression offers a therapy target for colorectal cancer. Gut 2019, 68, 118–129. [Google Scholar] [CrossRef]
- Ji, Q.; Zhang, L.; Liu, X.; Zhou, L.; Wang, W.; Han, Z.; Sui, H.; Tang, Y.; Wang, Y.; Liu, N.; et al. Long non-coding RNA MALAT1 promotes tumour growth and metastasis in colorectal cancer through binding to sfpq and releasing oncogene ptbp2 from sfpq/ptbp2 complex. Br. J. Cancer 2014, 111, 736–748. [Google Scholar] [CrossRef]
- Yang, P.; Chen, T.; Xu, Z.; Zhu, H.; Wang, J.; He, Z. Long noncoding RNA GAPLINC promotes invasion in colorectal cancer by targeting SNAI2 through binding with PSF and NONO. Oncotarget 2016, 7, 42183–42194. [Google Scholar] [CrossRef]
- Villamizar, O.; Chambers, C.B.; Riberdy, J.M.; Persons, D.A.; Wilber, A. Long noncoding RNA SAF and splicing factor 45 increase soluble FAS and resistance to apoptosis. Oncotarget 2016, 7, 13810–13826. [Google Scholar] [CrossRef]
- Yin, J.; Luo, W.; Zeng, X.; Zeng, L.; Li, Z.; Deng, X.; Tan, X.; Hu, W. UXT-AS1-induced alternative splicing of UXT is associated with tumor progression in colorectal cancer. Am. J. Cancer Res. 2017, 7, 462–472. [Google Scholar]
- Song, L.; Jia, J.; Peng, X.; Xiao, W.; Li, Y. The performance of the SEPT9 gene methylation assay and a comparison with other CRC screening tests: A meta-analysis. Sci. Rep. 2017, 7, 3032. [Google Scholar] [CrossRef]
- Liao, H.K.; Hatanaka, F.; Araoka, T.; Reddy, P.; Wu, M.Z.; Sui, Y.; Yamauchi, T.; Sakurai, M.; O’Keefe, D.D.; Nunez-Delicado, E.; et al. In vivo target gene activation via CRISPR/cas9-mediated trans-epigenetic modulation. Cell 2017, 171, 1495–1507.e15. [Google Scholar] [CrossRef]
- Stypula-Cyrus, Y.; Damania, D.; Kunte, D.P.; Cruz, M.D.; Subramanian, H.; Roy, H.K.; Backman, V. Hdac up-regulation in early colon field carcinogenesis is involved in cell tumorigenicity through regulation of chromatin structure. PLoS ONE 2013, 8, e64600. [Google Scholar] [CrossRef]
- Deng, X.; Ruan, H.; Zhang, X.; Xu, X.; Zhu, Y.; Peng, H.; Zhang, X.; Kong, F.; Guan, M. Long non-coding RNA CCAL transferred from fibroblasts by exosomes promotes chemoresistance of colorectal cancer cells. Int. J. Cancer 2019. [Google Scholar] [CrossRef]
- Chen, H.; Xu, J.; Hong, J.; Tang, R.; Zhang, X.; Fang, J.Y. Long noncoding rna profiles identify five distinct molecular subtypes of colorectal cancer with clinical relevance. Mol. Oncol. 2014, 8, 1393–1403. [Google Scholar] [CrossRef]
- James de Bony, E.; Bizet, M.; Van Grembergen, O.; Hassabi, B.; Calonne, E.; Putmans, P.; Bontempi, G.; Fuks, F. Comprehensive identification of long noncoding rnas in colorectal cancer. Oncotarget 2018, 9, 27605–27629. [Google Scholar]
- Surguchov, A.; Surgucheva, I.; Sharma, M.; Sharma, R.; Singh, V. Pore-forming proteins as mediators of novel epigenetic mechanism of epilepsy. Front. Neurol. 2017, 8, 3. [Google Scholar] [CrossRef]
- Srinivas Patnaik, A. Drugs targeting epigenetic modifications and plausible therapeutic strategies against colorectal cancer. Front. Pharmacol. 2019, 10, 588. [Google Scholar] [CrossRef]
- Matsui, M.; Corey, D.R. Non-coding RNAs as drug targets. Nat. Rev. Drug Discov. 2017, 16, 167–179. [Google Scholar] [CrossRef]
- Jothimani, G.; Sriramulu, S.; Chabria, Y.; Sun, X.F.; Banerjee, A.; Pathak, S. A review on theragnostic applications of micrornas and long non-coding rnas in colorectal cancer. Curr. Top. Med. Chem. 2018, 18, 2614–2629. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef]
- Wang, F.; Ma, Y.; Wang, H.; Qin, H. Reciprocal regulation between micrornas and epigenetic machinery in colorectal cancer. Oncol. Lett. 2017, 13, 1048–1057. [Google Scholar] [CrossRef]
- Xiang, S.; Zou, P.; Tang, Q.; Zheng, F.; Wu, J.; Chen, Z.; Hann, S.S. Hotair-mediated reciprocal regulation of EZH2 and DNMT1 contribute to polyphyllin I-inhibited growth of castration-resistant prostate cancer cells in vitro and in vivo. Biochim. Biophys. Acta. Gen. Subj. 2018, 1862, 589–599. [Google Scholar] [CrossRef]
- Li, Y.; Ren, Y.; Wang, Y.; Tan, Y.; Wang, Q.; Cai, J.; Zhou, J.; Yang, C.; Zhao, K.; Yi, K.; et al. A compound AC1Q3QWB selectively disrupts HOTAIR-mediated recruitment of PRC2 and enhances cancer therapy of DZNep. Theranostics 2019, 9, 4608–4623. [Google Scholar] [CrossRef]
- Sarma, K.; Levasseur, P.; Aristarkhov, A.; Lee, J.T. Locked nucleic acids (LNAS) reveal sequence requirements and kinetics of XIST RNA localization to the x chromosome. PNAS 2010, 107, 22196–22201. [Google Scholar] [CrossRef]
- Amodio, N.; Stamato, M.A.; Juli, G.; Morelli, E.; Fulciniti, M.; Manzoni, M.; Taiana, E.; Agnelli, L.; Cantafio, M.E.G.; Romeo, E.; et al. Drugging the lncRNA MALAT1 via lna gapmeR ASO inhibits gene expression of proteasome subunits and triggers anti-multiple myeloma activity. Leukemia 2018, 32, 1948–1957. [Google Scholar] [CrossRef]
- Naderi-Meshkin, H.; Lai, X.; Amirkhah, R.; Vera, J.; Rasko, J.E.J.; Schmitz, U. Exosomal lncrnas and cancer: Connecting the missing links. Bioinformatics 2019, 35, 352–360. [Google Scholar] [CrossRef]
- Wu, Y.; Yang, X.; Chen, Z.; Tian, L.; Jiang, G.; Chen, F.; Li, J.; An, P.; Lu, L.; Luo, N.; et al. M(6)a-induced lncRNA RP11 triggers the dissemination of colorectal cancer cells via upregulation of ZEB1. Mol. Cancer 2019, 18, 87. [Google Scholar] [CrossRef]
- Sun, T.; Wu, R.; Ming, L. The role of m6a RNA methylation in cancer. Biomed. Pharmacother. 2019, 112, 108613. [Google Scholar] [CrossRef]
- Pan, Y.; Ma, P.; Liu, Y.; Li, W.; Shu, Y. Multiple functions of m(6)a RNA methylation in cancer. J. Hematol. Oncol. 2018, 11, 48. [Google Scholar] [CrossRef]
- Jacob, R.; Zander, S.; Gutschner, T. The dark side of the epitranscriptome: Chemical modifications in long non-coding RNAs. Int. J. Mol. Sci. 2017, 18, 2387. [Google Scholar] [CrossRef]
- Cohen, A.J.; Saiakhova, A.; Corradin, O.; Luppino, J.M.; Lovrenert, K.; Bartels, C.F.; Morrow, J.J.; Mack, S.C.; Dhillon, G.; Beard, L.; et al. Hotspots of aberrant enhancer activity punctuate the colorectal cancer epigenome. Nat. Commun. 2017, 8, 14400. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-scale crispr-mediated control of gene repression and activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef]
- Blanas, A.; Cornelissen, L.A.M.; Kotsias, M.; van der Horst, J.C.; van de Vrugt, H.J.; Kalay, H.; Spencer, D.I.R.; Kozak, R.P.; van Vliet, S.J. Transcriptional activation of fucosyltransferase (FUT) genes using the CRISPR-dCas9-VPR technology reveals potent N-glycome alterations in colorectal cancer cells. Glycobiology 2019, 29, 137–150. [Google Scholar] [CrossRef]
- Bester, A.C.; Lee, J.D.; Chavez, A.; Lee, Y.R.; Nachmani, D.; Vora, S.; Victor, J.; Sauvageau, M.; Monteleone, E.; Rinn, J.L.; et al. An integrated genome-wide CRISPRA approach to functionalize lncRNAs in drug resistance. Cell 2018, 173, 649–664.e20. [Google Scholar] [CrossRef]
- Thakore, P.I.; D’Ippolito, A.M.; Song, L.; Safi, A.; Shivakumar, N.K.; Kabadi, A.M.; Reddy, T.E.; Crawford, G.E.; Gersbach, C.A. Highly specific epigenome editing by CRISPR-CAS9 repressors for silencing of distal regulatory elements. Nat. Methods 2015, 12, 1143–1149. [Google Scholar] [CrossRef]
- Zhang, X.; Choi, P.S.; Francis, J.M.; Gao, G.F.; Campbell, J.D.; Ramachandran, A.; Mitsuishi, Y.; Ha, G.; Shih, J.; Vazquez, F.; et al. Somatic superenhancer duplications and hotspot mutations lead to oncogenic activation of the KLF5 transcription factor. Cancer Discov. 2018, 8, 108–125. [Google Scholar] [CrossRef]
- Choudhury, S.R.; Cui, Y.; Lubecka, K.; Stefanska, B.; Irudayaraj, J. CRISPR-dCas9 mediated TET1 targeting for selective DNA demethylation at BRCA1 promoter. Oncotarget 2016, 7, 46545–46556. [Google Scholar] [CrossRef]
- Vojta, A.; Dobrinic, P.; Tadic, V.; Bockor, L.; Korac, P.; Julg, B.; Klasic, M.; Zoldos, V. Repurposing the crispr-cas9 system for targeted DNA methylation. Nucleic Acids Res. 2016, 44, 5615–5628. [Google Scholar] [CrossRef]
- Shechner, D.M.; Hacisuleyman, E.; Younger, S.T.; Rinn, J.L. Multiplexable, locus-specific targeting of long RNAs with CRISPR-display. Nat. Methods 2015, 12, 664–670. [Google Scholar] [CrossRef]
- Amirkhah, R.; Farazmand, A.; Wolkenhauer, O.; Schmitz, U. RNA systems biology for cancer: From diagnosis to therapy. Methods Mol. Biol. 2016, 1386, 305–330. [Google Scholar]
- Van Leeuwen, I.M.M.; Edwards, C.M.; Ilyas, M.; Byrne, H.M. Towards a multiscale model of colorectal cancer. World J. Gastroenterol. 2007, 13, 1399–1407. [Google Scholar] [CrossRef]
- De Matteis, G.; Graudenzi, A.; Antoniotti, M. A review of spatial computational models for multi-cellular systems, with regard to intestinal crypts and colorectal cancer development. J. Math. Biol. 2013, 66, 1409–1462. [Google Scholar] [CrossRef]
- Kirouac, D.C.; Schaefer, G.; Chan, J.; Merchant, M.; Orr, C.; Huang, S.-M.A.; Moffat, J.; Liu, L.; Gadkar, K.; Ramanujan, S. Clinical responses to ERK inhibition in BRAFV600e-mutant colorectal cancer predicted using a computational model. NPJ Syst. Biol. Appl. 2017, 3, 14. [Google Scholar] [CrossRef]
- Vafaee, F.; Diakos, C.; Kirschner, M.B.; Reid, G.; Michael, M.Z.; Horvath, L.G.; Alinejad-Rokny, H.; Cheng, Z.J.; Kuncic, Z.; Clarke, S. A data-driven, knowledge-based approach to biomarker discovery: Application to circulating microrna markers of colorectal cancer prognosis. NPJ Syst. Biol. Appl. 2018, 4, 20. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Non-Coding RNAs | Epigenetic Partner/Other Epigenetic Mediator | Target Gene | Tumorigenic Effects | Reference |
|---|---|---|---|---|
| LncRNA & DNA methylation | ||||
| DACOR1 | interaction with DNMT1 to reprogram genome-wide DNA methylation | DNA methylation at thousands of CpG sites | increased clonogenicity | [49,50] |
| HIF1A-AS2 | regulates miR-129-5p and DNMT3A expression | progression and EMT formation of CRC | [56] | |
| H19 | hypomethylation of the sixth CTCF-binding site in the differentially methylated region of IGF2/H19 | loss of imprinting of IGF2T → two forms of aberrant IGF2 expression | promotes microsatellite instability and oncogenesis | [62,63,64] |
| MicroRNA & DNA methylation | ||||
| miR-133b | promoter hypermethylation | HOXA9/ZEB1 pathway | inhibits migration and apoptosis; suppresses metastasis | [60,65] |
| miR-149 | epigenetically silenced by DNA methylation | Specificity Protein 1 (SP1) | independent prognostic factor for overall survival | [66] |
| miR-132 | downregulation by DNA hypermethylation | paxillin | associated with cell invasion | [59] |
| miR-345 | CpG island promoter hypermethylation | BCL2-associated athanogene 3 (BAG3) | suppresses colon cancer cell proliferation and invasiveness | [67] |
| miR-181a/135a/302c | DNA methylation-mediated repression | via repressing PLAG1/IGF2 signalling | promotes the microsatellite-unstable CRC development and 5-FU resistance | [58] |
| miR-203 | directly targets DNMT3B | causes ABCG2 promoter methylation | predisposing CRC development by lowering expression of ABCG2. | [61] |
| Non-Coding RNAs | Epigenetic Partner/Other Epigenetic Mediator | Target Gene | Tumorigenic Effects | Reference |
|---|---|---|---|---|
| Chromatin remodelling | ||||
| DLEU1 | recruits SMARCA1, an essential subunit of the NURF chromatin remodelling complex | activation of KPNA3 | CRC development and progression | [40] |
| CCAT1-L | regulates long-range chromatin interactions | activates the transcription of the MYC locus | both tumorigenesis and the metastatic process | [79] |
| HOTAIR | reprograms chromatin organization in cooperation with the PRC2 complex | global epigenetic regulation | contributes to liver metastases in stage IV CRC patients | [79] |
| Histone modification | ||||
| MALAT1 | EZH2 | represses E-cadherin | promotes chemoresistance | [80] |
| HULC | interacts with EZH2 | to repress NKD2 | oncogenic | [81] |
| SNHG1 | interacts with PRC2 in the nucleus and acts as a miR-154-5p sponge in the cytoplasm | modulates histone methylation of KLF2 and CDKN2B | tumour progression | [74] |
| CRNDE | binds to EZH2 | DUSP5/CDKN1A | positively correlates with advanced pathological stages and larger tumour sizes | [75] |
| SNHG17 | binds to the EZH2 | p57 | promotes cell proliferation | [82] |
| SH3PXD2A-AS1 | interacts with EZH2 | p57 and KLF2 | promotes cells proliferation, migration and invasion | [83] |
| SNHG6 | recruits EZH2 to the p21 promoter | p21 | positively correlates with advanced tumour stage | [84] |
| MEG3 | interacts with PRC2 and JARID2 to direct them to target promoters | Clusterin signalling pathway | inhibits cells proliferation and migration | [85] |
| PINT | interacts with PRC2 to silence genes | p53 autoregulatory negative mechanism | inhibits proliferation of tumour cells | [86] |
| PINT | interacts with PRC2 | EGR1 | inhibits tumour cell invasion | [87] |
| PCAT6 | forms a complex with EZH2 | activates anti-apoptotic ARC | inhibits colon cancer cell apoptosis | [88] |
| Histone modification/DNA methylation | ||||
| Lnc34a | recruits DNMT3A via PHB2 and HDAC1 to methylate and deacetylate the MIR34A promoter simultaneously | epigenetically silence miR-34a | Increase colon cancer stem cells (CSCs) proliferation in late-stage CRC s. | [77] |
| HOXA11-AS | scaffold for the chromatin modification factors PRC2, LSD1, and DNMT1 | lymph node metastasis | [89,90] | |
| Non-Coding RNAs | Mechanism of Action in AS | Target Gene | Tumorigenic Effect | Reference |
|---|---|---|---|---|
| LINC01133 | titrates SRSF6 away from its targets | inhibits EMT and metastasis | [141] | |
| GAPLINC | binds to PSF and NONO | SNAI2 | promotes invasion in CRC | [144] |
| MALAT1 | regulates SR splicing factor distribution in nuclear speckle domains | NA | [140] | |
| MALAT1 | binds to SFPQ and releases oncogene PTBP2 from the SFPQ/PTBP2 complex | promotes tumour growth and metastasis in CRC | [143] | |
| UXT-AS1 | isoform switching from UXT1 to UXT2 | UXT1 | promotes cell proliferation | [146] |
| miR-1296 | represses SFPQ expression | SFPQ | accelerates CRC progression | [132] |
| miR-92a | causing imbalanced expression of PTBP2 through AS-coupled nonsense mediated decay | RBM4 | contributes to progression and metabolic signature of CRC cells | [133] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amirkhah, R.; Naderi-Meshkin, H.; Shah, J.S.; Dunne, P.D.; Schmitz, U. The Intricate Interplay between Epigenetic Events, Alternative Splicing and Noncoding RNA Deregulation in Colorectal Cancer. Cells 2019, 8, 929. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8080929
Amirkhah R, Naderi-Meshkin H, Shah JS, Dunne PD, Schmitz U. The Intricate Interplay between Epigenetic Events, Alternative Splicing and Noncoding RNA Deregulation in Colorectal Cancer. Cells. 2019; 8(8):929. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8080929
Chicago/Turabian StyleAmirkhah, Raheleh, Hojjat Naderi-Meshkin, Jaynish S. Shah, Philip D. Dunne, and Ulf Schmitz. 2019. "The Intricate Interplay between Epigenetic Events, Alternative Splicing and Noncoding RNA Deregulation in Colorectal Cancer" Cells 8, no. 8: 929. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8080929