CHEK2 Germline Variants in Cancer Predisposition: Stalemate Rather than Checkmate

 , , and
, , and
Abstract
:1. Introduction
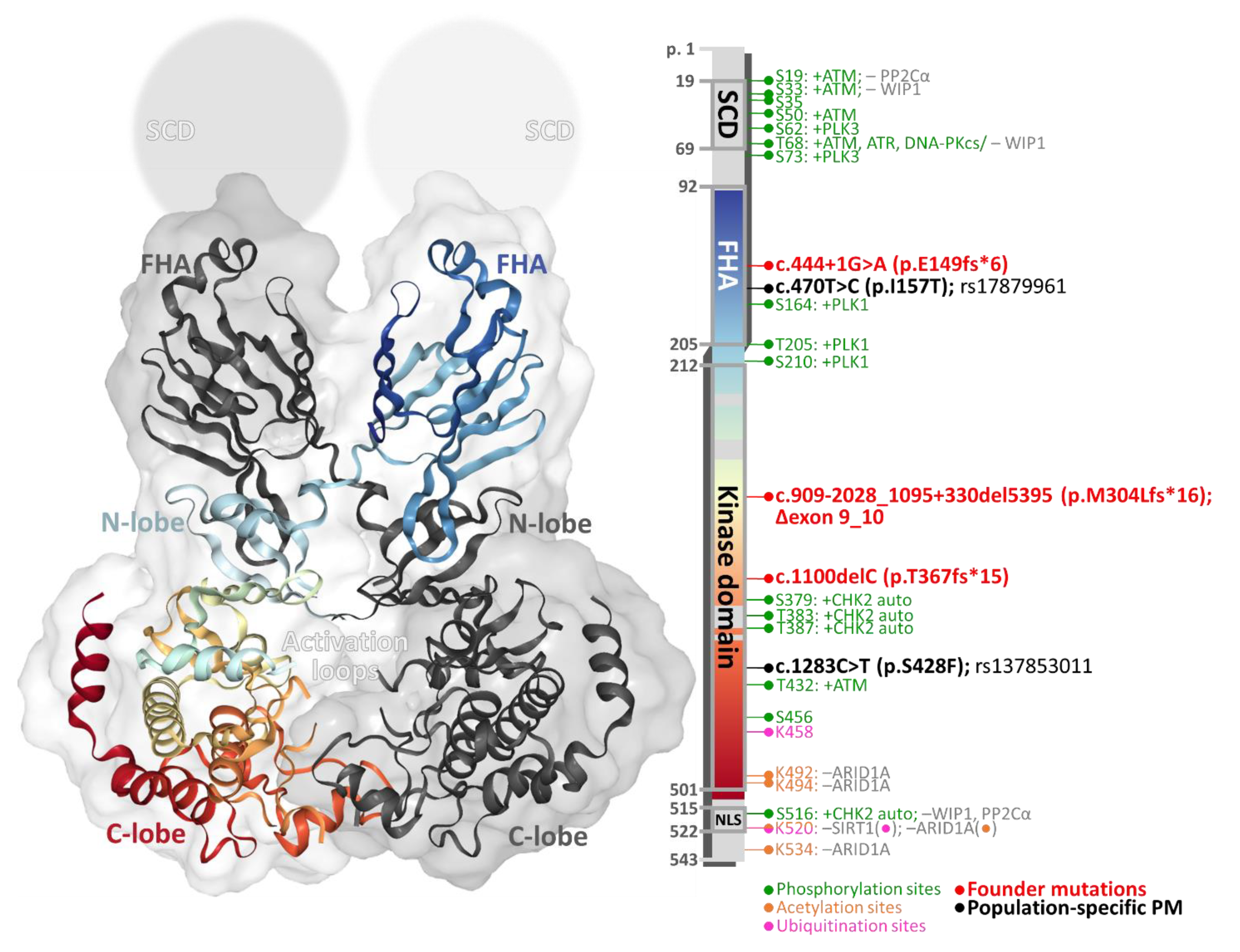
2. Structure and Function of CHK2 Kinase
2.1. The CHEK2 Gene
2.2. Structure of CHK2 Kinase Protein
2.3. Regulation of CHK2 Kinase Activity
2.3.1. Phosphorylation
2.3.2. Dephosphorylation
2.3.3. Ubiquitination
2.3.4. Acetylation
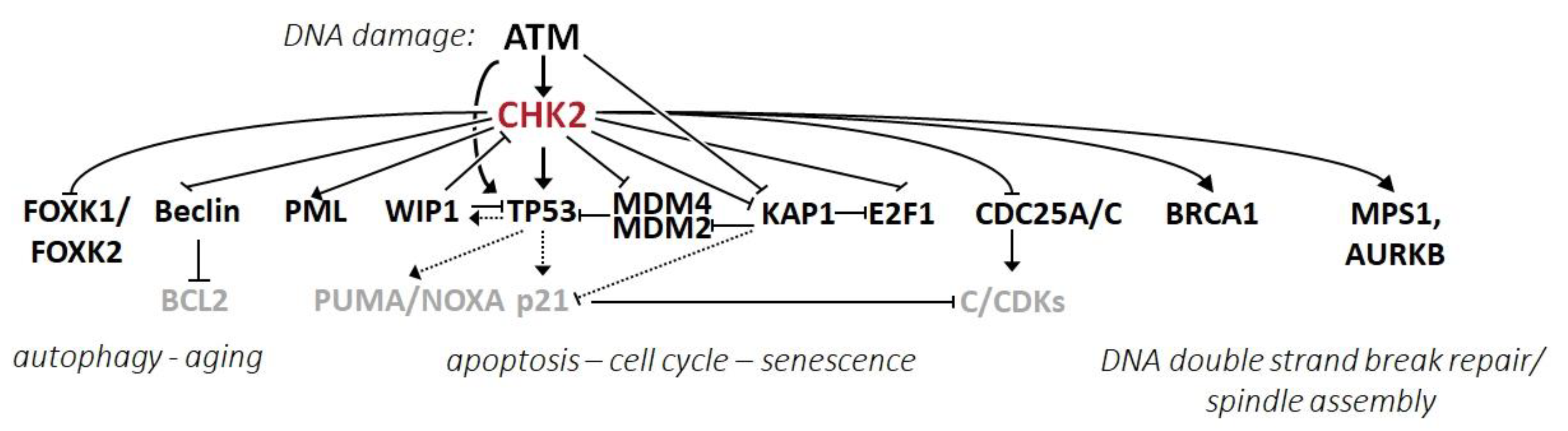
2.4. CHK2 Substrates and Its Effector Pathways
2.4.1. CHK2 in the Regulation of the Cell Cycle, Apoptosis, and Senescence
2.4.2. CHK2 in the Regulation of DNA Repair and Mitotic Spindle
2.4.3. CHK2 in the Regulation of Autophagy and Aging
2.4.4. CHK2 in the Regulation of Other Intracellular Pathways
3. Germline CHEK2 Variants
3.1. Ethnic and Geographical Differences in CHEK2 Mutation Frequency
3.2. Breast Cancer
3.3. Prostate Cancer
3.4. Kidney Cancer
3.5. Papillary Thyroid Cancer
3.6. Colorectal Cancer
3.7. Other Cancers
4. Concluding Remarks and Future Directions
Funding
Acknowledgments
Conflicts of Interest
References
- Pilleron, S.; Soto-Perez-de-Celis, E.; Vignat, J.; Ferlay, J.; Soerjomataram, I.; Bray, F.; Sarfati, D. Estimated global cancer incidence in the oldest adults in 2018 and projections to 2050. Int. J. Cancer 2020. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benada, J.; Macurek, L. Targeting the Checkpoint to Kill Cancer Cells. Biomolecules 2015, 5, 1912–1937. [Google Scholar] [CrossRef] [Green Version]
- Rahman, N. Realizing the promise of cancer predisposition genes. Nature 2014, 505, 302–308. [Google Scholar] [CrossRef] [Green Version]
- Hyman, D.M.; Taylor, B.S.; Baselga, J. Implementing Genome-Driven Oncology. Cell 2017, 168, 584–599. [Google Scholar] [CrossRef]
- Turnbull, C.; Sud, A.; Houlston, R.S. Cancer genetics, precision prevention and a call to action. Nat. Genet. 2018, 50, 1212–1218. [Google Scholar] [CrossRef]
- Easton, D.F.; Pharoah, P.D.; Antoniou, A.C.; Tischkowitz, M.; Tavtigian, S.V.; Nathanson, K.L.; Devilee, P.; Meindl, A.; Couch, F.J.; Southey, M.; et al. Gene-panel sequencing and the prediction of breast-cancer risk. N. Engl. J. Med. 2015, 372, 2243–2257. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.D.; Nathanson, K.L. Application of Panel-Based Tests for Inherited Risk of Cancer. Annu. Rev. Genom. Hum. Genet. 2017, 18, 201–227. [Google Scholar] [CrossRef]
- Kapoor, N.S.; Curcio, L.D.; Blakemore, C.A.; Bremner, A.K.; McFarland, R.E.; West, J.G.; Banks, K.C. Multigene Panel Testing Detects Equal Rates of Pathogenic BRCA1/2 Mutations and has a Higher Diagnostic Yield Compared to Limited BRCA1/2 Analysis Alone in Patients at Risk for Hereditary Breast Cancer. Ann. Surg. Oncol. 2015, 22, 3282–3288. [Google Scholar] [CrossRef]
- Federici, G.; Soddu, S. Variants of uncertain significance in the era of high-throughput genome sequencing: A lesson from breast and ovary cancers. J. Exp. Clin. Cancer Res. 2020, 39, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fackenthal, J.D.; Olopade, O.I. Breast cancer risk associated with BRCA1 and BRCA2 in diverse populations. Nat. Rev. Cancer 2007, 7, 937–948. [Google Scholar] [CrossRef] [PubMed]
- Belman, S.; Parsons, M.T.; Spurdle, A.B.; Goldgar, D.E.; Feng, B.J. Considerations in assessing germline variant pathogenicity using cosegregation analysis. Genet. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Mailand, N.; Syljuasen, R.G.; Bartek, J.; Lukas, J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001, 410, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Matsuoka, S.; Huang, M.; Elledge, S.J. Linkage of ATM to Cell Cycle Regulation by the Chk2 Protein Kinase. Science 1998, 282, 1893–1897. [Google Scholar] [CrossRef]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An Oncogene-Induced DNA Damage Model for Cancer Development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [Green Version]
- Bell, D.W.; Varley, J.M.; Szydlo, T.E.; Kang, D.H.; Wahrer, D.C.; Shannon, K.E.; Lubratovich, M.; Verselis, S.J.; Isselbacher, K.J.; Fraumeni, J.F.; et al. Heterozygous germ line hCHK2 mutations in Li-Fraumeni syndrome. Science 1999, 286, 2528–2531. [Google Scholar] [CrossRef]
- Cybulski, C.; Gorski, B.; Huzarski, T.; Masojc, B.; Mierzejewski, M.; Debniak, T.; Teodorczyk, U.; Byrski, T.; Gronwald, J.; Matyjasik, J.; et al. CHEK2 is a multiorgan cancer susceptibility gene. Am. J. Hum. Genet. 2004, 75, 1131–1135. [Google Scholar] [CrossRef] [Green Version]
- Delimitsou, A.; Fostira, F.; Kalfakakou, D.; Apostolou, P.; Konstantopoulou, I.; Kroupis, C.; Papavassiliou, A.G.; Kleibl, Z.; Stratikos, E.; Voutsinas, G.E.; et al. Functional characterization of CHEK2 variants in a Saccharomyces cerevisiae system. Hum. Mutat. 2019, 40, 631–648. [Google Scholar] [CrossRef] [PubMed]
- Caswell-Jin, J.L.; Gupta, T.; Hall, E.; Petrovchich, I.M.; Mills, M.A.; Kingham, K.E.; Koff, R.; Chun, N.M.; Levonian, P.; Lebensohn, A.P.; et al. Racial/ethnic differences in multiple-gene sequencing results for hereditary cancer risk. Genet. Med. 2018, 20, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.L.; Lee, C.H.; Schwarz, J.K.; Mitiku, N.; Piwnica-Worms, H.; Chung, J.H. A human Cds1-related kinase that functions downstream of ATM protein in the cellular response to DNA damage. Proc. Natl. Acad. Sci. USA 1999, 96, 3745–3750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaturvedi, P.; Eng, W.K.; Zhu, Y.; Mattern, M.R.; Mishra, R.; Hurle, M.R.; Zhang, X.; Annan, R.S.; Lu, Q.; Faucette, L.F.; et al. Mammalian Chk2 is a downstream effector of the ATM-dependent DNA damage checkpoint pathway. Oncogene 1999, 18, 4047–4054. [Google Scholar] [CrossRef] [PubMed]
- Melo, J.; Toczyski, D. A unified view of the DNA-damage checkpoint. Curr. Opin. Cell Biol. 2002, 14, 237–245. [Google Scholar] [CrossRef]
- Zoppoli, G.; Solier, S.; Reinhold, W.C.; Liu, H.; Connelly, J.W.; Monks, A.; Shoemaker, R.H.; Abaan, O.D.; Davis, S.R.; Meltzer, P.S.; et al. CHEK2 genomic and proteomic analyses reveal genetic inactivation or endogenous activation across the 60 cell lines of the US National Cancer Institute. Oncogene 2012, 31, 403–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tominaga, K.; Morisaki, H.; Kaneko, Y.; Fujimoto, A.; Tanaka, T.; Ohtsubo, M.; Hirai, M.; Okayama, H.; Ikeda, K.; Nakanishi, M. Role of human Cds1 (Chk2) kinase in DNA damage checkpoint and its regulation by p53. J. Biol. Chem. 1999, 274, 31463–31467. [Google Scholar] [CrossRef] [Green Version]
- Staalesen, V.; Falck, J.; Geisler, S.; Bartkova, J.; Borresen-Dale, A.L.; Lukas, J.; Lillehaug, J.R.; Bartek, J.; Lonning, P.E. Alternative splicing and mutation status of CHEK2 in stage III breast cancer. Oncogene 2004. [Google Scholar] [CrossRef] [Green Version]
- Matsui, T.; Katsuno, Y.; Inoue, T.; Fujita, F.; Joh, T.; Niida, H.; Murakami, H.; Itoh, M.; Nakanishi, M. Negative regulation of Chk2 expression by p53 is dependent on the CCAAT-binding transcription factor NF-Y. J. Biol. Chem. 2004, 279, 25093–25100. [Google Scholar] [CrossRef] [Green Version]
- Williams, L.H.; Choong, D.; Johnson, S.A.; Campbell, I.G. Genetic and epigenetic analysis of CHEK2 in sporadic breast, colon, and ovarian cancers. Clin. Cancer Res. 2006, 12, 6967–6972. [Google Scholar] [CrossRef] [Green Version]
- Sodha, N.; Williams, R.; Mangion, J.; Bullock, S.L.; Yuille, M.R.; Eeles, R.A.; Bell, D.W.; Wahrer, D.C.R.; Varley, J.M.; Haber, D.A. Screening hCHK2 for Mutations. Science 2000, 289, 359a. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munch, C.; Kirsch, S.; Fernandes, A.M.; Schempp, W. Evolutionary analysis of the highly dynamic CHEK2 duplicon in anthropoids. BMC Evol. Biol. 2008, 8, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, J.; Urist, M.; Prives, C. The Chk2 protein kinase. DNA Repair 2004, 3, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Williams, B.L.; Haire, L.F.; Goldberg, M.; Wilker, E.; Durocher, D.; Yaffe, M.B.; Jackson, S.P.; Smerdon, S.J. Structural and functional versatility of the FHA domain in DNA-damage signaling by the tumor suppressor kinase Chk2. Mol. Cell 2002, 9, 1045–1054. [Google Scholar] [CrossRef]
- Cai, Z.; Chehab, N.H.; Pavletich, N.P. Structure and activation mechanism of the CHK2 DNA damage checkpoint kinase. Mol. Cell. 2009, 35, 818–829. [Google Scholar] [CrossRef]
- Wybenga-Groot, L.E.; Ho, C.S.; Sweeney, F.D.; Ceccarelli, D.F.; McGlade, C.J.; Durocher, D.; Sicheri, F. Structural basis of Rad53 kinase activation by dimerization and activation segment exchange. Cell Signal 2014, 26, 1825–1836. [Google Scholar] [CrossRef]
- Matsuoka, S.; Rotman, G.; Ogawa, A.; Shiloh, Y.; Tamai, K.; Elledge, S.J. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 10389–10394. [Google Scholar] [CrossRef] [Green Version]
- Ouchi, M.; Ouchi, T. Distinct DNA damage determines differential phosphorylation of Chk2. Cancer Biol. Ther. 2014, 15, 1700–1704. [Google Scholar] [CrossRef] [Green Version]
- Zannini, L.; Lecis, D.; Lisanti, S.; Benetti, R.; Buscemi, G.; Schneider, C.; Delia, D. Karyopherin-alpha2 protein interacts with Chk2 and contributes to its nuclear import. J. Biol. Chem. 2003, 278, 42346–42351. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.K.; Lovly, C.M.; Piwnica-Worms, H. Regulation of the Chk2 Protein Kinase by Oligomerization-Mediated cis- and trans-Phosphorylation. Mol. Cancer Res. 2003, 1, 598–609. [Google Scholar]
- Kurz, E.U.; Douglas, P.; Lees-Miller, S.P. Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J. Biol. Chem. 2004, 279, 53272–53281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kass, E.M.; Ahn, J.; Tanaka, T.; Freed-Pastor, W.A.; Keezer, S.; Prives, C. Stability of checkpoint kinase 2 is regulated via phosphorylation at serine 456. J. Biol. Chem. 2007, 282, 30311–30321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabant, G.; Lorphelin, A.; Nozerand, N.; Marchetti, C.; Bellanger, L.; Dedieu, A.; Quemeneur, E.; Alpha-Bazin, B. Autophosphorylated residues involved in the regulation of human chk2 in vitro. J. Mol. Biol. 2008, 380, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.W.; Paul, A.; Boxall, K.J.; Barrie, S.E.; Aherne, G.W.; Garrett, M.D.; Mittnacht, S.; Pearl, L.H. Trans-activation of the DNA-damage signalling protein kinase Chk2 by T-loop exchange. EMBO J. 2006, 25, 3179–3190. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, J. Autophosphorylation of checkpoint kinase 2 at serine 516 is required for radiation-induced apoptosis. J. Biol. Chem. 2003, 278, 36163–36168. [Google Scholar] [CrossRef] [Green Version]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, Z.; Yu, L.; Lin, Y.F.; Matsunaga, S.; Shen, C.Y.; Chen, B.P. DNA-PKcs activates the Chk2-Brca1 pathway during mitosis to ensure chromosomal stability. Oncogenesis 2014, 3, e85. [Google Scholar] [CrossRef] [Green Version]
- Aquino Perez, C.; Palek, M.; Stolarova, L.; von Morgen, P.; Macurek, L. Phosphorylation of PLK3 Is Controlled by Protein Phosphatase 6. Cells 2020, 9, 1506. [Google Scholar] [CrossRef]
- Bahassiel, M.; Myer, D.L.; McKenney, R.J.; Hennigan, R.F.; Stambrook, P.J. Priming phosphorylation of Chk2 by polo-like kinase 3 (Plk3) mediates its full activation by ATM and a downstream checkpoint in response to DNA damage. Mutat. Res. 2006, 596, 166–176. [Google Scholar] [CrossRef]
- Van Vugt, M.A.; Gardino, A.K.; Linding, R.; Ostheimer, G.J.; Reinhardt, H.C.; Ong, S.E.; Tan, C.S.; Miao, H.; Keezer, S.M.; Li, J.; et al. A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to inactivate the G(2)/M DNA damage checkpoint. PLoS Biol. 2010, 8, e1000287. [Google Scholar] [CrossRef]
- Benada, J.; Burdová, K.; Lidak, T.; von Morgen, P.; Macurek, L. Polo-like kinase 1 inhibits DNA damage response during mitosis. Cell Cycle 2015, 14, 219–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouinard, G.; Clement, I.; Lafontaine, J.; Rodier, F.; Schmitt, E. Cell cycle-dependent localization of CHK2 at centrosomes during mitosis. Cell Div. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva-Trastoy, M.; Berthonaud, V.; Chevalier, A.; Ducrot, C.; Marsolier-Kergoat, M.C.; Mann, C.; Leteurtre, F. The Wip1 phosphatase (PPM1D) antagonizes activation of the Chk2 tumour suppressor kinase. Oncogene 2007, 26, 1449–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujimoto, H.; Onishi, N.; Kato, N.; Takekawa, M.; Xu, X.Z.; Kosugi, A.; Kondo, T.; Imamura, M.; Oishi, I.; Yoda, A.; et al. Regulation of the antioncogenic Chk2 kinase by the oncogenic Wip1 phosphatase. Cell Death Differ. 2006, 13, 1170–1180. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.K.; Dapic, V.; Monteiro, A.N. Negative regulation of CHK2 activity by protein phosphatase 2A is modulated by DNA damage. Cell Cycle 2010, 9, 736–747. [Google Scholar] [CrossRef] [PubMed]
- Leroy, C.; Lee, S.E.; Vaze, M.B.; Ochsenbien, F.; Guerois, R.; Haber, J.E.; Marsolier-Kergoat, M.-C. PP2C Phosphatases Ptc2 and Ptc3 Are Required for DNA Checkpoint Inactivation after a Double-Strand Break. Mol. Cell 2003, 11, 827–835. [Google Scholar] [CrossRef]
- Heikkinen, K.; Rapakko, K.; Karppinen, S.M.; Erkko, H.; Knuutila, S.; Lundan, T.; Mannermaa, A.; Borresen-Dale, A.L.; Borg, A.; Barkardottir, R.B.; et al. RAD50 and NBS1 are breast cancer susceptibility genes associated with genomic instability. Carcinogenesis 2006, 27, 1593–1599. [Google Scholar] [CrossRef] [Green Version]
- Carlessi, L.; Buscemi, G.; Fontanella, E.; Delia, D. A protein phosphatase feedback mechanism regulates the basal phosphorylation of Chk2 kinase in the absence of DNA damage. Biochim. Biophys. Acta 2010, 1803, 1213–1223. [Google Scholar] [CrossRef] [Green Version]
- Bolton, K.L.; Ptashkin, R.N.; Gao, T.; Braunstein, L.; Devlin, S.M.; Kelly, D.; Patel, M.; Berthon, A.; Syed, A.; Yabe, M.; et al. Cancer therapy shapes the fitness landscape of clonal hematopoiesis. Nat. Genet. 2020. [Google Scholar] [CrossRef]
- Bohgaki, M.; Hakem, A.; Halaby, M.J.; Bohgaki, T.; Li, Q.; Bissey, P.A.; Shloush, J.; Kislinger, T.; Sanchez, O.; Sheng, Y.; et al. The E3 ligase PIRH2 polyubiquitylates CHK2 and regulates its turnover. Cell Death Differ. 2013, 20, 812–822. [Google Scholar] [CrossRef] [Green Version]
- Kass, E.M.; Poyurovsky, M.V.; Zhu, Y.; Prives, C. Mdm2 and PCAF increase Chk2 ubiquitination and degradation independently of their intrinsic E3 ligase activities. Cell Cycle 2009, 8, 430–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Limones, C.; Lara-Chica, M.; Jimenez-Jimenez, C.; Perez, M.; Moreno, P.; Munoz, E.; Calzado, M.A. CHK2 stability is regulated by the E3 ubiquitin ligase SIAH2. Oncogene 2016, 35, 4289–4301. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.M.; Yan, L.; Ryan, C.E.; Takada, S.; Piwnica-Worms, H. Regulation of Chk2 ubiquitination and signaling through autophosphorylation of serine 379. Mol. Cell. Biol. 2008, 28, 5874–5885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Yang, L.; Wang, C.; Zhao, W.; Ju, Z.; Zhang, W.; Shen, J.; Peng, Y.; An, C.; Luu, Y.T.; et al. Inhibition of the ATM/Chk2 axis promotes cGAS/STING signaling in ARID1A-deficient tumors. J. Clin. Investig. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Zaugg, K.; Mak, T.W.; Elledge, S.J. A role for the deubiquitinating enzyme USP28 in control of the DNA-damage response. Cell 2006, 126, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Chen, Y.; Geng, G.; Li, L.; Yin, P.; Nowsheen, S.; Li, Y.; Wu, C.; Liu, J.; Zhao, F.; et al. USP39 regulates DNA damage response and chemo-radiation resistance by deubiquitinating and stabilizing CHK2. Cancer Lett. 2019, 449, 114–124. [Google Scholar] [CrossRef]
- Alves-Fernandes, D.K.; Jasiulionis, M.G. The Role of SIRT1 on DNA Damage Response and Epigenetic Alterations in Cancer. Int. J. Mol. Sci. 2019, 20, 3153. [Google Scholar] [CrossRef] [Green Version]
- Seo, G.J.; Kim, S.E.; Lee, Y.M.; Lee, J.W.; Lee, J.R.; Hahn, M.J.; Kim, S.T. Determination of substrate specificity and putative substrates of Chk2 kinase. Biochem. Biophys. Res. Commun. 2003, 304, 339–343. [Google Scholar] [CrossRef]
- Zannini, L.; Delia, D.; Buscemi, G. CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 2014, 6, 442–457. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef]
- Sulli, G.; Di Micco, R.; d’Adda di Fagagna, F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat. Rev. Cancer 2012, 12, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Bartkova, J.; Lukas, J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007, 26, 7773–7779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neizer-Ashun, F.; Bhattacharya, R. Reality CHEK: Understanding the biology and clinical potential of CHK1. Cancer Lett. 2020, 497, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Chen, J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 2010, 9, 472–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirao, A.; Kong, Y.Y.; Matsuoka, S.; Wakeham, A.; Ruland, J.; Yoshida, H.; Liu, D.; Elledge, S.J.; Mak, T.W. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science 2000, 287, 1824–1827. [Google Scholar] [CrossRef]
- Chen, L.; Gilkes, D.M.; Pan, Y.; Lane, W.S.; Chen, J. ATM and Chk2-dependent phosphorylation of MDMX contribute to p53 activation after DNA damage. EMBO J. 2005, 24, 3411–3422. [Google Scholar] [CrossRef] [Green Version]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef]
- Waldman, T.; Kinzler, K.W.; Vogelstein, B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995, 55, 5187–5190. [Google Scholar]
- Kastan, M.B.; Zhan, Q.; el-Deiry, W.S.; Carrier, F.; Jacks, T.; Walsh, W.V.; Plunkett, B.S.; Vogelstein, B.; Fornace, A.J., Jr. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell 1992, 71, 587–597. [Google Scholar] [CrossRef]
- Jack, M.T.; Woo, R.A.; Hirao, A.; Cheung, A.; Mak, T.W.; Lee, P.W. Chk2 is dispensable for p53-mediated G1 arrest but is required for a latent p53-mediated apoptotic response. Proc. Natl. Acad. Sci. USA 2002, 99, 9825–9829. [Google Scholar] [CrossRef] [Green Version]
- Jallepalli, P.V.; Lengauer, C.; Vogelstein, B.; Bunz, F. The Chk2 tumor suppressor is not required for p53 responses in human cancer cells. J. Biol. Chem. 2003, 278, 20475–20479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaltiel, I.A.; Aprelia, M.; Saurin, A.T.; Chowdhury, D.; Kops, G.J.; Voest, E.E.; Medema, R.H. Distinct phosphatases antagonize the p53 response in different phases of the cell cycle. Proc. Natl. Acad. Sci. USA 2014, 111, 7313–7318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Zhang, S.; Gao, X.; Gao, X.; Xu, X.; Lv, Y.; Zhang, Y.; Zhu, Z.; Zhang, C.; Li, Q.; et al. Roles of Kruppel-associated Box (KRAB)-associated Co-repressor KAP1 Ser-473 Phosphorylation in DNA Damage Response. J. Biol. Chem. 2012, 287, 18937–18952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.H.; Goodarzi, A.A.; Adelmant, G.O.; Pan, Y.; Jeggo, P.A.; Marto, J.A.; Chowdhury, D. Phosphoproteomic analysis reveals that PP4 dephosphorylates KAP-1 impacting the DNA damage response. EMBO J. 2012, 31, 2403–2415. [Google Scholar] [CrossRef] [PubMed]
- Stevens, C.; Smith, L.; La Thangue, N.B. Chk2 activates E2F-1 in response to DNA damage. Nat. Cell Biol. 2003, 5, 401–409. [Google Scholar] [CrossRef]
- Donzelli, M.; Draetta, G.F. Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 2003, 4, 671–677. [Google Scholar] [CrossRef] [Green Version]
- Chen, J. The Cell-Cycle Arrest and Apoptotic Functions of p53 in Tumor Initiation and Progression. Cold Spring Harb. Perspect. Med. 2016, 6, a026104. [Google Scholar] [CrossRef]
- Peng, C.Y.; Graves, P.R.; Thoma, R.S.; Wu, Z.; Shaw, A.S.; Piwnica-Worms, H. Mitotic and G2 checkpoint control: Regulation of 14-3-3 protein binding by phosphorylation of Cdc25C on serine-216. Science 1997, 277, 1501–1505. [Google Scholar] [CrossRef]
- Yang, S.; Kuo, C.; Bisi, J.E.; Kim, M.K. PML-dependent apoptosis after DNA damage is regulated by the checkpoint kinase hCds1/Chk2. Nat. Cell Biol. 2002, 4, 865–870. [Google Scholar] [CrossRef]
- Di Masi, A.; Cilli, D.; Berardinelli, F.; Talarico, A.; Pallavicini, I.; Pennisi, R.; Leone, S.; Antoccia, A.; Noguera, N.I.; Lo-Coco, F.; et al. PML nuclear body disruption impairs DNA double-strand break sensing and repair in APL. Cell Death Dis. 2016, 7, e2308. [Google Scholar] [CrossRef]
- Yang, S.; Jeong, J.H.; Brown, A.L.; Lee, C.H.; Pandolfi, P.P.; Chung, J.H.; Kim, M.K. Promyelocytic leukemia activates Chk2 by mediating Chk2 autophosphorylation. J. Biol. Chem. 2006, 281, 26645–26654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Willers, H.; Feng, Z.; Ghosh, J.C.; Kim, S.; Weaver, D.T.; Chung, J.H.; Powell, S.N.; Xia, F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol. Cell Biol. 2004, 24, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.S.; Collins, K.M.; Brown, A.L.; Lee, C.H.; Chung, J.H. hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature 2000, 404, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Petsalaki, E.; Zachos, G. DNA damage response proteins regulating mitotic cell division: Double agents preserving genome stability. FEBS J. 2020, 287, 1700–1721. [Google Scholar] [CrossRef] [PubMed]
- Sankaran, S.; Starita, L.M.; Groen, A.C.; Ko, M.J.; Parvin, J.D. Centrosomal microtubule nucleation activity is inhibited by BRCA1-dependent ubiquitination. Mol. Cell Biol. 2005, 25, 8656–8668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chabalier-Taste, C.; Racca, C.; Dozier, C.; Larminat, F. BRCA1 is regulated by Chk2 in response to spindle damage. Biochim. Biophys. Acta 2008, 1783, 2223–2233. [Google Scholar] [CrossRef]
- Stolz, A.; Ertych, N.; Bastians, H. Tumor suppressor CHK2: Regulator of DNA damage response and mediator of chromosomal stability. Clin. Cancer Res. 2011, 17, 401–405. [Google Scholar] [CrossRef] [Green Version]
- Nai, S.; Shi, Y.; Ru, H.; Ding, Y.; Geng, Q.; Li, Z.; Dong, M.Q.; Xu, X.; Li, J. Chk2-dependent phosphorylation of myosin phosphatase targeting subunit 1 (MYPT1) regulates centrosome maturation. Cell Cycle 2019, 18, 2651–2659. [Google Scholar] [CrossRef]
- Guo, Q.Q.; Wang, S.S.; Zhang, S.S.; Xu, H.D.; Li, X.M.; Guan, Y.; Yi, F.; Zhou, T.T.; Jiang, B.; Bai, N.; et al. ATM-CHK2-Beclin 1 axis promotes autophagy to maintain ROS homeostasis under oxidative stress. EMBO J. 2020, 39, e103111. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, J.; Liang, G.; Geng, G.; Zhao, F.; Yin, P.; Nowsheen, S.; Wu, C.; Li, Y.; Li, L.; et al. CHK2-FOXK axis promotes transcriptional control of autophagy programs. Sci. Adv. 2020, 6, eaax5819. [Google Scholar] [CrossRef] [Green Version]
- Takai, H.; Naka, K.; Okada, Y.; Watanabe, M.; Harada, N.; Saito, S.; Anderson, C.W.; Appella, E.; Nakanishi, M.; Suzuki, H.; et al. Chk2-deficient mice exhibit radioresistance and defective p53-mediated transcription. EMBO J. 2002, 21, 5195–5205. [Google Scholar] [CrossRef] [PubMed]
- Niida, H.; Murata, K.; Shimada, M.; Ogawa, K.; Ohta, K.; Suzuki, K.; Fujigaki, H.; Khaw, A.K.; Banerjee, B.; Hande, M.P.; et al. Cooperative functions of Chk1 and Chk2 reduce tumour susceptibility in vivo. EMBO J. 2010, 29, 3558–3570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paperna, T.; Sharon-Shwartzman, N.; Kurolap, A.; Goldberg, Y.; Moustafa, N.; Carasso, Y.; Feinstien, M.; Mory, A.; Reznick-Levi, G.; Gonzaga-Jauregui, C.; et al. Homozygosity for CHEK2 p.Gly167Arg leads to a unique cancer syndrome with multiple complex chromosomal translocations in peripheral blood karyotype. J. Med. Genet. 2019. [Google Scholar] [CrossRef]
- Rainville, I.; Hatcher, S.; Rosenthal, E.; Larson, K.; Bernhisel, R.; Meek, S.; Gorringe, H.; Mundt, E.; Manley, S. High risk of breast cancer in women with biallelic pathogenic variants in CHEK2. Breast Cancer Res. Treat 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Jaarsveld, M.T.M.; Deng, D.; Ordoñez-Rueda, D.; Paulsen, M.; Wiemer, E.A.C.; Zi, Z. Cell-type-specific role of CHK2 in mediating DNA damage-induced G2 cell cycle arrest. Oncogenesis 2020, 9, 35. [Google Scholar] [CrossRef]
- Wu, X.; Webster, S.R.; Chen, J. Characterization of tumor-associated Chk2 mutations. J. Biol. Chem. 2001, 276, 2971–2974. [Google Scholar] [CrossRef] [Green Version]
- Allinen, M.; Huusko, P.; Mantyniemi, S.; Launonen, V.; Winqvist, R. Mutation analysis of the CHK2 gene in families with hereditary breast cancer. Br. J. Cancer 2001, 85, 209–212. [Google Scholar] [CrossRef]
- Bougeard, G.; Limacher, J.M.; Martin, C.; Charbonnier, F.; Killian, A.; Delattre, O.; Longy, M.; Jonveaux, P.; Fricker, J.P.; Stoppa-Lyonnet, D.; et al. Detection of 11 germline inactivating TP53 mutations and absence of TP63 and HCHK2 mutations in 17 French families with Li-Fraumeni or Li-Fraumeni-like syndrome. J. Med. Genet. 2001, 38, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Sodha, N.; Houlston, R.S.; Bullock, S.; Yuille, M.A.; Chu, C.; Turner, G.; Eeles, R.A. Increasing evidence that germline mutations in CHEK2 do not cause Li-Fraumeni syndrome. Hum. Mutat. 2002, 20, 460–462. [Google Scholar] [CrossRef]
- Vahteristo, P.; Bartkova, J.; Eerola, H.; Syrjakoski, K.; Ojala, S.; Kilpivaara, O.; Tamminen, A.; Kononen, J.; Aittomaki, K.; Heikkila, P.; et al. A CHEK2 genetic variant contributing to a substantial fraction of familial breast cancer. Am. J. Hum. Genet. 2002, 71, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, R.; Onel, K.; Facio, F.; Nafa, K.; Diaz, L.R.; Kauff, N.; Huang, H.; Robson, M.; Ellis, N.; Offit, K. The TP53 mutational spectrum and frequency of CHEK2*1100delC in Li-Fraumeni-like kindreds. Fam. Cancer 2005, 4, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Hogervorst, F.B.; Cornelis, R.S.; Bout, M.; van Vliet, M.; Oosterwijk, J.C.; Olmer, R.; Bakker, B.; Klijn, J.G.; Vasen, H.F.; Meijers-Heijboer, H.; et al. Rapid detection of BRCA1 mutations by the protein truncation test. Nat. Genet. 1995, 10, 208–212. [Google Scholar] [CrossRef] [PubMed]
- Aloraifi, F.; McCartan, D.; McDevitt, T.; Green, A.J.; Bracken, A.; Geraghty, J. Protein-truncating variants in moderate-risk breast cancer susceptibility genes: A meta-analysis of high-risk case-control screening studies. Cancer Genet. 2015, 208, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, E.G.; Stettner, A.R.; Miller, S.A.; Solomon, S.R.; Marshall, M.L.; Roberts, M.E.; Susswein, L.R.; Arvai, K.J.; Klein, R.T.; Murphy, P.D.; et al. Differences in cancer prevalence among CHEK2 carriers identified via multi-gene panel testing. Cancer Genet. 2020, 246–247, 12–17. [Google Scholar] [CrossRef]
- Kleiblova, P.; Stolarova, L.; Krizova, K.; Lhota, F.; Hojny, J.; Zemankova, P.; Havranek, O.; Vocka, M.; Cerna, M.; Lhotova, K.; et al. Identification of deleterious germline CHEK2 mutations and their association with breast and ovarian cancer. Int. J. Cancer. J. Int. Cancer 2019. [Google Scholar] [CrossRef]
- Apostolou, P.; Fostira, F.; Mollaki, V.; Delimitsou, A.; Vlassi, M.; Pentheroudakis, G.; Faliakou, E.; Kollia, P.; Fountzilas, G.; Yannoukakos, D.; et al. Characterization and prevalence of two novel CHEK2 large deletions in Greek breast cancer patients. J. Hum. Genet. 2018, 63, 877–886. [Google Scholar] [CrossRef]
- Soukupova, J.; Zemankova, P.; Kleiblova, P.; Janatova, M.; Kleibl, Z. CZECANCA: CZEch CAncer paNel for Clinical Application-- Design and Optimization of the Targeted Sequencing Panel for the Identification of Cancer Susceptibility in High-risk Individuals from the Czech Republic. Klin. Onkol. 2016, 29 (Suppl. 1), S46–S54. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Young, E.L.; Feng, B.J.; Stark, A.W.; Damiola, F.; Durand, G.; Forey, N.; Francy, T.C.; Gammon, A.; Kohlmann, W.K.; Kaphingst, K.A.; et al. Multigene testing of moderate-risk genes: Be mindful of the missense. J. Med. Genet. 2016, 53, 366–376. [Google Scholar] [CrossRef]
- Manoukian, S.; Peissel, B.; Frigerio, S.; Lecis, D.; Bartkova, J.; Roversi, G.; Radice, P.; Bartek, J.; Delia, D. Two new CHEK2 germ-line variants detected in breast cancer/sarcoma families negative for BRCA1, BRCA2, and TP53 gene mutations. Breast Cancer Res. Treat 2011, 130, 207–215. [Google Scholar] [CrossRef]
- Bell, D.W.; Kim, S.H.; Godwin, A.K.; Schiripo, T.A.; Harris, P.L.; Haserlat, S.M.; Wahrer, D.C.; Haiman, C.A.; Daly, M.B.; Niendorf, K.B.; et al. Genetic and functional analysis of CHEK2 (CHK2) variants in multiethnic cohorts. Int. J. Cancer 2007, 121, 2661–2667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desrichard, A.; Bidet, Y.; Uhrhammer, N.; Bignon, Y.J. CHEK2 contribution to hereditary breast cancer in non-BRCA families. Breast Cancer Res. 2011, 13, R119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roeb, W.; Higgins, J.; King, M.C. Response to DNA damage of CHEK2 missense mutations in familial breast cancer. Hum. Mol. Genet. 2012, 21, 2738–2744. [Google Scholar] [CrossRef] [Green Version]
- Tischkowitz, M.D.; Yilmaz, A.; Chen, L.Q.; Karyadi, D.M.; Novak, D.; Kirchhoff, T.; Hamel, N.; Tavtigian, S.V.; Kolb, S.; Bismar, T.A.; et al. Identification and characterization of novel SNPs in CHEK2 in Ashkenazi Jewish men with prostate cancer. Cancer Lett. 2008, 270, 173–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaag, A.; Walsh, T.; Renbaum, P.; Kirchhoff, T.; Nafa, K.; Shiovitz, S.; Mandell, J.B.; Welcsh, P.; Lee, M.K.; Ellis, N.; et al. Functional and genomic approaches reveal an ancient CHEK2 allele associated with breast cancer in the Ashkenazi Jewish population. Hum. Mol. Genet. 2005, 14, 555–563. [Google Scholar] [CrossRef] [Green Version]
- Hauke, J.; Horvath, J.; Gross, E.; Gehrig, A.; Honisch, E.; Hackmann, K.; Schmidt, G.; Arnold, N.; Faust, U.; Sutter, C.; et al. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: Results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med. 2018, 7, 1349–1358. [Google Scholar] [CrossRef]
- Lhota, F.; Zemankova, P.; Kleiblova, P.; Soukupova, J.; Vocka, M.; Stranecky, V.; Janatova, M.; Hartmannova, H.; Hodanova, K.; Kmoch, S.; et al. Hereditary truncating mutations of DNA repair and other genes in BRCA1/BRCA2/PALB2-negatively tested breast cancer patients. Clin. Genet. 2016, 90, 324–333. [Google Scholar] [CrossRef]
- Kurian, A.W.; Ward, K.C.; Howlader, N.; Deapen, D.; Hamilton, A.S.; Mariotto, A.; Miller, D.; Penberthy, L.S.; Katz, S.J. Genetic Testing and Results in a Population-Based Cohort of Breast Cancer Patients and Ovarian Cancer Patients. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef] [Green Version]
- Kleibl, Z.; Novotny, J.; Bezdickova, D.; Malik, R.; Kleiblova, P.; Foretova, L.; Petruzelka, L.; Ilencikova, D.; Cinek, P.; Pohlreich, P. The CHEK2 c.1100delC germline mutation rarely contributes to breast cancer development in the Czech Republic. Breast Cancer Res. Treat 2005, 90, 165–167. [Google Scholar] [CrossRef]
- Caligo, M.A.; Agata, S.; Aceto, G.; Crucianelli, R.; Manoukian, S.; Peissel, B.; Scaini, M.C.; Sensi, E.; Veschi, S.; Cama, A.; et al. The CHEK2 c.1100delC mutation plays an irrelevant role in breast cancer predisposition in Italy. Hum. Mutat. 2004, 24, 100–101. [Google Scholar] [CrossRef]
- Fachal, L.; Santamarina, M.; Blanco, A.; Carracedo, A.; Vega, A. CHEK2 c.1100delC mutation among non-BRCA1/2 Spanish hereditary breast cancer families. Clin. Transl. Oncol. 2013, 15, 164–165. [Google Scholar] [CrossRef] [PubMed]
- Apostolou, P.; Fostira, F.; Papamentzelopoulou, M.; Michelli, M.; Panopoulos, C.; Fountzilas, G.; Konstantopoulou, I.; Voutsinas, G.E.; Yannoukakos, D. CHEK2 c.1100delC allele is rarely identified in Greek breast cancer cases. Cancer Genet. 2015, 208, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Irmejs, A.; Miklasevics, E.; Boroschenko, V.; Gardovskis, A.; Vanags, A.; Melbarde-Gorkusa, I.; Bitina, M.; Suchy, J.; Gardovskis, J. Pilot study on low penetrance breast and colorectal cancer predisposition markers in latvia. Hered. Cancer Clin. Pract. 2006, 4, 48–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, P.; McKay, J.; Moore, L.; Zaridze, D.; Mukeria, A.; Szeszenia-Dabrowska, N.; Lissowska, J.; Rudnai, P.; Fabianova, E.; Mates, D.; et al. Uncommon CHEK2 mis-sense variant and reduced risk of tobacco-related cancers: Case control study. Hum. Mol. Genet. 2007, 16, 1794–1801. [Google Scholar] [CrossRef] [PubMed]
- Bermisheva, M.A.; Takhirova, Z.R.; Bogdanova, N.; Khusnutdinova, E.K. Frequency of CHEK2 gene mutations in breast cancer patients from Republic of Bashkortostan. Mol. Biol. 2014, 48, 46–51. [Google Scholar] [CrossRef]
- Kleibl, Z.; Havranek, O.; Novotny, J.; Kleiblova, P.; Soucek, P.; Pohlreich, P. Analysis of CHEK2 FHA domain in Czech patients with sporadic breast cancer revealed distinct rare genetic alterations. Breast Cancer Res. Treat 2008, 112, 159–164. [Google Scholar] [CrossRef]
- Kaufman, B.; Laitman, Y.; Gronwald, J.; Winqvist, R.; Irmejs, A.; Lubinski, J.; Pylkas, K.; Gardovskis, J.; Miklasevics, E.; Friedman, E. Haplotypes of the I157T CHEK2 germline mutation in ethnically diverse populations. Fam. Cancer 2009, 8, 473–478. [Google Scholar] [CrossRef]
- Walsh, T.; Casadei, S.; Coats, K.H.; Swisher, E.; Stray, S.M.; Higgins, J.; Roach, K.C.; Mandell, J.; Lee, M.K.; Ciernikova, S.; et al. Spectrum of mutations in BRCA1, BRCA2, CHEK2, and TP53 in families at high risk of breast cancer. JAMA J. Am. Med. Assoc. 2006, 295, 1379–1388. [Google Scholar] [CrossRef] [Green Version]
- Cybulski, C.; Huzarski, T.; Byrski, T.; Gronwald, J.; Debniak, T.; Jakubowska, A.; Gorski, B.; Wokolorczyk, D.; Masojc, B.; Narod, S.A.; et al. Estrogen receptor status in CHEK2-positive breast cancers: Implications for chemoprevention. Clin Genet. 2009, 75, 72–78. [Google Scholar] [CrossRef]
- Plonis, J.; Kalniete, D.; Nakazawa-Miklasevica, M.; Irmejs, A.; Vjaters, E.; Gardovskis, J.; Miklasevics, E. The CHEK2 del5395 is a founder mutation without direct effects for cancer risk in the latvian population. Balk. J. Med. Genet. 2015, 18, 33–36. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Meng, H.; Yao, L.; Lv, M.; Bai, J.; Zhang, J.; Wang, L.; Ouyang, T.; Li, J.; Wang, T.; et al. Germline Mutations in Cancer Susceptibility Genes in a Large Series of Unselected Breast Cancer Patients. Clin. Cancer Res. 2017, 23, 6113–6119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Z.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; Lin, B.; Xu, Y.; Xie, Y. Identification and analysis of CHEK2 germline mutations in Chinese BRCA1/2-negative breast cancer patients. Breast Cancer Res. Treat 2018, 169, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Guo, X.; Wen, W.; Shi, J.; Long, J.; Cai, Q.; Shu, X.O.; Xiang, Y.; Zheng, W. Evaluation of pathogenetic mutations in breast cancer predisposition genes in population-based studies conducted among Chinese women. Breast Cancer Res. Treat 2020, 181, 465–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Momozawa, Y.; Iwasaki, Y.; Hirata, M.; Liu, X.; Kamatani, Y.; Takahashi, A.; Sugano, K.; Yoshida, T.; Murakami, Y.; Matsuda, K.; et al. Germline Pathogenic Variants in 7636 Japanese Patients With Prostate Cancer and 12 366 Controls. J. Natl. Cancer Inst. 2020, 112, 369–376. [Google Scholar] [CrossRef] [Green Version]
- Momozawa, Y.; Iwasaki, Y.; Parsons, M.T.; Kamatani, Y.; Takahashi, A.; Tamura, C.; Katagiri, T.; Yoshida, T.; Nakamura, S.; Sugano, K.; et al. Germline pathogenic variants of 11 breast cancer genes in 7,051 Japanese patients and 11,241 controls. Nat. Commun. 2018, 9, 4083. [Google Scholar] [CrossRef]
- Fostira, F.; Kostantopoulou, I.; Apostolou, P.; Papamentzelopoulou, M.S.; Papadimitriou, C.; Faliakou, E.; Christodoulou, C.; Boukovinas, I.; Razis, E.; Tryfonopoulos, D.; et al. One in three highly selected Greek patients with breast cancer carries a loss-of-function variant in a cancer susceptibility gene. J. Med. Genet. 2020, 57, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Kurian, A.W.; Bernhisel, R.; Larson, K.; Caswell-Jin, J.L.; Shadyab, A.H.; Ochs-Balcom, H.; Stefanick, M.L. Prevalence of Pathogenic Variants in Cancer Susceptibility Genes Among Women With Postmenopausal Breast Cancer. JAMA J. Am. Med. Assoc. 2020, 323, 995–997. [Google Scholar] [CrossRef]
- Rogoża-Janiszewska, E.; Malińska, K.; Cybulski, C.; Jakubowska, A.; Gronwald, J.; Huzarski, T.; Lener, M.; Górski, B.; Kluźniak, W.; Rudnicka, H.; et al. Prevalence of Recurrent Mutations Predisposing to Breast Cancer in Early-Onset Breast Cancer Patients from Poland. Cancers 2020, 12, 2321. [Google Scholar] [CrossRef]
- Cybulski, C.; Kluźniak, W.; Huzarski, T.; Wokołorczyk, D.; Kashyap, A.; Rusak, B.; Stempa, K.; Gronwald, J.; Szymiczek, A.; Bagherzadeh, M.; et al. The spectrum of mutations predisposing to familial breast cancer in Poland. Int. J. Cancer 2019, 145, 3311–3320. [Google Scholar] [CrossRef]
- Nurmi, A.; Muranen, T.A.; Pelttari, L.M.; Kiiski, J.I.; Heikkinen, T.; Lehto, S.; Kallioniemi, A.; Schleutker, J.; Butzow, R.; Blomqvist, C.; et al. Recurrent moderate-risk mutations in Finnish breast and ovarian cancer patients. Int. J. Cancer 2019. [Google Scholar] [CrossRef]
- Girard, E.; Eon-Marchais, S.; Olaso, R.; Renault, A.L.; Damiola, F.; Dondon, M.G.; Barjhoux, L.; Goidin, D.; Meyer, V.; Le Gal, D.; et al. Familial breast cancer and DNA repair genes: Insights into known and novel susceptibility genes from the GENESIS study, and implications for multigene panel testing. Int. J. Cancer 2019, 144, 1962–1974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, B.; Allen, J.; Luccarini, C.; Pooley, K.A.; Shah, M.; Bolla, M.K.; Wang, Q.; Ahmed, S.; Baynes, C.; Conroy, D.M.; et al. Rare, protein-truncating variants in ATM, CHEK2 and PALB2, but not XRCC2, are associated with increased breast cancer risks. J. Med. Genet. 2017, 54, 732–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slavin, T.P.; Maxwell, K.N.; Lilyquist, J.; Vijai, J.; Neuhausen, S.L.; Hart, S.N.; Ravichandran, V.; Thomas, T.; Maria, A.; Villano, D.; et al. The contribution of pathogenic variants in breast cancer susceptibility genes to familial breast cancer risk. NPJ Breast Cancer 2017, 3, 22. [Google Scholar] [CrossRef] [PubMed]
- Couch, F.J.; Shimelis, H.; Hu, C.; Hart, S.N.; Polley, E.C.; Na, J.; Hallberg, E.; Moore, R.; Thomas, A.; Lilyquist, J.; et al. Associations Between Cancer Predisposition Testing Panel Genes and Breast Cancer. JAMA Oncol. 2017, 3, 1190–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.K.; Hogervorst, F.; van Hien, R.; Cornelissen, S.; Broeks, A.; Adank, M.A.; Meijers, H.; Waisfisz, Q.; Hollestelle, A.; Schutte, M.; et al. Age-and Tumor Subtype-Specific Breast Cancer Risk Estimates for CHEK2*1100delC Carriers. J. Clin. Oncol. 2016, 34, 2750–2760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naslund-Koch, C.; Nordestgaard, B.G.; Bojesen, S.E. Increased Risk for Other Cancers in Addition to Breast Cancer for CHEK2*1100delC Heterozygotes Estimated From the Copenhagen General Population Study. J. Clin. Oncol. 2016, 34, 1208–1216. [Google Scholar] [CrossRef]
- Southey, M.C.; Goldgar, D.E.; Winqvist, R.; Pylkas, K.; Couch, F.; Tischkowitz, M.; Foulkes, W.D.; Dennis, J.; Michailidou, K.; van Rensburg, E.J.; et al. PALB2, CHEK2 and ATM rare variants and cancer risk: Data from COGS. J. Med. Genet. 2016, 53, 800–811. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Liao, J.; Xu, Y.; Chen, W.; Liu, D.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; et al. A recurrent CHEK2 p.H371Y mutation is associated with breast cancer risk in Chinese women. Hum. Mutat. 2011, 32, 1000–1003. [Google Scholar] [CrossRef]
- Cybulski, C.; Wokolorczyk, D.; Jakubowska, A.; Huzarski, T.; Byrski, T.; Gronwald, J.; Masojc, B.; Deebniak, T.; Gorski, B.; Blecharz, P.; et al. Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J. Clin. Oncol. 2011, 29, 3747–3752. [Google Scholar] [CrossRef]
- Le Calvez-Kelm, F.; Lesueur, F.; Damiola, F.; Vallee, M.; Voegele, C.; Babikyan, D.; Durand, G.; Forey, N.; McKay-Chopin, S.; Robinot, N.; et al. Rare, evolutionarily unlikely missense substitutions in CHEK2 contribute to breast cancer susceptibility: Results from a breast cancer family registry case-control mutation-screening study. Breast Cancer Res. 2011, 13, R6. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, O.; Johnson, N.; Dos Santos Silva, I.; Kilpivaara, O.; Aittomaki, K.; Blomqvist, C.; Nevanlinna, H.; Wasielewski, M.; Meijers-Heijerboer, H.; Broeks, A.; et al. Family history, genetic testing, and clinical risk prediction: Pooled analysis of CHEK2 1100delC in 1,828 bilateral breast cancers and 7030 controls. Cancer Epidemiol. Biomark. Prev. 2009, 18, 230–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weischer, M.; Bojesen, S.E.; Tybjaerg-Hansen, A.; Axelsson, C.K.; Nordestgaard, B.G. Increased risk of breast cancer associated with CHEK2*1100delC. J. Clin. Oncol. 2007, 25, 57–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cybulski, C.; Gorski, B.; Huzarski, T.; Byrski, T.; Gronwald, J.; Debniak, T.; Wokolorczyk, D.; Jakubowska, A.; Kowalska, E.; Oszurek, O.; et al. CHEK2-positive breast cancers in young Polish women. Clin. Cancer Res. 2006, 12, 4832–4835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chekmariova, E.V.; Sokolenko, A.P.; Buslov, K.G.; Iyevleva, A.G.; Ulibina, Y.M.; Rozanov, M.E.; Mitiushkina, N.V.; Togo, A.V.; Matsko, D.E.; Voskresenskiy, D.A.; et al. CHEK2 1100delC mutation is frequent among Russian breast cancer patients. Breast Cancer Res. Treat 2006, 100, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Dufault, M.R.; Betz, B.; Wappenschmidt, B.; Hofmann, W.; Bandick, K.; Golla, A.; Pietschmann, A.; Nestle-Kramling, C.; Rhiem, K.; Huttner, C.; et al. Limited relevance of the CHEK2 gene in hereditary breast cancer. Int. J. Cancer 2004, 110, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Consortium, C.B.C.C.-C. CHEK2*1100delC and susceptibility to breast cancer: A collaborative analysis involving 10,860 breast cancer cases and 9,065 controls from 10 studies. Am. J. Hum. Genet. 2004, 74, 1175–1182. [Google Scholar] [CrossRef] [Green Version]
- Meijers-Heijboer, H.; van den, O.A.; Klijn, J.; Wasielewski, M.; de Snoo, A.; Oldenburg, R.; Hollestelle, A.; Houben, M.; Crepin, E.; Veghel-Plandsoen, M.; et al. Low-penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat. Genet. 2002, 31, 55–59. [Google Scholar]
- Liang, M.; Zhang, Y.; Sun, C.; Rizeq, F.K.; Min, M.; Shi, T.; Sun, Y. Association between CHEK2*1100delC and Breast Cancer: A Systematic Review and Meta-Analysis. Mol. Diagn. 2018, 22, 397–407. [Google Scholar] [CrossRef]
- Hallamies, S.; Pelttari, L.M.; Poikonen-Saksela, P.; Jekunen, A.; Jukkola-Vuorinen, A.; Auvinen, P.; Blomqvist, C.; Aittomaki, K.; Mattson, J.; Nevanlinna, H. CHEK2 c.1100delC mutation is associated with an increased risk for male breast cancer in Finnish patient population. BMC Cancer 2017, 17, 620. [Google Scholar] [CrossRef]
- Wasielewski, M.; den Bakker, M.A.; van den, O.A.; Meijer-van Gelder, M.E.; Portengen, H.; Klijn, J.G.; Meijers-Heijboer, H.; Foekens, J.A.; Schutte, M. CHEK2 1100delC and male breast cancer in the Netherlands. Breast Cancer Res. Treat. 2009, 116, 397–400. [Google Scholar] [CrossRef]
- Yang, Y.; Shu, X.; Shu, X.O.; Bolla, M.K.; Kweon, S.S.; Cai, Q.; Michailidou, K.; Wang, Q.; Dennis, J.; Park, B.; et al. Re-evaluating genetic variants identified in candidate gene studies of breast cancer risk using data from nearly 280,000 women of Asian and European ancestry. EBioMedicine 2019, 48, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Han, F.F.; Guo, C.L.; Liu, L.H. The effect of CHEK2 variant I157T on cancer susceptibility: Evidence from a meta-analysis. DNA Cell Biol. 2013, 32, 329–335. [Google Scholar] [CrossRef]
- Liu, C.; Wang, Y.; Wang, Q.S.; Wang, Y.J. The CHEK2 I157T variant and breast cancer susceptibility: A systematic review and meta-analysis. Asian Pac. J. Cancer Prev. APJCP 2012, 13, 1355–1360. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, F.; Wang, Y.; Liu, S.C. CHEK2 1100delC variant and breast cancer risk in Caucasians: A meta-analysis based on 25 studies with 29,154 cases and 37,064 controls. Asian Pac. J. Cancer Prev. 2012, 13, 3501–3505. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Beeghly-Fadiel, A.; Long, J.; Zheng, W. Genetic variants associated with breast-cancer risk: Comprehensive research synopsis, meta-analysis, and epidemiological evidence. Lancet. Oncol. 2011, 12, 477–488. [Google Scholar] [CrossRef] [Green Version]
- Weischer, M.; Bojesen, S.E.; Ellervik, C.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. CHEK2*1100delC genotyping for clinical assessment of breast cancer risk: Meta-analyses of 26,000 patient cases and 27,000 controls. J. Clin. Oncol. 2008, 26, 542–548. [Google Scholar] [CrossRef] [Green Version]
- Johnson, N.; Fletcher, O.; Naceur-Lombardelli, C.; dos Santos Silva, I.; Ashworth, A.; Peto, J. Interaction between CHEK2*1100delC and other low-penetrance breast-cancer susceptibility genes: A familial study. Lancet 2005, 366, 1554–1557. [Google Scholar] [CrossRef]
- Muranen, T.A.; Greco, D.; Blomqvist, C.; Aittomaki, K.; Khan, S.; Hogervorst, F.; Verhoef, S.; Pharoah, P.D.P.; Dunning, A.M.; Shah, M.; et al. Genetic modifiers of CHEK2*1100delC-associated breast cancer risk. Genet. Med. 2017, 19, 599–603. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.; Mavaddat, N.; Wilcox, A.N.; Cunningham, A.P.; Carver, T.; Hartley, S.; Babb de Villiers, C.; Izquierdo, A.; Simard, J.; Schmidt, M.K.; et al. BOADICEA: A comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet. Med. 2019, 21, 1708–1718. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, S.; Hughes, E.; Wagner, S.; Tshiaba, P.; Rosenthal, E.; Roa, B.B.; Kurian, A.W.; Domchek, S.M.; Garber, J.; Lancaster, J.; et al. Association of a Polygenic Risk Score With Breast Cancer Among Women Carriers of High- and Moderate-Risk Breast Cancer Genes. JAMA Netw. Open 2020, 3, e208501. [Google Scholar] [CrossRef]
- Akdeniz, D.; Schmidt, M.K.; Seynaeve, C.M.; McCool, D.; Giardiello, D.; van den Broek, A.J.; Hauptmann, M.; Steyerberg, E.W.; Hooning, M.J. Risk factors for metachronous contralateral breast cancer: A systematic review and meta-analysis. Breast 2019, 44, 1–14. [Google Scholar] [CrossRef]
- Nizic-Kos, T.; Krajc, M.; Blatnik, A.; Stegel, V.; Skerl, P.; Novakovic, S.; Gazic, B.; Besic, N. Bilateral Disease Common among Slovenian CHEK2-Positive Breast Cancer Patients. Ann. Surg. Oncol. 2020. [Google Scholar] [CrossRef]
- De Bock, G.H.; Schutte, M.; Krol-Warmerdam, E.M.; Seynaeve, C.; Blom, J.; Brekelmans, C.T.; Meijers-Heijboer, H.; van Asperen, C.J.; Cornelisse, C.J.; Devilee, P.; et al. Tumour characteristics and prognosis of breast cancer patients carrying the germline CHEK2*1100delC variant. J. Med. Genet. 2004, 41, 731–735. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.; Dork, T.; Sohn, C.; Karstens, J.H.; Bremer, M. Breast cancer in patients carrying a germ-line CHEK2 mutation: Outcome after breast conserving surgery and adjuvant radiotherapy. Radiother. Oncol. J. 2007, 82, 349–353. [Google Scholar] [CrossRef]
- Weischer, M.; Nordestgaard, B.G.; Pharoah, P.; Bolla, M.K.; Nevanlinna, H.; Van’t Veer, L.J.; Garcia-Closas, M.; Hopper, J.L.; Hall, P.; Andrulis, I.L.; et al. CHEK2*1100delC heterozygosity in women with breast cancer associated with early death, breast cancer-specific death, and increased risk of a second breast cancer. J. Clin. Oncol. 2012, 30, 4308–4316. [Google Scholar] [CrossRef] [Green Version]
- Kriege, M.; Hollestelle, A.; Jager, A.; Huijts, P.E.; Berns, E.M.; Sieuwerts, A.M.; Meijer-van Gelder, M.E.; Collee, J.M.; Devilee, P.; Hooning, M.J.; et al. Survival and contralateral breast cancer in CHEK2 1100delC breast cancer patients: Impact of adjuvant chemotherapy. Br. J. Cancer 2014, 111, 1004–1013. [Google Scholar] [CrossRef]
- Muranen, T.A.; Blomqvist, C.; Dork, T.; Jakubowska, A.; Heikkila, P.; Fagerholm, R.; Greco, D.; Aittomaki, K.; Bojesen, S.E.; Shah, M.; et al. Patient survival and tumor characteristics associated with CHEK2:p.I157T-findings from the Breast Cancer Association Consortium. Breast Cancer Res. 2016, 18, 98. [Google Scholar] [CrossRef] [Green Version]
- De Bock, G.H.; Mourits, M.J.; Schutte, M.; Krol-Warmerdam, E.M.; Seynaeve, C.; Blom, J.; Brekelmans, C.T.; Meijers-Heijboer, H.; van Asperen, C.J.; Cornelisse, C.J.; et al. Association between the CHEK2*1100delC germ line mutation and estrogen receptor status. Int. J Gynecol. Cancer 2006, 16 (Suppl. 2), 552–555. [Google Scholar] [CrossRef]
- Nagel, J.H.; Peeters, J.K.; Smid, M.; Sieuwerts, A.M.; Wasielewski, M.; de Weerd, V.; Trapman-Jansen, A.M.; van den Ouweland, A.; Bruggenwirth, H.; van, I.J.W.F.; et al. Gene expression profiling assigns CHEK2 1100delC breast cancers to the luminal intrinsic subtypes. Breast Cancer Res. Treat 2012, 132, 439–448. [Google Scholar] [CrossRef]
- Couch, F.J.; Hart, S.N.; Sharma, P.; Toland, A.E.; Wang, X.; Miron, P.; Olson, J.E.; Godwin, A.K.; Pankratz, V.S.; Olswold, C.; et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J. Clin. Oncol. 2015, 33, 304–311. [Google Scholar] [CrossRef]
- Honrado, E.; Osorio, A.; Palacios, J.; Benitez, J. Pathology and gene expression of hereditary breast tumors associated with BRCA1, BRCA2 and CHEK2 gene mutations. Oncogene 2006, 25, 5837–5845. [Google Scholar] [CrossRef]
- Kilpivaara, O.; Bartkova, J.; Eerola, H.; Syrjakoski, K.; Vahteristo, P.; Lukas, J.; Blomqvist, C.; Holli, K.; Heikkila, P.; Sauter, G.; et al. Correlation of CHEK2 protein expression and c.1100delC mutation status with tumor characteristics among unselected breast cancer patients. Int. J. Cancer 2005, 113, 575–580. [Google Scholar] [CrossRef]
- Bahassi el, M.; Robbins, S.B.; Yin, M.; Boivin, G.P.; Kuiper, R.; van Steeg, H.; Stambrook, P.J. Mice with the CHEK2*1100delC SNP are predisposed to cancer with a strong gender bias. Proc. Natl. Acad. Sci. USA 2009, 106, 17111–17116. [Google Scholar] [CrossRef] [Green Version]
- Huzarski, T.; Cybulski, C.; Domagala, W.; Gronwald, J.; Byrski, T.; Szwiec, M.; Woyke, S.; Narod, S.A.; Lubinski, J. Pathology of breast cancer in women with constitutional CHEK2 mutations. Breast Cancer Res. Treat. 2005, 90, 187–189. [Google Scholar] [CrossRef]
- Angelova, S.G.; Krasteva, M.E.; Gospodinova, Z.I.; Georgieva, E.I. CHEK2 gene alterations independently increase the risk of death from breast cancer in Bulgarian patients. Neoplasma 2012, 59, 622–630. [Google Scholar] [CrossRef]
- Boughey, J.C.; Attai, D.J.; Chen, S.L.; Cody, H.S.; Dietz, J.R.; Feldman, S.M.; Greenberg, C.C.; Kass, R.B.; Landercasper, J.; Lemaine, V.; et al. Contralateral Prophylactic Mastectomy Consensus Statement from the American Society of Breast Surgeons: Additional Considerations and a Framework for Shared Decision Making. Ann. Surg. Oncol. 2016, 23, 3106–3111. [Google Scholar] [CrossRef] [Green Version]
- Wood, M.E.; McKinnon, W.; Garber, J. Risk for breast cancer and management of unaffected individuals with non-BRCA hereditary breast cancer. Breast J. 2020, 26, 1528–1534. [Google Scholar] [CrossRef]
- Kukita, Y.; Okami, J.; Yoneda-Kato, N.; Nakamae, I.; Kawabata, T.; Higashiyama, M.; Kato, J.; Kodama, K.; Kato, K. Homozygous inactivation of CHEK2 is linked to a familial case of multiple primary lung cancer with accompanying cancers in other organs. Cold Spring Harb. Mol. Case Stud. 2016, 2, a001032. [Google Scholar] [CrossRef] [Green Version]
- Van Puijenbroek, M.; van Asperen, C.J.; van Mil, A.; Devilee, P.; van Wezel, T.; Morreau, H. Homozygosity for a CHEK2*1100delC mutation identified in familial colorectal cancer does not lead to a severe clinical phenotype. J. Pathol. 2005, 206, 198–204. [Google Scholar] [CrossRef]
- Dong, X.; Wang, L.; Taniguchi, K.; Wang, X.; Cunningham, J.M.; McDonnell, S.K.; Qian, C.; Marks, A.F.; Slager, S.L.; Peterson, B.J.; et al. Mutations in CHEK2 associated with prostate cancer risk. Am. J. Hum. Genet. 2003, 72, 270–280. [Google Scholar] [CrossRef] [Green Version]
- Zhen, J.T.; Syed, J.; Nguyen, K.A.; Leapman, M.S.; Agarwal, N.; Brierley, K.; Llor, X.; Hofstatter, E.; Shuch, B. Genetic testing for hereditary prostate cancer: Current status and limitations. Cancer 2018. [Google Scholar] [CrossRef] [Green Version]
- Brandão, A.; Paulo, P.; Maia, S.; Pinheiro, M.; Peixoto, A.; Cardoso, M.; Silva, M.P.; Santos, C.; Eeles, R.A.; Kote-Jarai, Z.; et al. The CHEK2 Variant C.349A>G Is Associated with Prostate Cancer Risk and Carriers Share a Common Ancestor. Cancers 2020, 12, 3254. [Google Scholar] [CrossRef]
- Conti, D.V.; Wang, K.; Sheng, X.; Bensen, J.T.; Hazelett, D.J.; Cook, M.B.; Ingles, S.A.; Kittles, R.A.; Strom, S.S.; Rybicki, B.A.; et al. Two Novel Susceptibility Loci for Prostate Cancer in Men of African Ancestry. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef]
- Wang, Y.; Dai, B.; Ye, D. CHEK2 mutation and risk of prostate cancer: A systematic review and meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 15708–15715. [Google Scholar]
- Hale, V.; Weischer, M.; Park, J.Y. CHEK2 (*) 1100delC Mutation and Risk of Prostate Cancer. Prostate Cancer 2014, 2014, 294575. [Google Scholar] [CrossRef] [Green Version]
- Cybulski, C.; Wokolorczyk, D.; Huzarski, T.; Byrski, T.; Gronwald, J.; Gorski, B.; Debniak, T.; Masojc, B.; Jakubowska, A.; Gliniewicz, B.; et al. A large germline deletion in the Chek2 kinase gene is associated with an increased risk of prostate cancer. J. Med. Genet. 2006, 43, 863–866. [Google Scholar] [CrossRef]
- Seppala, E.H.; Ikonen, T.; Mononen, N.; Autio, V.; Rokman, A.; Matikainen, M.P.; Tammela, T.L.; Schleutker, J. CHEK2 variants associate with hereditary prostate cancer. Br. J. Cancer 2003, 89, 1966–1970. [Google Scholar] [CrossRef] [Green Version]
- Abramson, J.H. WINPEPI updated: Computer programs for epidemiologists, and their teaching potential. Epidemiol. Perspect. Innov. Ep+I 2011, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Isaacsson Velho, P.; Silberstein, J.L.; Markowski, M.C.; Luo, J.; Lotan, T.L.; Isaacs, W.B.; Antonarakis, E.S. Intraductal/ductal histology and lymphovascular invasion are associated with germline DNA-repair gene mutations in prostate cancer. Prostate 2018. [Google Scholar] [CrossRef]
- Giri, V.N.; Hegarty, S.E.; Hyatt, C.; O’Leary, E.; Garcia, J.; Knudsen, K.E.; Kelly, W.K.; Gomella, L.G. Germline genetic testing for inherited prostate cancer in practice: Implications for genetic testing, precision therapy, and cascade testing. Prostate 2019, 79, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Yu, H.; Zheng, S.L.; Na, R.; Mamawala, M.; Landis, T.; Wiley, K.; Petkewicz, J.; Shah, S.; Shi, Z.; et al. A comprehensive evaluation of CHEK2 germline mutations in men with prostate cancer. Prostate 2018. [Google Scholar] [CrossRef]
- Yadav, S.; Hu, C.; Hart, S.N.; Boddicker, N.; Polley, E.C.; Na, J.; Gnanaolivu, R.; Lee, K.Y.; Lindstrom, T.; Armasu, S.; et al. Evaluation of Germline Genetic Testing Criteria in a Hospital-Based Series of Women With Breast Cancer. J. Clin. Oncol. 2020, 38, 1409–1418. [Google Scholar] [CrossRef]
- Cybulski, C.; Wokolorczyk, D.; Kluzniak, W.; Kashyap, A.; Golab, A.; Slojewski, M.; Sikorski, A.; Puszynski, M.; Soczawa, M.; Borkowski, T.; et al. A personalised approach to prostate cancer screening based on genotyping of risk founder alleles. Br. J. Cancer 2013, 108, 2601–2609. [Google Scholar] [CrossRef] [Green Version]
- Zlowocka-Perlowska, E.; Narod, S.A.; Cybulski, C. CHEK2 Alleles Predispose to Renal Cancer in Poland. JAMA Oncol. 2019, 5, 576. [Google Scholar] [CrossRef]
- Carlo, M.I.; Mukherjee, S.; Mandelker, D.; Vijai, J.; Kemel, Y.; Zhang, L.; Knezevic, A.; Patil, S.; Ceyhan-Birsoy, O.; Huang, K.C.; et al. Prevalence of Germline Mutations in Cancer Susceptibility Genes in Patients With Advanced Renal Cell Carcinoma. JAMA Oncol. 2018, 4, 1228–1235. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Wang, Y.; Shao, W.; Jin, J.; Du, M.; Ma, G.; Chu, H.; Wang, M.; Zhang, Z. Rare variants in BRCA2 and CHEK2 are associated with the risk of urinary tract cancers. Sci. Rep. 2016, 6, 33542. [Google Scholar] [CrossRef] [Green Version]
- Ged, Y.; Chaim, J.L.; DiNatale, R.G.; Knezevic, A.; Kotecha, R.R.; Carlo, M.I.; Lee, C.H.; Foster, A.; Feldman, D.R.; Teo, M.Y.; et al. DNA damage repair pathway alterations in metastatic clear cell renal cell carcinoma and implications on systemic therapy. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef]
- Hartman, T.R.; Demidova, E.V.; Lesh, R.W.; Hoang, L.; Richardson, M.; Forman, A.; Kessler, L.; Speare, V.; Golemis, E.A.; Hall, M.J.; et al. Prevalence of pathogenic variants in DNA damage response and repair genes in patients undergoing cancer risk assessment and reporting a personal history of early-onset renal cancer. Sci. Rep. 2020, 10, 13518. [Google Scholar] [CrossRef]
- Smith, P.S.; West, H.; Whitworth, J.; Castle, B.; Sansbury, F.H.; Warren, A.Y.; Woodward, E.R.; Tischkowitz, M.; Maher, E.R. Pathogenic germline variants in patients with features of hereditary renal cell carcinoma: Evidence for further locus heterogeneity. Genes Chromosomes Cancer 2020. [Google Scholar] [CrossRef]
- Gadd, S.; Huff, V.; Walz, A.L.; Ooms, A.; Armstrong, A.E.; Gerhard, D.S.; Smith, M.A.; Auvil, J.M.G.; Meerzaman, D.; Chen, Q.R.; et al. A Children’s Oncology Group and TARGET initiative exploring the genetic landscape of Wilms tumor. Nat. Genet. 2017, 49, 1487–1494. [Google Scholar] [CrossRef] [Green Version]
- Ciceri, S.; Gamba, B.; Corbetta, P.; Mondini, P.; Terenziani, M.; Catania, S.; Nantron, M.; Bianchi, M.; D’Angelo, P.; Torri, F.; et al. Genetic and epigenetic analyses guided by high resolution whole-genome SNP array reveals a possible role of CHEK2 in Wilms tumour susceptibility. Oncotarget 2018, 9, 34079–34089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczmarek-Rys, M.; Ziemnicka, K.; Hryhorowicz, S.T.; Gorczak, K.; Hoppe-Golebiewska, J.; Skrzypczak-Zielinska, M.; Tomys, M.; Golab, M.; Szkudlarek, M.; Budny, B.; et al. The c.470 T > C CHEK2 missense variant increases the risk of differentiated thyroid carcinoma in the Great Poland population. Hered. Cancer Clin. Pract. 2015, 13, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siolek, M.; Cybulski, C.; Gasior-Perczak, D.; Kowalik, A.; Kozak-Klonowska, B.; Kowalska, A.; Chlopek, M.; Kluzniak, W.; Wokolorczyk, D.; Palyga, I.; et al. CHEK2 mutations and the risk of papillary thyroid cancer. Int. J. Cancer 2015, 137, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Wojcicka, A.; Czetwertynska, M.; Swierniak, M.; Dlugosinska, J.; Maciag, M.; Czajka, A.; Dymecka, K.; Kubiak, A.; Kot, A.; Ploski, R.; et al. Variants in the ATM-CHEK2-BRCA1 axis determine genetic predisposition and clinical presentation of papillary thyroid carcinoma. Genes Chromosomes Cancer 2014, 53, 516–523. [Google Scholar] [CrossRef] [Green Version]
- Pekova, B.; Dvorakova, S.; Sykorova, V.; Vacinova, G.; Vaclavikova, E.; Moravcova, J.; Katra, R.; Vlcek, P.; Sykorova, P.; Kodetova, D.; et al. Somatic genetic alterations in a large cohort of pediatric thyroid nodules. Endocr. Connect. 2019, 8, 796–805. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Yu, T.; Chen, L.; Xie, D.; Wang, F.; Fu, L.; Cheng, C.; Li, Y.; Zhu, X.; Miao, G. A Germline CHEK2 Mutation in a Family with Papillary Thyroid Cancer. Thyroid 2020, 30, 924–930. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Meijers-Heijboer, H.; Wijnen, J.; Vasen, H.; Wasielewski, M.; Wagner, A.; Hollestelle, A.; Elstrodt, F.; van den, B.R.; de Snoo, A.; Fat, G.T.; et al. The CHEK2 1100delC mutation identifies families with a hereditary breast and colorectal cancer phenotype. Am. J. Hum. Genet. 2003, 72, 1308–1314. [Google Scholar] [CrossRef] [Green Version]
- Naseem, H.; Boylan, J.; Speake, D.; Leask, K.; Shenton, A.; Lalloo, F.; Hill, J.; Trump, D.; Evans, D.G. Inherited association of breast and colorectal cancer: Limited role of CHEK2 compared with high-penetrance genes. Clin. Genet. 2006, 70, 388–395. [Google Scholar] [CrossRef]
- Katona, B.W.; Yang, Y.X. Colorectal cancer risk associated with the CHEK2 1100delC variant. Eur. J. Cancer 2017, 83, 103–105. [Google Scholar] [CrossRef]
- Xiang, H.P.; Geng, X.P.; Ge, W.W.; Li, H. Meta-analysis of CHEK2 1100delC variant and colorectal cancer susceptibility. Eur. J. Cancer 2011, 47, 2546–2551. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Zhang, B.; Zheng, W. Genetic variants associated with colorectal cancer risk: Comprehensive research synopsis, meta-analysis, and epidemiological evidence. Gut 2014, 63, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wang, Q.S.; Wang, Y.J. The CHEK2 I157T variant and colorectal cancer susceptibility: A systematic review and meta-analysis. Asian Pac. J. Cancer Prev. 2012, 13, 2051–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchy, J.; Cybulski, C.; Wokolorczyk, D.; Oszurek, O.; Gorski, B.; Debniak, T.; Jakubowska, A.; Gronwald, J.; Huzarski, T.; Byrski, T.; et al. CHEK2 mutations and HNPCC-related colorectal cancer. Int. J. Cancer 2010, 126, 3005–3009. [Google Scholar] [CrossRef]
- Kleibl, Z.; Havranek, O.; Hlavata, I.; Novotny, J.; Sevcik, J.; Pohlreich, P.; Soucek, P. The CHEK2 gene I157T mutation and other alterations in its proximity increase the risk of sporadic colorectal cancer in the Czech population. Eur. J. Cancer 2009, 45, 618–624. [Google Scholar] [CrossRef]
- Cybulski, C.; Wokolorczyk, D.; Kladny, J.; Kurzawski, G.; Suchy, J.; Grabowska, E.; Gronwald, J.; Huzarski, T.; Byrski, T.; Gorski, B.; et al. Germline CHEK2 mutations and colorectal cancer risk: Different effects of a missense and truncating mutations? Eur. J. Hum. Genet. 2007, 15, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Djureinovic, T.; Lindblom, A.; Dalen, J.; Dedorson, S.; Edler, D.; Hjern, F.; Holm, J.; Lenander, C.; Lindforss, U.; Lundqvist, N.; et al. The CHEK2 1100delC variant in Swedish colorectal cancer. Anticancer Res. 2006, 26, 4885–4888. [Google Scholar]
- Cragun, D.; Radford, C.; Dolinsky, J.S.; Caldwell, M.; Chao, E.; Pal, T. Panel-based testing for inherited colorectal cancer: A descriptive study of clinical testing performed by a US laboratory. Clin. Genet. 2014, 86, 510–520. [Google Scholar] [CrossRef]
- Pearlman, R.; Frankel, W.L.; Swanson, B.; Zhao, W.; Yilmaz, A.; Miller, K.; Bacher, J.; Bigley, C.; Nelsen, L.; Goodfellow, P.J.; et al. Prevalence and Spectrum of Germline Cancer Susceptibility Gene Mutations Among Patients With Early-Onset Colorectal Cancer. JAMA Oncol. 2017, 3, 464–471. [Google Scholar] [CrossRef]
- You, Y.N.; Borras, E.; Chang, K.; Price, B.A.; Mork, M.; Chang, G.J.; Rodriguez-Bigas, M.A.; Bednarski, B.K.; Meric-Bernstam, F.; Vilar, E. Detection of Pathogenic Germline Variants Among Patients With Advanced Colorectal Cancer Undergoing Tumor Genomic Profiling for Precision Medicine. Dis. Colon Rectum 2019, 62, 429–437. [Google Scholar] [CrossRef]
- Rosenthal, E.T.; Evans, B.; Kidd, J.; Brown, K.; Gorringe, H.; van Orman, M.; Manley, S. Increased Identification of Candidates for High-Risk Breast Cancer Screening Through Expanded Genetic Testing. J. Am. Coll. Radiol. 2017, 14, 561–568. [Google Scholar] [CrossRef] [Green Version]
- Weischer, M.; Heerfordt, I.M.; Bojesen, S.E.; Eigentler, T.; Garbe, C.; Rocken, M.; Holmich, L.R.; Schmidt, H.; Klyver, H.; Bastholt, L.; et al. CHEK2*1100delC and risk of malignant melanoma: Danish and German studies and meta-analysis. J. Investig. Derm. 2012, 132, 299–303. [Google Scholar] [CrossRef]
- Konstantinova, D.V.; Kadiyska, T.K.; Kaneva, R.P.; Tosheva, E.G.; Guseva, V.T.; Dimitrov, B.H.; Dimitrov, R.G.; Doganov, N.I.; Ivanov, S.I.; Kremensky, I.M.; et al. CHEK2 I157T and endometrial cancer. DNA Cell Biol. 2009, 28, 9–12. [Google Scholar] [CrossRef]
- Ring, K.L.; Bruegl, A.S.; Allen, B.A.; Elkin, E.P.; Singh, N.; Hartman, A.R.; Daniels, M.S.; Broaddus, R.R. Germline multi-gene hereditary cancer panel testing in an unselected endometrial cancer cohort. Mod. Pathol. 2016, 29, 1381–1389. [Google Scholar] [CrossRef]
- AlDubayan, S.H.; Pyle, L.C.; Gamulin, M.; Kulis, T.; Moore, N.D.; Taylor-Weiner, A.; Hamid, A.A.; Reardon, B.; Wubbenhorst, B.; Godse, R.; et al. Association of Inherited Pathogenic Variants in Checkpoint Kinase 2 (CHEK2) With Susceptibility to Testicular Germ Cell Tumors. JAMA Oncol. 2019. [Google Scholar] [CrossRef]
- Bartsch, D.K.; Krysewski, K.; Sina-Frey, M.; Fendrich, V.; Rieder, H.; Langer, P.; Kress, R.; Schneider, M.; Hahn, S.A.; Slater, E.P. Low Frequency of CHEK2 Mutations in Familial Pancreatic Cancer. Fam. Cancer 2006, 5, 305–308. [Google Scholar] [CrossRef]
- Mohelnikova-Duchonova, B.; Havranek, O.; Hlavata, I.; Foretova, L.; Kleibl, Z.; Pohlreich, P.; Soucek, P. CHEK2 gene alterations in the forkhead-associated domain, 1100delC and del5395 do not modify the risk of sporadic pancreatic cancer. Cancer Epidemiol. 2010, 34, 656–658. [Google Scholar] [CrossRef]
- Obazee, O.; Archibugi, L.; Andriulli, A.; Soucek, P.; Malecka-Panas, E.; Ivanauskas, A.; Johnson, T.; Gazouli, M.; Pausch, T.; Lawlor, R.T.; et al. Germline BRCA2 K3326X and CHEK2 I157T mutations increase risk for sporadic pancreatic ductal adenocarcinoma. Int. J. Cancer 2019. [Google Scholar] [CrossRef]
- Hu, C.; Hart, S.N.; Bamlet, W.R.; Moore, R.M.; Nandakumar, K.; Eckloff, B.W.; Lee, Y.K.; Petersen, G.M.; McWilliams, R.R.; Couch, F.J. Prevalence of Pathogenic Mutations in Cancer Predisposition Genes among Pancreatic Cancer Patients. Cancer Epidemiol. Biomark. Prev. 2016, 25, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Yurgelun, M.B.; Chittenden, A.B.; Morales-Oyarvide, V.; Rubinson, D.A.; Dunne, R.F.; Kozak, M.M.; Qian, Z.R.; Welch, M.W.; Brais, L.K.; Da Silva, A.; et al. Germline cancer susceptibility gene variants, somatic second hits, and survival outcomes in patients with resected pancreatic cancer. Genet. Med. 2019, 21, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Lovecek, M.; Janatova, M.; Skalicky, P.; Zemanek, T.; Havlik, R.; Ehrmann, J.; Strouhal, O.; Zemankova, P.; Lhotova, K.; Borecka, M.; et al. Genetic analysis of subsequent second primary malignant neoplasms in long-term pancreatic cancer survivors suggests new potential hereditary genetic alterations. Cancer Manag. Res. 2019, 11, 599–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pazderová, N.; Urbán, V.; Makovník, M.; Macák, D.; Janega, P.; Chovanec, M.; Rejleková, K.; Mardiak, J.; Mego, M. Complete Response to Chemotherapy in Metastatic Pancreatic Carcinoma Associated with Double Heterozygous Germline Mutation in BRCA2 and CHEK2 Genes—A Case Report. Klin. Onkol. 2020, 33, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.B.; Zhao, L.; Wang, X.; Ghelman, Y.; Overman, M.J.; Javle, M.M.; Shroff, R.T.; Varadhachary, G.R.; Wolff, R.A.; McAllister, F.; et al. Germline DNA Sequencing Reveals Novel Mutations Predictive of Overall Survival in a Cohort of Patients with Pancreatic Cancer. Clin. Cancer Res. 2020, 26, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; McKay, J.D.; Rafnar, T.; Wang, Z.; Timofeeva, M.N.; Broderick, P.; Zong, X.; Laplana, M.; Wei, Y.; Han, Y.; et al. Rare variants of large effect in BRCA2 and CHEK2 affect risk of lung cancer. Nat. Genet. 2014, 46, 736–741. [Google Scholar] [CrossRef]
- Hangaishi, A.; Ogawa, S.; Qiao, Y.; Wang, L.; Hosoya, N.; Yuji, K.; Imai, Y.; Takeuchi, K.; Miyawaki, S.; Hirai, H. Mutations of Chk2 in primary hematopoietic neoplasms. Blood 2002, 99, 3075–3077. [Google Scholar] [CrossRef] [Green Version]
- Rudd, M.F.; Sellick, G.S.; Webb, E.L.; Catovsky, D.; Houlston, R.S. Variants in the ATM-BRCA2-CHEK2 axis predispose to chronic lymphocytic leukemia. Blood 2006, 108, 638–644. [Google Scholar] [CrossRef]
- Janiszewska, H.; Bak, A.; Pilarska, M.; Heise, M.; Junkiert-Czarnecka, A.; Kuliszkiewicz-Janus, M.; Calbecka, M.; Jazwiec, B.; Wolowiec, D.; Kuliczkowski, K.; et al. A risk of essential thrombocythemia in carriers of constitutional CHEK2 gene mutations. Haematologica 2012, 97, 366–370. [Google Scholar] [CrossRef]
- Havranek, O.; Kleiblova, P.; Hojny, J.; Lhota, F.; Soucek, P.; Trneny, M.; Kleibl, Z. Association of Germline CHEK2 Gene Variants with Risk and Prognosis of Non-Hodgkin Lymphoma. PLoS ONE 2015, 10, e0140819. [Google Scholar] [CrossRef] [Green Version]
- Havranek, O.; Spacek, M.; Hubacek, P.; Mocikova, H.; Markova, J.; Trneny, M.; Kleibl, Z. Alterations of CHEK2 forkhead-associated domain increase the risk of Hodgkin lymphoma. Neoplasma 2011, 58, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Szymanska-Pasternak, J.; Szymanska, A.; Medrek, K.; Imyanitov, E.N.; Cybulski, C.; Gorski, B.; Magnowski, P.; Dziuba, I.; Gugala, K.; Debniak, B.; et al. CHEK2 variants predispose to benign, borderline and low-grade invasive ovarian tumors. Gynecol. Oncol. 2006, 102, 429–431. [Google Scholar] [CrossRef]
- Lilyquist, J.; LaDuca, H.; Polley, E.; Davis, B.T.; Shimelis, H.; Hu, C.; Hart, S.N.; Dolinsky, J.S.; Couch, F.J.; Goldgar, D.E. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol. Oncol. 2017, 147, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.J.; Marshall, M.L.; Susswein, L.R.; Zorn, K.K.; Hiraki, S.; Arvai, K.J.; Torene, R.I.; McGill, A.K.; Yackowski, L.; Murphy, P.D.; et al. Germline pathogenic variants identified in women with ovarian tumors. Gynecol. Oncol. 2018, 151, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Koczkowska, M.; Krawczynska, N.; Stukan, M.; Kuzniacka, A.; Brozek, I.; Sniadecki, M.; Debniak, J.; Wydra, D.; Biernat, W.; Kozlowski, P.; et al. Spectrum and Prevalence of Pathogenic Variants in Ovarian Cancer Susceptibility Genes in a Group of 333 Patients. Cancers 2018, 10, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Reference | Population | P: Patients C: Controls | Analysis * | Odds Ratio (95% Confidence Interval); p—Remark (Statistically Insignificant in Italics) |
|---|---|---|---|---|
| Female breast cancer | ||||
| Fostira 2020 [146] | GR | P: 1382 high-risk BC patients C: ExAC/FLOSSIES | CHEK2 (panel NGS) | 1.7 (0.98–2.7); 0.11—all LoF variants/ExAC 2.6 (1.44–4.68); 0.003—all LoF variants/FLOSSIES 3.8 (1.86–7.12); 1.2 × 10−3—missense deleterious/ExAC 5.9 (2.38–14.8); 1.2 × 10−4—missense deleterious/FLOSSIES |
| Kurian 2020 [147] | US (66% white) | P: 2,195 postmenopausal BC C: 2322 age-matched PMC | CHEK2 (panel NGS) | N.D.; CHEK2 PV found in 0.59% P and 0.26% C |
| Rogoza-Janiszewska 2020 [148] | PL | P. 2,464 BC diagnosed at <41 C: from Cybulski 2019 | c.1100delC; c.444+1G>A; del5395 | 3.8 (2.53–5.58); <0.0001—BC at < 41 y; all truncations 4.6 (2.44–8.80); <0.0001—BC at < 31 y; all truncations |
| Kleiblova 2019 [115] | CZ | P: 1526 high-risk female BC C: 3360 PMC | CHEK2 (panel NGS) | 7.94 (3.90–17.47); 4.1 × 10−11—unilat. BC: truncations 3.90 (1.24–13.35); 0.009—unilat. BC: deleterious missense 8.39 (1.92–28.74); 0.003—bilat. BC: truncations 3.77 (0.08–31.42); 0.26—bilat BC: deleterious missense |
| Cybulski 2019 [149] | PL | P: 1,018 hereditary BC C: 4346 PMC | c.1100delC c.444+1G>A del5395 | 6.9 (3.2–14.7); <0.0001—for c.1100delC 8.4 (3.0–23.3); <0.0001—for c.444+1G>A 6.5 (3.2–13.4); <0.0001—for del53957.2 (4.5–11.6); <0.0001—for all above truncations |
| Nurmi 2019 [150] | FI | P: 3156 BC C: 2089 PMC | c.319+2T>A; c.444+1G>A; c.1100delC | 5.40 (1.58–18.45); 0.007—for c.319+2T>A unselected BC 6.04 (1.65–22.10); 0.007—for c.319+2T>A familial BC |
| Girard 2019 [151] | FR | P: 1207 BRCA1/2−ve BC pts having sister with BC C: 1199 non-cancer PMC | CHEK2 (WES + panel NGS) | 3.0 (1.9–5.0); 1 × 10−5—any rare variant 5.8 (2.0–16.9); 0.001—LoF variants 2.4 (1.4–4.3); 0.002—likely-deleterious missense |
| Hauke 2018 [126] | DE | P: 5589 BRCA1/2−ve BC C: 2189 non-cancer PMC | CHEK2 (panel NGS) | 3.72 (1.99–6.94); <0.0001—truncations |
| Momozawa 2018 [145] | JP | P: 7051 BC C: 11,241 PMC | CHEK2 (panel NGS) | 3.2 (1.6–6.8); 3.2 × 10−4 |
| Decker 2017 [152] | UK | P: 13,087 BC C: 5488 PMC | CHEK2 (& 3 other genes) | 3.11 (2.15–4.69); 5.6 × 10−11—truncations 1.36 (0.99–1.87); 0.066—all rare missense 1.51 (1.02–2.24); 0.047—rare missense in any domain 3.27 (1.66–5.83); 0.0014—bilateral BC 3.42 (2.33–5.21); 1.5 × 10−11—ER+ve BC 3.98 (2.62–6.21)—age at dg < 50 years 3.37 (2.24–5.22)—age at dg = 50–60 years 2.12 (1.35–3.41)—age at dg > 60 years |
| Slavin 2017 [153] | US (80% white) | P: 2266 BRCA1/2−ve fam. BC C: ExAC | CHEK2 (panel NGS) | 1.62 (1.03–2.51); 0.004 – truncations |
| Couch 2017 [154] | US (white) | P: 29,090 BC C: 25,215 ExAC-NFE | CHEK2 (panel NGS) | 2.31 (1.88–2.85); 3.04 × 10−17—c.1100delC 2.26 (1.89–2.72); 1.75 × 10−20—PVs (w/o p.I157T, p.S428F) 1.48 (1.31–1.67); 1.11 × 10−10—any var (w p.I157T, p.S428F) 1.35 (1.1–1.63); 0.0002; bilateral BC |
| Schmidt 2016 [155] | BCAC | P: 44,777 population+ hospital-based BC C: 42,977 PMC | c.1100delC | 2.26 (1.90–2.69); 2.3×10−20—invasive BC 2.55 (2.10–3.10); 4.9 × 10−21—ER+ve BC 1.32 (0.93–1.88); 0.12—ER−ve BC |
| Naslund-Koch 2016 [156] | DK | 2442 BC pts /86,975 individ. (longitudinal study); | c.1100delC | 2.08 (1.51–2.85); <0.001 |
| Southey 2016 [157] | BCAC | P: 42,671 C: 42,164 PMC | iCOGS array incl. 6 rare CHEK2 variants | 2.26 (1.29–3.95); 0.003—for p.R117G 1.33 (1.05–1.67); 0.016—for p.R180C 1.70 (0.73–3.93); 0.210—for p.E239K 5.06 (1.09–23.5); 0.017—for p.R346C 1.03 (0.62–1.71); 0.910—for p.D438Y |
| Liu Y 2011 [158] | CN (Han) | P: 118 familial BC P: 909 unselected BC C: 1228 healthy PMC | CHEK2 (dHPLC) for familial BC | 5.99 (1.98–18.11); 0.002—for p.H371Y familial BC 2.43 (1.07–5.52); 0.034—for p.H371Y unselected BC |
| Cybulski 2011 [159] | PL | P: 7494 BRCA1−ve BC C: 4346 PMC | c.1100delC; c.444+1G>A; del5395 | 3.6 (2.6–5.1)—all BC 3.3 (2.3–4.7)—patients with no BC family history 5.0 (3.3–7.6)—patients with BC in 1° or 2° relative 7.3 (3.2–16.8)—patients with BC in 1° and 2° relatives |
| Desrichard 2011 [122] | FR | P: 507 BRCA1/2−ve BC C: 513 non-cancer PMC | CHEK2 (sequencing) | 4.15 (1.38–12.50); 0.007—any variant 5.18 (1.49–18.00); 0.004—deleterious (p.K244R ex) |
| Le Calvez-Kelm 2011 [160] | US/CA/AU | P: 1242 BC ≤ 45y C: 1109 non-ca PMC female | CHEK2 (HRM) | 6.18 (1.76–21.8)—truncations/splice mutations 2.20 (1.20–4.01)—rare missense |
| Fletcher 2009 [161] | UK/FI/NL/ RU/DE | P: 1828 bilateral BC C: 7030 PMC | c.1100delC | 6.43 (4.33–9.53); <0.0001—second primary for mut. carriers |
| Weischer 2007 [162] | DK | P + C: 9231 (prospective) P: 1101 BC/4665 PMC (case-control) | c.1100delC | 3.2 (1.0–9.9)—BC (prospective study) 2.6 (1.3–5.4)—BC (case-control study) |
| Cybulski 2006 [163] | PL | P: 3228 BC diagnosed at ≤50 C: 5496 PMC | c.1100delC c.444+1G>A p.I157T | 2.3 (1.1–4.8); 0.04—for c.1100delC 2.4 (1.4–4.2); 0.002—for c.444+1G>A 2.4 (1.5–3.7); 0.0001—for any truncation 1.4 (1.1–1.6); 0.002—for p. I157T |
| Chekmariova 2006 [164] | RU | P: 660 unilat; 155 bilat BC C: 448 middle aged females; | c.1100delC (ASO PCR) | 9.8 (1.34–198.26); 0.007 - early onset/bilat BC/C carriers frequencies: 3.4/5.2/0.2% |
| Cybulski 2004 [20] | PL | P: 1017 BC C: 4000 PMC | c.1100delC; c.444+1G>A; p.I157T | 2.2; p = 0.02—for c.1100delC and c.444+1G>A 1.4; p = 0.02—for p.I157T |
| Caligo 2004 [130] | IT | P: 939 BC (incl. BRCA1/2+ve) C: 334 PMC | c.1100delC | N.S.; frequency of carriers 0.11% (95% CI 0.00–0.59%) |
| Dufault 2004 [165] | DE | P: 516 BRCA1/2−ve BC C: 500 PMC (1,315 PMC for c.1100delC) | CHEK2 | 3.44 (1.19–9.95); 0.016—c.1100delC 3.9 (1.3–10.9)—c.1100delC and c.1214del4 |
| CHEK2 BC consortium 2004 [166] | UK/NL/FI/ DE/AU | P: 10,860 BC C: 9065 multinatl. | c.1100delC | 2.34(1.72–3.20); 1 × 10−7—all BC 2.23 (1.60–3.11)—BC w/o BC in 1° relative 3.12 (1.90–5.15)—BC with 1 BC in 1° relative 4.17 (1.26–13.75)—BC with ≥2 BC in 1° relatives |
| CHEK2 BC consortium 2002 [167] | UK/NL/ US/CA | P: 636 unselected BC P: 718 BRCA1/2-ve BC C: 1620 multinatl. | c.1100delC | 2.52 (0.78–8.18)—unselected BC 1.70 (1.32–3.38)—BRCA1/2−ve BC |
| Vahteristo 2002 [110] | FI | P: 1035 unselected BC C: 1885 PMC (blood donors) | c.1100delC | 1.48 (0.83–2.65); 0.182—unselected BC 2.27 (1.11–4.63); 0.021—familial BC 6.17 (1.87–20.32); 0.007 bilat. vs. unilat. BC |
| Male breast cancer | ||||
| Kleiblova 2019 [115] | CZ | P: 48 male BC C: 3360 PMC | CHEK2 (panel NGS) | 20.21 (3.50–80.00); 8.6 × 10−4—truncations 11.87 (0.25–100.83); 0.1—deleterious missense |
| Liang 2018 [168] | meta | P: 1063 male BC C: 31,571 | c.1100delC | 3.13 (1.94–5.07) |
| Hallamies 2017 [169] | FI | P: 68 male BC C: 1885 from [110] | c.1100delC | 4.47 (1.51–13.18); 0.019 |
| Wasielewski 2009 [170] | NL | P: 71 male BC C: 1692 | c.1100delC | 4.1 (1.2–14.3); 0.05 |
| CHEK2 consortium 2002 [167] | UK/NL/US/CA | P: 52 male BC families C: 1620 multinatl. | c.1100delC | 10.28 (3.54–29.87) |
| Meta-analyses | ||||
| Yang 2019 [171] | BCAC+ ABCC meta | P: 122,977 + 24,206 BC C: 105,974 + 24,775 PMC | p.I157T | 1.28 (1.17–1.39); 9.66 × 10−9—for Europeans only 1.35 (1.18–1.54); 9.82 × 10−6—for ER+ve BC 0.95 (0.81–1.12); 0.55—for ER−ve BC |
| Liang 2018 [168] | meta | P: 118,735 BC C: 195,807 | c.1100delC | 2.88 (2.65–3.22)—female BC 2.87 (1.85–4.47)—early-onset BC 3.21 (2.41–4.29)—familial BC 3.13 (1.94–5.07)—male BC |
| Aloraifi 2015 [113] | meta | P. 7283 C: 13,785 | CHEK2 truncations | 3.25 (2.55–4.13) |
| Han 2013 [172] | meta | P: 15,985 BC C: 18,609 | p.I157T | 1.58 (1.42–1.75); <0.0001 |
| Liu 2012 [173] | meta | P: 19,621 BC C: 27,001 | p.I157T | 1.48 (1.31–1.68); <0.0001—unselected BC 1.48 (1.16–1.89); <0.0001—familiar BC 1.47 (1.29–1.66); <0.0001—early onset BC 4.17 (2.89–6.03); <0.0001—lobular BC |
| Yang 2012 [174] | meta | P: 29,154 BC C: 37,064 | c.1100delC | 2.33 (1.79–3.05)—unselected BC 3.72 (2.61–5.31)—familiar BC 2.78 (2.28–3.39)—early onset BC |
| Zhang 2011 [175] | meta | P: 9970/C:7526 P: 13,331/C: 10,817 P: 10,543/C:10,817 P: 41,791/C: 50,910 | c.444+1G>A del5395 c.1100delC p.I157T | 3.07 (2.03–4.63); 9.82 × 10−8—for variant c.444+1G>A 2.53 (1.61–3.97); 6.33 × 10−5—for variant del5395 3.10 (2.59–3.71); <10−20—for variant c.1100delC 1.52 (1.31–1.77); 4.76 × 10−8—for variant p.I157T |
| Weischer 2008 [176] | meta | P: 26,488 C: 27,402 | c.1100delC | 2.7 (2.1–3.4)—unselected BC 2.6 (1.3–5.5)—early onset BC 4.8 (3.3–7.2)—familial BC |
| Reference | Population | P: Patients C: Controls | Analysis ** | Odds Ratio (95% Confidence Interval); p—Remark (Statistically Insignificant in Italics) |
|---|---|---|---|---|
| Brandao 2020 [202] | PT PRACTICAL | P: 462 early-onset/familial PrC C: 710 PMC P: 55,162 PrC/C: 36,147 | c.349A>G (p.R117G) | 7.7 (0.9–66.6); 0.06—PT PrC p.R117G 1.9 (1.1–3.2); 0.04—PRACTICAL PrC p.R117G |
| Momozawa 2018 [144] | JP | P: 7636 C: 12,366 | Panel NGS (8 genes) | 2.43 (0.91–6.86); 0.06 |
| Conti 2017 [203] | AAPC, GH | AAPC–P:4,853 PrC; C: 4678 GH–P: 474; C: 458 | GWAS array (rs78554043 = rs17886163; CHEK2 c.1343T>G; p.I448S) | 1.60 (1.27–2.00); 5.02 × 10−5—for AAPC PrC 2.45 (1.33–4.52); 0.004—for Ghana PrC |
| Naslund-Koch 2016 [156] | DK | 86,975 individuals (longitudinal study); 1340 developed PrC | c.1100delC | 1.60 (1.00–2.56); 0.05 |
| Southey 2016 [157] | OCAC | P: 22,301 PrC C: 22,320 PMC | iCOGS array incl. 6 rare CHEK2 variants | 1.46 (0.71–3.02); 0.3—for p.R117G 1.02 (0.73–1.44); 0.9—for p.R180C 1.47 (0.41–5.35); 0.6—for p.E239K 1.07 (0.28–4.07); 0.9—for p.D438Y 2.21 (1.06–4.63); 0.03—for p.D438Y 3.03 (1.53–6.03); 0.001—for I448S in Africans |
| Pritchard 2016 [118] | US, UK | P: 692 metastat. PrC C: ExAC/TCGA | Panel NGS | 3.1 (1.5–5.6); 0.002—vs. ExAC (excl. p.I157T) 4.7 (2.2–8.5); <0.001—vs. TCGA (excl. p.I157T) |
| Wang 2015 [204] | meta | P: 6409 PrC C: 11,634 | c.1100delC c.444+1G>A p.I157T | 3.29 (1.85–5.85); <0.001—c.1100delC 1.59 (0.79–3.20); 0.20—c.1100delC, familial 1.58 (0.93–2.71); 0.09—c.444+1G>A 1.80 (1.51–2.14); <0.001—p.I157T |
| Hale 2014 [205] | meta | P: 5,124 PrC C: 9,258 | c.1100delC | 1.98 (1.23–3.18); 0.004—unselected 3.39 (1.78–6.47); 0.0001—familial |
| Cybulski 2006 [206] | PL | P: 1864 PrC (incl. 249 famil.) C: 5496 | c.1100delC; c.444+1G>A; 5395del; p.I157T | 2.3 (1.1–3.9); <0.001—truncations, sporadic 4.7 (2.5–9.0); <0.001—truncations, familial 1.6 (1.3–2.0); <0.001—p.I157T, sporadic 2.7 (1.8–4.1); <0.001—p.I157T, familial |
| Weischer 2007 [162] | DK | P: 116 PrC (prospective) C: 3999 PMC men (prospect.) | c.1100delC | 2.3 (0.6–9.5) PrC prospective study |
| Johnson 2005 [177] | UK | P: 469 bilat. BC | c.1100delC | 2.41 (1.67–3.36)—risk of PrC for relatives of patients with bilateral BC |
| Cybulski 2004 [20] | PL | P: 690 PrC C: 4000 PMC | c.1100delC; c.444+1G>A; p.I157T | 2.2; 0.04—truncations 1.7; 0.002—p.I157T |
| Seppala 2003 [207] | FI | P1: 537 unselected PrC; P2: 120 hereditary PrC C: 510 non-PrC men | CHEK2 (SSCP: heredit. PrC) c.1100delC/p.I157T | 3.14 (0.65–15.16); 0.15—c.1100delC, sporadic 8.24 (1.49–45.54); 0.02—c.1100delC, hereditary 1.48 (0.89–2.46); 0.13—p.I157T, sporadic 2.12 (1.06–4.27); 0.04—p.I157T, hereditary |
| Dong 2003 [200] | US | P1: 400 sporadic PrC; P2: 298 familial PrCC: 510 non-PrC men | CHEK2 (DHPLC) | 2.71 (1.04–7.04); 0.049 *—sporadic PrC 2.66 (0.98–7.28); 0.078 *– familial PrC 6.84 (0.86–54.1); 0.05 *—sporadic (w/o p.I157T) 5.74 (0.64–51.5); 0.17 *—familial (w/o p.I157T) |
| Reference | Population | P: Patients C: Controls | Analysis * | Odds Ratio (95% Confidence Interval); p—Remark (Statistically Insignificant in Italics) |
|---|---|---|---|---|
| Zlowocka-Perlowska 2019 [214] | PL | P: 835 invasive RCC C: 8304 non-cancer | c.1100delC; c.444+1A>G; 5395del; c.I157T | 2.5 (1.5–4.1); 0.0003—for truncations 2.0 (1.6–2.6); <0.001—for p.I157T |
| Carlo 2018 [215] | US | P: 254 RCC (stage III-IV) C: ExAC | CHEK2 | 3.0 (1.3–5.8); 0.003 |
| Ge 2016 [216] | GWAS | P: 1322 C: 3428 | p.I157T | 0.63 (0.44–0.89); 0.01 |
| Naslund-Koch 2016 [156] | DK | 138/86,975 individuals developed RCC | c.1100delC | 3.61 (1.33–9.79); 0.01 |
| Weischer 2007 [162] | DK | P: 33 RCC (prospective) C: 9166 PMC (prospect.) | c.1100delC | 9.8 (2.3–41.2) RCC prospective study |
| Cybulski 2004 [20] | PL | P: 264 RCC C: 4000 PMC | c.1100delC; c.444+1G>A; p.I157T | 1.0; p = 0.8—truncations 2.1; p = 0.0006—for p.I157T |
| Reference | Population | P: Patients C: Controls | Analysis of Specific CHEK2 Variants | Odds Ratio (95% Confidence Interval); p—Remark (Statistically Insignificant in Italics) |
|---|---|---|---|---|
| Kaczmarek-Rys 2015 [222] | PL | P: 602 differentiated PTC C: 829 PMC | p.I157T | 1.81 (1.20–2.72); 0.004—p.I157T heterozygote 12.81 (0.6–248.46); 0.02—p.I157T homozygote |
| Siolek 2015 [223] | PL | P: 468 unselected PTC C: 468 matched non-cancer | c.1100delC; c.444+1G>A; 5395del; p.I157T | 5.7 (1.7–19.3); 0.006—truncations 2.8 (1.7–4.6); 0.0001—p.I157T |
| Wojcicka 2014 [224] | PL | P: 1781 PTC C: 2081 healthy PMC | p.I157T | 2.21 (1.69–2.88); 2.37 × 10−10—for p.I157T |
| Cybulski 2004 [20] | PL | P: 173 PTC C: 4000 PMC | c.1100delC; c.444+1G>A; p.I157T | 4.9; 0.006—truncations 1.9; 0.04—p.I157T |
| Reference | Population | P: Patients C: Controls | Analysis | Odds Ratio (95% Confidence Interval); p—Remark (Statistically Insignificant in Italics) |
|---|---|---|---|---|
| Xiang 2017 [230] | meta | Revision of the analysis from [231] | 1.8 (1.2–2.7)—unselected CRC 2.4 (0.9–6.6)—familial CRC 1.7 (0.9–2.9)–sporadic CRC | |
| Naslund-Koch 2016 [156] | DK | 1131/86,975 individuals developed CRC | c.1100delC | 0.86 (0.43–1.72);0.68 |
| Ma 2014 [232] | meta | P: 3874 CRC/ C:11,630 P: 6042 CRC/ C:17,051 | c.1100delC p.I157T | 1.88 (1.29–2.73); 0.001 1.56 (1.32–1.84); 1.22 × 10−7 |
| Han 2013 [172] | meta | P: 3166 CRC C: 9844 | p.I157T | 1.67 (1.24–2.26); 0.0008 |
| Liu 2012 [233] | meta | P: 4029 CRC C: 13,844 | p.I157T | 1.61 (1.40–1.87); <0.001—unselected CRC 1.48 (1.23–1.77); <0.001—sporadic CRC 1.97 (1.41–2.74); <0.001—familial CRC |
| Xiang 2011 [231] | meta | P: 4,194 CRC C: 10,010 | c.1100delC | 2.11 (1.41–3.16); 0.0003—unselected CRC 2.80 (1.74–4.51); <0.0001—familial CRC 1.45 (0.49–4.30); 0.5—sporadic CRC |
| Suchy 2010 [234] | PL | P: 463 HNPCC-related C: 5496 PMC | c.1100delC; c.444+1G>A; del5395; p.I157T | 1.0 (0.4–2.6); 1.0—truncations 1.7 (1.2–2.4); 0.007—p.I157T |
| Kleibl 2009 [235] | CZ | P: 631 CRC C: 683 PMC | FHA-coding region c.1100delC;5395del | 2.3 (1.3–4.0); 0.003—all variants in FHA domain 2.0 (1.1–3.6); 0.03—p.I157T only 2.3 (0.4–12.8); 0.4—c.1100delC; zero 5395del carriers |
| Weischer 2007 [162] | DK | P: 210 (prospective) C: 9007 PMC (prospect.) | c.1100delC | 1.6 (0.4–6.5)—prospective CRC |
| Cybulski 2007 [236] | PL | P. 1085 unselected CRC C: 5496 controls | c.1100delC; c.444+1G>A; del5395; p.I157T | 1.0 (0.5–1.8); 0.9—truncations 1.5 (1.2–2.0); 0.002—p.I157T |
| Djureinovic 2006 [237] | SE | P: 174 familial CRC P: 644 unselected CRC C: 760 PMC | c.1100delC | 1.76 (0.34–9.09); 0.6—familial CRC 1.42 (0.43–4.67); 0.7—sporadic CRC |
| Irmejs 2006 [133] | LV | P: 235 C: 978 newborn PMC | p.I157T | 1.7 (1.01–2.70); p < 0.05 |
| Meijers-Heijboer 2003 [228] | UK/NL/ US/CA | P: 329 CRC C: 1620 [167] | c.1100delC (ASO) | 2.34 (0.95–5.79); 0.07—HNPCC-like families 5.19 (2.17–12.4); < 0.001—HBCC families |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stolarova, L.; Kleiblova, P.; Janatova, M.; Soukupova, J.; Zemankova, P.; Macurek, L.; Kleibl, Z. CHEK2 Germline Variants in Cancer Predisposition: Stalemate Rather than Checkmate. Cells 2020, 9, 2675. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9122675
Stolarova L, Kleiblova P, Janatova M, Soukupova J, Zemankova P, Macurek L, Kleibl Z. CHEK2 Germline Variants in Cancer Predisposition: Stalemate Rather than Checkmate. Cells. 2020; 9(12):2675. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9122675
Chicago/Turabian StyleStolarova, Lenka, Petra Kleiblova, Marketa Janatova, Jana Soukupova, Petra Zemankova, Libor Macurek, and Zdenek Kleibl. 2020. "CHEK2 Germline Variants in Cancer Predisposition: Stalemate Rather than Checkmate" Cells 9, no. 12: 2675. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9122675