The Relationship between Leptin, the Leptin Receptor and FGFR1 in Primary Human Breast Tumors

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Preparation of Fat Tissue Filtrates
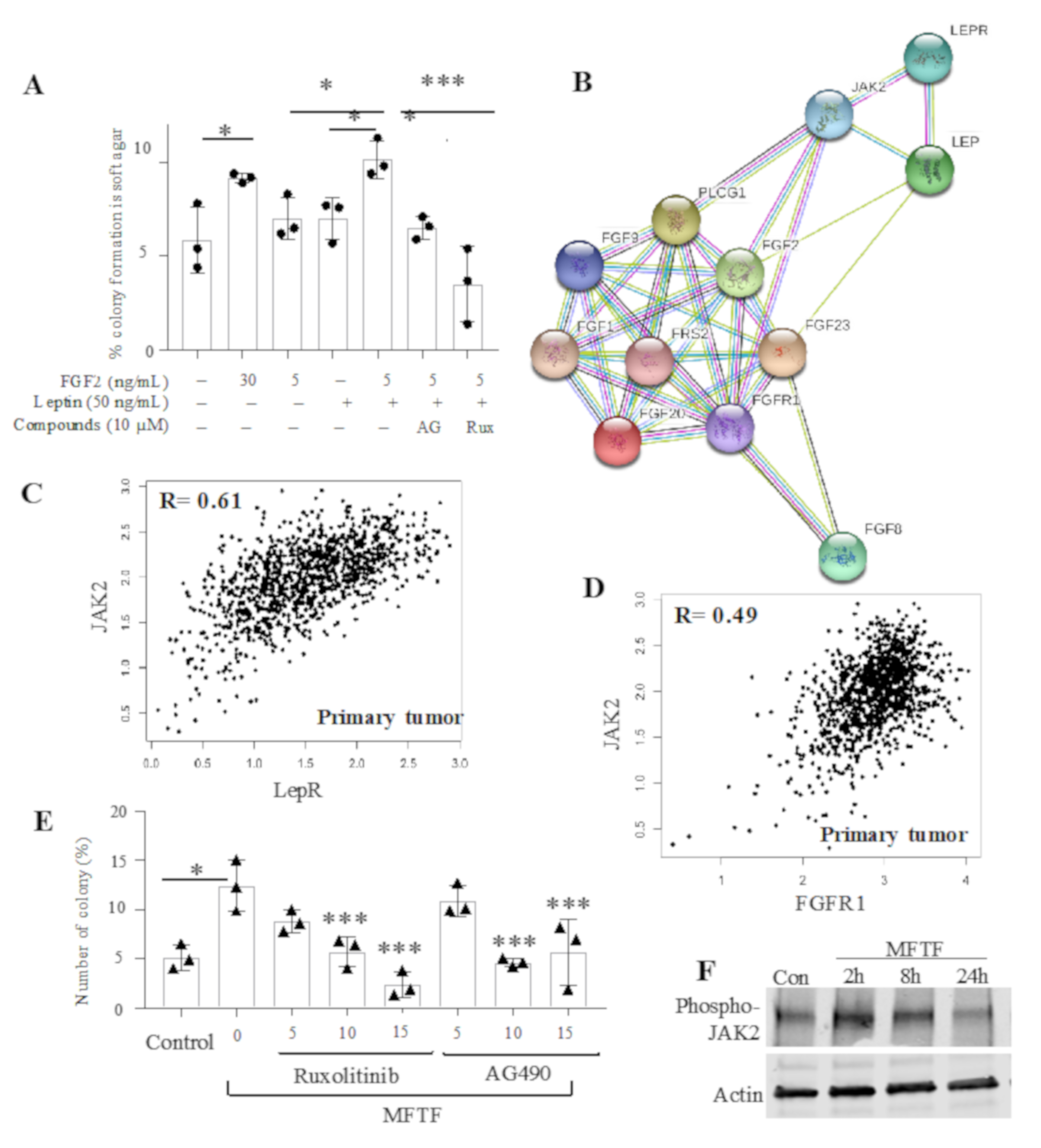
2.3. Anchorage-Independent Colony Formation Soft Agar Assay
2.4. Western Blotting
2.5. Statistics
3. Results
3.1. Positive Correlation between FGFR1 mRNA and Leptin mRNA in Primary Breast Cancer
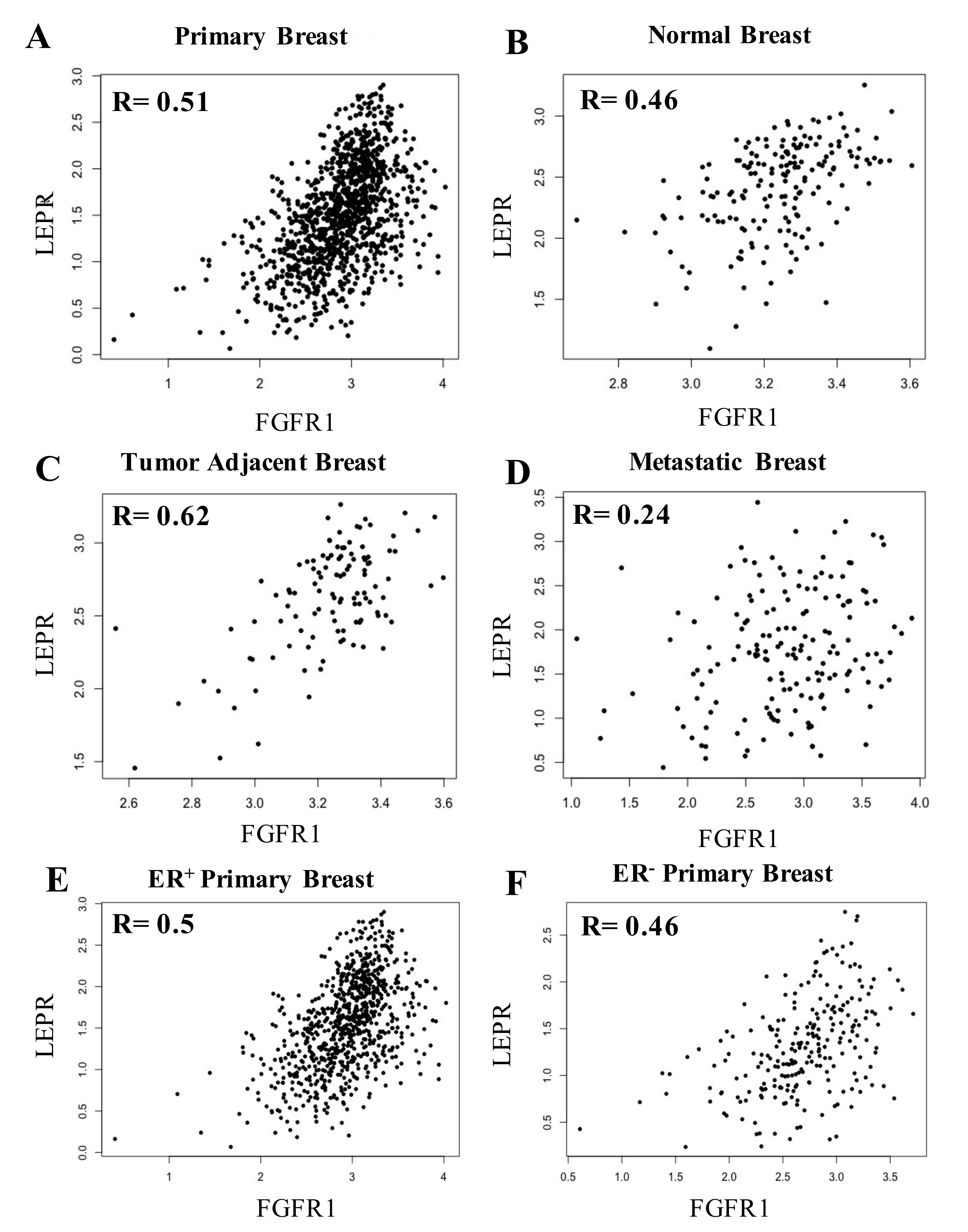
3.2. Relationship between Leptin and FGFR1 mRNA in Normal Breast Tissue, Primary Breast Cancer Tumor Tissue and Breast Cancer-Adjacent Tissue
3.3. Leptin Levels in FGFR1 Amplification, Gain, and Diploid ER- Tumors Compared to ER+ Breast Tumors
3.4. LepR mRNA and FGFR1 mRNA Were Positively Correlated in Breast Tissue
3.5. Role of Jak2 Activation in Adipokine-Stimulated Mammary Epithelial Cell Transformation
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Collaborators, G.B.D.O.; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [CrossRef]
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of Obesity Among Adults and Youth: United States, 2015-2016; NCHS Data Brief; NCHS-National Center for Health Statistics: Washington, DC, USA, 2017; pp. 1–8.
- Malik, V.S.; Willett, W.C.; Hu, F.B. Global obesity: Trends, risk factors and policy implications. Nat. Rev. Endocrinol. 2013, 9, 13–27. [Google Scholar] [CrossRef]
- Calle, E.E.; Rodriguez, C.; Walker-Thurmond, K.; Thun, M.J. Overweight, Obesity, and Mortality from Cancer in a Prospectively Studied Cohort of U.S. Adults. N. Engl. J. Med. 2003, 348, 1625–1638. [Google Scholar] [CrossRef] [Green Version]
- James, W.P.T.; McPherson, K. The costs of overweight. Lancet Public Health 2017, 2, e203–e204. [Google Scholar] [CrossRef] [Green Version]
- Pierobon, M.; Frankenfeld, C.L. Obesity as a risk factor for triple-negative breast cancers: A systematic review and meta-analysis. Breast Cancer Res. Treat. 2013, 137, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Colditz, G.A.; Peterson, L.L. Obesity and Cancer: Evidence, Impact, and Future Directions. Clin. Chem. 2018, 64, 154–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, H.R.; Willett, W.C.; Terry, K.L.; Michels, K.B. Body fat distribution and risk of premenopausal breast cancer in the Nurses’ Health Study II. J. Natl. Cancer Inst. 2011, 103, 273–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millikan, R.C.; Newman, B.; Tse, C.K.; Moorman, P.G.; Conway, K.; Dressler, L.G.; Smith, L.V.; Labbok, M.H.; Geradts, J.; Bensen, J.T.; et al. Epidemiology of basal-like breast cancer. Breast Cancer Res. Treat. 2008, 109, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Perou, C.M.; Livasy, C.A.; Dressler, L.G.; Cowan, D.; Conway, K.; Karaca, G.; Troester, M.A.; Tse, C.K.; Edmiston, S.; et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA 2006, 295, 2492–2502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phipps, A.I.; Chlebowski, R.T.; Prentice, R.; McTiernan, A.; Stefanick, M.L.; Wactawski-Wende, J.; Kuller, L.H.; Adams-Campbell, L.L.; Lane, D.; Vitolins, M.; et al. Body size, physical activity, and risk of triple-negative and estrogen receptor-positive breast cancer. Cancer Epidemiol. Biomarkers Prev. 2011, 20, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, J.L.; Bluher, S.; Yiannakouris, N.; Suchard, M.A.; Kratzsch, J.; Mantzoros, C.S. Regulation of circulating soluble leptin receptor levels by gender, adiposity, sex steroids, and leptin: Observational and interventional studies in humans. Diabetes 2002, 51, 2105–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Perez, R.R.; Xu, Y.; Guo, S.; Watters, A.; Zhou, W.; Leibovich, S.J. Leptin upregulates VEGF in breast cancer via canonic and non-canonical signalling pathways and NFkappaB/HIF-1alpha activation. Cell Signal. 2010, 22, 1350–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malvi, P.; Chaube, B.; Singh, S.V.; Mohammad, N.; Vijayakumar, M.V.; Singh, S.; Chouhan, S.; Bhat, M.K. Elevated circulatory levels of leptin and resistin impair therapeutic efficacy of dacarbazine in melanoma under obese state. Cancer Metab. 2018, 6, 2. [Google Scholar] [CrossRef]
- Gorrab, A.; Pagano, A.; Ayed, K.; Chebil, M.; Derouiche, A.; Kovacic, H.; Gati, A. Leptin Promotes Prostate Cancer Proliferation and Migration by Stimulating STAT3 Pathway. Nutr. Cancer 2020, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Modzelewska, P.; Chludzinska, S.; Lewko, J.; Reszec, J. The influence of leptin on the process of carcinogenesis. Contemp Oncol. (Pozn.) 2019, 23, 63–68. [Google Scholar] [CrossRef]
- Pond, A.C.; Bin, X.; Batts, T.; Roarty, K.; Hilsenbeck, S.; Rosen, J.M. Fibroblast growth factor receptor signaling is essential for normal mammary gland development and stem cell function. Stem Cells 2013, 31, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Hao, R.H.; Guo, Y.; Dong, S.S.; Weng, G.Z.; Yan, H.; Zhu, D.L.; Chen, X.F.; Chen, J.B.; Yang, T.L. Associations of Plasma FGF2 Levels and Polymorphisms in the FGF2 Gene with Obesity Phenotypes in Han Chinese Population. Sci. Rep. 2016, 6, 19868. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, D.; Benham, V.; Bullard, B.; Kearney, T.; Hsia, H.C.; Gibbon, D.; Demireva, E.Y.; Lunt, S.Y.; Bernard, J.J. Fibroblast growth factor receptor is a mechanistic link between visceral adiposity and cancer. Oncogene 2017. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, D.; Benham, V.; Bernard, J.J. Elucidating the role of adipose tissue secreted factors in malignant transformation. Adipocyte 2017, 1–4. [Google Scholar] [CrossRef]
- Benham, V.; Chakraborty, D.; Bullard, B.; Bernard, J.J. A role for FGF2 in visceral adiposity-associated mammary epithelial transformation. Adipocyte 2018, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, Y.; Liu, J.; Chen, Q.; Pang, D.; Jiang, Y. Effects of FGFR1 Gene Polymorphisms on the Risk of Breast Cancer and FGFR1 Protein Expression. Cell Physiol. Biochem. 2018, 47, 2569–2578. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.; Chemi, F.; Panza, S.; Barone, I.; Bonofiglio, D.; Lanzino, M.; Cordella, A.; Campana, A.; Hashim, A.; Rizza, P.; et al. Leptin as a mediator of tumor-stromal interactions promotes breast cancer stem cell activity. Oncotarget 2016, 7, 1262–1275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, Y.; Funahashi, T.; Tanaka, S.; Taguchi, T.; Tamaki, Y.; Shimomura, I.; Noguchi, S. High expression of leptin receptor mRNA in breast cancer tissue predicts poor prognosis for patients with high, but not low, serum leptin levels. Int. J. Cancer 2006, 118, 1414–1419. [Google Scholar] [CrossRef]
- Chang, C.C.; Wu, M.J.; Yang, J.Y.; Camarillo, I.G.; Chang, C.J. Leptin-STAT3-G9a Signaling Promotes Obesity-Mediated Breast Cancer Progression. Cancer Res. 2015, 75, 2375–2386. [Google Scholar] [CrossRef] [Green Version]
- Zeng, B.; Glicksberg, B.S.; Newbury, P.; Xing, J.; Liu, K.; Wen, A.; Chow, C.; Chen, B. OCTAD: An open workplace for virtually screening therapeutics targeting precise cancer patient groups using gene expression features. bioRxiv 2019, 821546. [Google Scholar] [CrossRef]
- Wagner, K.U.; Rui, H. Jak2/Stat5 signaling in mammogenesis, breast cancer initiation and progression. J. Mammary Gland Biol. Neoplasia 2008, 13, 93–103. [Google Scholar] [CrossRef]
- Stover, D.G.; Gil Del Alcazar, C.R.; Brock, J.; Guo, H.; Overmoyer, B.; Balko, J.; Xu, Q.; Bardia, A.; Tolaney, S.M.; Gelman, R.; et al. Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple-negative breast cancer. NPJ Breast Cancer 2018, 4, 10. [Google Scholar] [CrossRef]
- O’Shaughnessy, J.; DeMichele, A.; Ma, C.X.; Richards, P.; Yardley, D.A.; Wright, G.S.; Kalinsky, K.; Steis, R.; Diab, S.; Kennealey, G.; et al. A randomized, double-blind, phase 2 study of ruxolitinib or placebo in combination with capecitabine in patients with advanced HER2-negative breast cancer and elevated C-reactive protein, a marker of systemic inflammation. Breast Cancer Res. Treat. 2018, 170, 547–557. [Google Scholar] [CrossRef]
- Bernard, J.J.; Lou, Y.R.; Peng, Q.Y.; Li, T.; Vakil, P.R.; Ding, N.; Laskin, J.D.; Dong, Z.; Conney, A.H.; Lu, Y.P. Parametrial fat tissue from high fat diet-treated SKH-1 mice stimulates transformation of mouse epidermal JB6 cells. J. Carcinog. Mutagen. 2014, 5, 2157–2518. [Google Scholar] [CrossRef] [Green Version]
- Neuhouser, M.L.; Aragaki, A.K.; Prentice, R.L.; Manson, J.E.; Chlebowski, R.; Carty, C.L.; Ochs-Balcom, H.M.; Thomson, C.A.; Caan, B.J.; Tinker, L.F.; et al. Overweight, Obesity, and Postmenopausal Invasive Breast Cancer Risk: A Secondary Analysis of the Women’s Health Initiative Randomized Clinical Trials. JAMA Oncol. 2015, 1, 611–621. [Google Scholar] [CrossRef] [PubMed]
- Elbauomy Elsheikh, S.; Green, A.R.; Lambros, M.B.; Turner, N.C.; Grainge, M.J.; Powe, D.; Ellis, I.O.; Reis-Filho, J.S. FGFR1 amplification in breast carcinomas: A chromogenic in situ hybridisation analysis. Breast Cancer Res. 2007, 9, R23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomiguchi, M.; Yamamoto, Y.; Yamamoto-Ibusuki, M.; Goto-Yamaguchi, L.; Fujiki, Y.; Fujiwara, S.; Sueta, A.; Hayashi, M.; Takeshita, T.; Inao, T.; et al. Fibroblast growth factor receptor-1 protein expression is associated with prognosis in estrogen receptor-positive/human epidermal growth factor receptor-2-negative primary breast cancer. Cancer Sci. 2016, 107, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Santolla, M.F.; Vivacqua, A.; Lappano, R.; Rigiracciolo, D.C.; Cirillo, F.; Galli, G.R.; Talia, M.; Brunetti, G.; Miglietta, A.M.; Belfiore, A.; et al. GPER Mediates a Feedforward FGF2/FGFR1 Paracrine Activation Coupling CAFs to Cancer Cells toward Breast Tumor Progression. Cells 2019, 8, 223. [Google Scholar] [CrossRef] [Green Version]
- Formisano, L.; Stauffer, K.M.; Young, C.D.; Bhola, N.E.; Guerrero-Zotano, A.L.; Jansen, V.M.; Estrada, M.V.; Hutchinson, K.E.; Giltnane, J.M.; Schwarz, L.J.; et al. Association of FGFR1 with ERα Maintains Ligand-Independent ER Transcription and Mediates Resistance to Estrogen Deprivation in ER+ Breast Cancer. Clin. Cancer Res. 2017, 23, 6138–6150. [Google Scholar] [CrossRef] [Green Version]
- Qian, Y.; Shi, D.; Qiu, J.; Zhu, F.; Qian, J.; He, S.; Shu, Y.; Yin, Y.; Chen, X. ObRb downregulation increases breast cancer cell sensitivity to tamoxifen. Tumor Biol. 2015, 36, 6813–6821. [Google Scholar] [CrossRef]
- Lipsey, C.C.; Harbuzariu, A.; Robey, R.W.; Huff, L.M.; Gottesman, M.M.; Gonzalez-Perez, R.R. Leptin Signaling Affects Survival and Chemoresistance of Estrogen Receptor Negative Breast Cancer. Int. J. Mol. Sci. 2020, 21, 3794. [Google Scholar] [CrossRef]
- Newman, G.; Gonzalez-Perez, R.R. Leptin-cytokine crosstalk in breast cancer. Mol. Cell Endocrinol. 2014, 382, 570–582. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Primary Cancer Type | Rho | p-Value | Samples | Average Leptin mRNA | Average FGFR1 mRNA |
|---|---|---|---|---|---|
| White Blood Cell | 0.3421 | 4.08×10−6 | 200 | 0.1364 | 2.3125 |
| Colon | 0.2846 | 8.32×10−7 | 463 | 0.1169 | 2.1402 |
| Pancreas | 0.2837 | 0.0001 | 186 | 0.4168 | 2.8082 |
| Rectum | 0.2665 | 0.0098 | 168 | 0.1098 | 2.2070 |
| Bile Duct | 0.2494 | 0.1424 | 36 | 0.0294 | 2.8067 |
| Testis | 0.2363 | 0.0032 | 156 | 0.3647 | 3.0769 |
| Stomach | 0.1915 | 8.83×10−5 | 478 | 0.1513 | 2.4725 |
| Breast | 0.1774 | 3.17E-09 | 1108 | 0.6713 | 2.8852 |
| Bladder | 0.1736 | 0.0004 | 413 | 0.2391 | 2.0195 |
| Ovary | 0.1450 | 0.0027 | 613 | 0.1288 | 2.8148 |
| Skin | 0.1408 | 0.0022 | 478 | 0.1434 | 2.6506 |
| Thyroid Gland | 0.1405 | 0.0014 | 515 | 0.0528 | 2.4212 |
| Body Cavity | 0.1350 | 0.2126 | 87 | 0.1555 | 3.0533 |
| Liver | 0.1177 | 0.0234 | 379 | 0.0363 | 1.4615 |
| Lung | 0.1138 | 0.0003 | 1091 | 0.2550 | 2.5369 |
| Eye | 0.1073 | 0.3464 | 80 | 0.0082 | 2.1440 |
| Brain | 0.0878 | 0.0211 | 1138 | 0.0274 | 2.8951 |
| Adrenal Gland | 0.0866 | 0.4539 | 92 | 0.0539 | 2.5385 |
| Bone | 0.0661 | 0.2867 | 265 | 0.1456 | 3.2758 |
| Prostate | 0.0615 | 0.1716 | 499 | 0.0247 | 2.5691 |
| Lymphatic Tissue | 0.0540 | 0.7183 | 48 | 0.3418 | 2.1315 |
| Esophagus | 0.0238 | 0.7500 | 186 | 0.1591 | 2.5131 |
| Thymus | 0.0193 | 0.8350 | 124 | 0.0940 | 2.3847 |
| Uterus | 0.0130 | 0.9237 | 57 | 0.0749 | 2.9587 |
| Paraganglia | −0.0053 | 0.9435 | 184 | 0.0781 | 3.0194 |
| Kidney | −0.0084 | 0.8023 | 895 | 0.1258 | 2.7034 |
| Head and Neck | −0.0190 | 0.6657 | 530 | 0.2131 | 2.2329 |
| Cervix | −0.0498 | 0.3856 | 310 | 0.2104 | 2.0481 |
| Endometrium | −0.0903 | 0.2268 | 548 | 0.0413 | 2.6931 |
| Breast Tissue Type | Rho | p-Value | Average Leptin mRNA | Average FGFR1 mRNA |
|---|---|---|---|---|
| Normal | −0.27 | 0.0002 | 3.02 ± 0.09 | 2.16 ± 0.97 |
| Primary Cancer | 0.16 | 1.36 × 10-7 | 2.63 ± 0.32 | 0.63 ± 0.71 |
| Breast Tumor Adjacent | 0.26 | 0.0059 | 2.94 ± 0.1 | 2.55 ± 0.77 |
| Metastatic Cancer | −0.01 | 0.97 | 2.63 ± 0.35 | 0.38 ± 0.65 |
| Breast Tumor FGFR1 Copy Number Status | FGFR1 Amplification | FGFR1 Gain | FGFR1 Diploid | FGFR1 Shallow Deletion | FGFR1 Deep Deletion |
|---|---|---|---|---|---|
| Average leptin relative copy number | 0.24071 | 0.13977 | 0.0444 | −0.0278 | −0.11275 |
| Average leptin relative copy number in ER+ tumors | 0.15647 * | 0.08372 * | 0.01266 * | −0.04446 | −0.2005 |
| Average leptin relative copy number in ER− tumors | 0.65223 * | 0.33081 * | 0.14237 * | 0.00842 | −0.025 |
| Breast Tumor FGFR1 Copy Number Status | FGFR1 Amplification | FGFR1 Gain | FGFR1 Diploid | FGFR1 Shallow Deletion | FGFR1 Deep Deletion |
|---|---|---|---|---|---|
| Standard deviation of ER+ Samples | 0.2879 | 0.3001 | 0.3239 | 0.3152 | 0.2963 |
| Standard deviation of ER- Samples | 1.2884 | 0.2193 | 0.4196 | 0.5715 | 0.2524 |
| P-value of one-sided t-test comparing average ER+ and ER- relative copy number | 0.0025 | 0.0041 | 0.0003 | 0.1972 | 0.2668 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boothby-Shoemaker, W.; Benham, V.; Paithankar, S.; Shankar, R.; Chen, B.; Bernard, J.J. The Relationship between Leptin, the Leptin Receptor and FGFR1 in Primary Human Breast Tumors. Cells 2020, 9, 2224. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9102224
Boothby-Shoemaker W, Benham V, Paithankar S, Shankar R, Chen B, Bernard JJ. The Relationship between Leptin, the Leptin Receptor and FGFR1 in Primary Human Breast Tumors. Cells. 2020; 9(10):2224. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9102224
Chicago/Turabian StyleBoothby-Shoemaker, Wyatt, Vanessa Benham, Shreya Paithankar, Rama Shankar, Bin Chen, and Jamie J. Bernard. 2020. "The Relationship between Leptin, the Leptin Receptor and FGFR1 in Primary Human Breast Tumors" Cells 9, no. 10: 2224. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9102224