The Role of gp130 Cytokines in Tuberculosis


1
Infection Immunology, Research Centre Borstel, D-23845 Borstel, Germany
2
German Centre for Infection Research (DZIF), Partner Site Hamburg-Borstel-Lübeck-Riems, D-23845 Borstel, Germany
*
Author to whom correspondence should be addressed.
Cells 2020, 9(12), 2695; https://0-doi-org.brum.beds.ac.uk/10.3390/cells9122695
Submission received: 14 October 2020
/
Revised: 1 December 2020
/
Accepted: 10 December 2020
/
Published: 15 December 2020
(This article belongs to the Special Issue Regulation of Cytokine Signaling in Immunity)
Abstract
:Protective immune responses to Mycobacterium tuberculosis (Mtb) infection substantially depend on a delicate balance within cytokine networks. Thus, immunosuppressive therapy by cytokine blockers, as successfully used in the management of various chronic inflammatory diseases, is often connected with an increased risk for tuberculosis (TB) reactivation. Hence, identification of alternative therapeutics which allow the treatment of inflammatory diseases without compromising anti-mycobacterial immunity remains an important issue. On the other hand, in the context of novel therapeutic approaches for the management of TB, host-directed adjunct therapies, which combine administration of antibiotics with immunomodulatory drugs, play an increasingly important role, particularly to reduce the duration of treatment. In both respects, cytokines/cytokine receptors related to the common receptor subunit gp130 may serve as promising target candidates. Within the gp130 cytokine family, interleukin (IL)-6, IL-11 and IL-27 are most explored in the context of TB. This review summarizes the differential roles of these cytokines in protection and immunopathology during Mtb infection and discusses potential therapeutic implementations with respect to the aforementioned approaches.
1. Introduction
Tuberculosis (TB) is still the leading cause of death from an infectious agent and thus represents a major health problem, with one quarter of the global population infected with Mycobacterium tuberculosis (Mtb) [1]. In 2018, about 1.5 million people died from TB and nearly 10 million people worldwide fell ill with TB [1]. Infection with Mtb is mainly initiated by aerogenic exposure to a patient with active pulmonary TB [2]. Inhaled bacteria are quickly phagocytized by alveolar macrophages and a small granulomatous lesion, containing neutrophils, macrophages, multinucleated giant cells and lymphocytes, develops which in most cases prevents the systemic spread and limits the growth of Mtb [2,3]. The majority of infected individuals (>90%) remain latently infected without developing any symptoms. However, because containment of Mtb in these individuals is facilitated by an active immune response, anti-inflammatory therapies to treat autoimmune and chronic inflammatory diseases such as rheumatoid arthritis, psoriasis and Crohn’s disease increase the risk of reactivation of latent TB [4,5,6,7]. Eventually, 5 to 10% of infected individuals develop active TB caused by reactivation of latent TB accompanied by chronic inflammation [3,8]. These active TB patients require at least 6 months of treatment with multiple drugs, but the spread of multi-drug resistant (MDR-TB) and extremely drug-resistant (XDR-TB) strains has made the management of TB more challenging because of the poor, expensive, less-effective and toxic alternatives to the first-line drugs [9,10]. New treatment regimens interconnecting TB drugs and immunomodulation as adjunct therapy (host-directed therapy, HDT) may help to shorten the treatment duration and thereby prevent the development of drug resistant Mtb [8,9,11]. In order to develop novel immunomodulatory interventions (1) for the anti-inflammatory therapy of potentially latently Mtb-infected patients suffering from autoimmune or chronic inflammatory diseases or (2) for the adjunct treatment of TB, the understanding of the mechanisms that mediate protection but also pathogenesis in TB is mandatory.
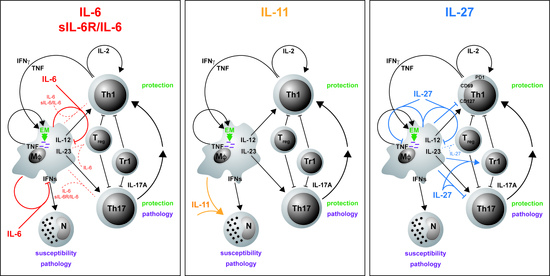
Dysregulated secretion of cytokines or the lack of cytokines/cytokine receptors and their subsequent signaling pathways contribute to susceptibility and/or pathogenesis of infectious diseases in humans and various animal models [12,13,14]. In this context, cytokines were shown to be in host defense against Mtb by supporting a cellular immune response required for the control of mycobacterial growth [15,16] but also prevent a detrimental inflammatory immune response [17,18]. A type 1 or T helper 1 (TH1) immune response is instructed by the stimulation of naïve CD4+ T cells through interaction with antigen-presenting cells (APCs) that express cytokines, costimulatory molecules and other polarizing signals that promote the differentiation into effector TH1 cells [15,19]. In particular, interleukin (IL)-12, produced after phagocytosis of Mtb by macrophages and dendritic cells (DC), is needed for the induction of TH1 cells (Figure 1). These cells typically secret interferon (IFN)γ and tumor necrosis factor (TNF), leading to a synergistic activation of anti-mycobacterial effector mechanisms in macrophages [20,21] (Figure 1) and an elevated production of pro-inflammatory cytokines such as IL-1β, IL-6 and TNF [12,22,23].
In recent decades, IFNγ-producing CD4+ T cells were considered to be the main arm of a protective cellular immune response by conveying granuloma organization and bacterial killing of macrophages in TB patients and animal models of TB [24,25,26]. However, this view is currently under debate due to a poor correlation between the levels of IFNγ and the degree of protection against the infection [27,28,29]. In addition to the IFNγ-dominated TH1 immune response, another cell population, IL-17A-producing TH17 cells, came into focus that supports protection against Mtb [27,28,30,31] (Figure 1).
TH17 cells differentiate from naïve T cells through the interaction with various cytokines such as IL-6 and are maintained by IL-23 [32]. In TB, IL-17A contributes indirectly to granuloma formation and the recruitment of IFNγ/TNF/IL-2-producing multifunctional T cells to the site of infection by the induction of various chemokines [17,33,34] (Figure 1). However, because TH1 and TH17 cells are both strong inducers of inflammatory immune responses, both also contribute to chronic inflammation and tissue destruction in experimental Mtb infection [17,18] (Figure 1). Hence, a balanced activation of TH1 and TH17 cells is needed to restrict mycobacterial growth and to limit immunopathology [28,31,35]. Checkpoints of these TH1 and TH17 immune responses are regulatory T cells (Treg) and type 1 regulatory (Tr1) cells, which accumulate in the lungs at an early stage of the infection and thus delay the migration of effector T cells [36,37]. However, it can also be assumed that these regulatory cells keep a pathological inflammatory reaction in check in the course of TB (Figure 1). While TH1 and TH17 cells are rather implicated in protective immune responses, susceptibility to TB and the pathology of this disease are facilitated by the activity of neutrophils which strikingly depend on type 1 interferons [38] (Figure 1). In respect to novel adjunct therapeutic approaches for the management of active TB, immunomodulation has to promote protective immune responses without the risk of developing a pathological inflammation.
In addition to IFNγ and IL-17A, TNF also plays a crucial role in the containment of Mtb infection by sustaining the granuloma structural integrity and promoting anti-mycobacterial effector mechanisms in macrophages [2,12,39]. Accordingly, anti-TNF immunotherapy, which is used to treat autoimmune and chronic inflammatory diseases, disrupts effective immunity against Mtb and eventually increases the risk of reactivation of latent TB [4,5,6,7]. TNF blockers are therefore an important example of how immunomodulatory therapies may adversely affect host immunity in TB. The development of novel anti-inflammatory treatment strategies for autoimmune or chronic inflammatory diseases must always take the risk of reactivation of latent TB into account.
Altogether, cytokine-directed interventions in the host immune system may open two major perspectives to advance prevention and control of TB: the identification of improved approaches to treat inflammatory diseases without compromising anti-mycobacterial immunity and the development of new therapeutic strategies in the treatment of TB, which especially include HDTs. In both respects, cytokines/cytokine receptors which are linked to the signal-transducing receptor gp130 may serve as promising target candidates. This review therefore summarizes the role of the gp130-related cytokines in the context of Mtb infection and discusses potential therapeutic implementations of these cytokines with regard to the aforementioned approaches.
2. gp130-Related Cytokines
The gp130 cytokine receptor family is defined by shared structural features of both ligands and receptors. These receptors are utilized by a variety of functionally and structurally related cytokines within the IL-6 and IL-12 family including IL-6, IL-11, IL-27, IL-35, IL-39, leukemia inhibitory factor (LIF), oncostatin M (OSM), ciliary neurotrophic factor (CNTF), cardiotrophin-1 (CT-1), novel neutrophin-1/B cell stimulating factor-3 or cardiotrophin-like cytokine (CLC) and neuropoetin (NP) [40,41,42,43,44,45,46,47,48,49,50,51,52,53] (Table 1). Structurally related to the IL-6 cytokine family is the newly discovered cytokine IL-31 [54]. This cytokine is gp130-independent but utilizes the ortholog of gp130, IL-31Rα chain and OSM receptor β (OSMR) [54,55,56].
The subunit gp130 is a common receptor component belonging to the type I cytokine receptor family. The shared usage of gp130 within the gp130 cytokine receptor family results in functional redundancy [52,57]. While gp130 is ubiquitously expressed, specificity and distinctness of action can be attributed to the use of ligand-specific receptor components that show a more limited expression pattern [52,58,59]. All members of the gp130 cytokine family induce Janus Kinase/Signal Transducer and Activator of Transcription (JAK/STAT) molecules, mostly STAT3 but also, to a lesser extent, STAT1 [60]. Alternatively, also other signal transduction pathways can be activated, however, in all receptor complexes, gp130 is required for signaling. In TB, IL-6, IL-11 and IL-27 are the most explored gp130 cytokines and should therefore be discussed in this review.
2.1. IL-6
IL-6 is the representative member of the IL-6 cytokine family. It has a four a-helix bundle structure [61] and, initially, many names were attributed to this cytokine due to its diverse functions, such as B cell stimulatory factor 2 [62], hybridoma growth factor [63], plasmacytomas growth factor [64], hepatocyte-stimulatory factor [65] and cytotoxic T cell differentiation factor [66], until it became clear that all of these properties were attributable to a common cytokine. The names already indicate its broad-ranging effects in hematopoiesis, tissue homeostasis, metabolism, neurogenesis and immunology. IL-6-mediated immune mechanisms include, inter alia, the induction of various acute phase proteins, the induction of immunoglobulin production in B cells and the proliferation and differentiation of T lymphocytes [52,67,68]. After infection, in essence, IL-6 is produced by many cell types of which the most important are monocytes and macrophages and is believed to play a central role in hosts defense mechanisms against various infectious agents [69,70,71].
It is now known that IL-6 mediates different mechanisms via two fundamentally different pathways. It can signal via a membrane-bound receptor (cis) or, after proteolytic cleavage of the IL-6 receptor (R) from the cell membrane, via a soluble receptor (trans) (Figure 2) [72].
2.1.1. Cis-Signaling
In cis-signaling—also called classical signaling—IL-6 binds to the membrane-bound IL-6Rα chain (mIL-6Rα) (Figure 2). The complex consisting of IL-6/mIL-6Rα recruits homodimers of gp130 which triggers a downstream JAK/STAT signaling cascade [68,73] (Figure 2). Consequently, because gp130 is universally expressed, cis-signaling is only relevant for cells that express mIL-6Rα (i.e., hepatocytes, neutrophils, monocytes/macrophages and CD4+ T cells [65,72]). By using IL-6-deficient (-/-) mice, IL-6 has been shown in the 1990s to mediate the acute phase response during inflammation [74] and to promote TH1 immune responses [75]. More recently, IL-6 cis-signaling acts in a pro-inflammatory fashion by tipping the balance from the development of Treg towards a TH17 immune response. IL-6 favors the differentiation of IL-17A-producing TH17 cells and inhibits the generation of Treg [68,76,77,78,79]. Since dysregulation of the TH17/Treg balance is linked to the development of autoimmunity and chronic inflammatory diseases [80,81], the induction of a TH17-type of inflammation characterizes a major pro-inflammatory function of IL-6 cis-signaling.
In contrast to the pro-inflammatory role of IL-6 cis-signaling in direct interaction with CD4+ T cells in TH1 immune responses and during the development of TH17 cells, IL-6 can also act in an anti-inflammatory fashion through the mIL-6Rα on macrophages because IL-6 cis-signaling inhibits the release of pro-inflammatory cytokines such as IL-12 and IL-23 by activated macrophages in vitro [82,83] and is able to promote alternative macrophage activation during obesity-related inflammation in mice leading to an ameliorated disease outcome [84].
Hence, IL-6 cis-signaling can act in a pro-inflammatory as well as in an anti-inflammatory fashion apparently depending on the type of target cell.
2.1.2. Trans-Signaling
In addition to cis-signaling, there is an alternative signaling pathway called trans-signaling. Here, IL-6 binds to a soluble IL-6Rα chain (sIL-6Rα) that is secreted after proteolytic cleavage of mIL-6Rα by the metalloprotease ADAM17 in a process called shedding [85,86] (Figure 2). Trans-signaling can evoke downstream signaling in cells, which do not express mIL-6Rα and, as a result, do not respond to IL-6, by binding of the IL-6/sIL-6Rα complex to the ubiquitously expressed gp130 subunit [87] (Figure 1). The development of a fusion protein of sIL-6Rα and IL-6, linked by a flexible peptide chain, called hyper-IL-6 allowed discriminating between trans-signaling and cis-signaling [88]. Binding of IL-6/sIL-6Rα promotes mainly pro-inflammatory immune responses characterized by the recruitment of immune cells [89,90], prevention of apoptosis [91] and inhibition of Treg differentiation [92]. The naturally occurring sgp130 serves as a potent antagonist of IL-6 trans-signaling [93] and selective inhibition of IL-6 trans-signaling by recombinant sgp130 fused to the Fc region of human IgG1 (sgp130Fc) suppresses a detrimental outcome of many inflammatory diseases [91,94,95,96,97,98,99]. Hence, treatment of autoimmune and chronic inflammatory disorders with sgp130Fc represents a valuable anti-inflammatory therapeutic strategy.
2.1.3. Trans-Presentation
Recently, another mode of IL-6 signaling called trans-presentation was described [100]. In this condition, in the DC–T cells interaction zone, the IL-6/mIL-6Rα complex on DCs is presented to gp130 expressed on T cells. This trans-presentation stimulates the STAT3-dependent development of pathogenic TH17 cells [100].
2.2. IL-11
The cytokine IL-11 belongs to the IL-6 cytokine family and is a monomeric cytokine. IL-11-induced signaling is mediated by the formation of the receptor complex, composed of the ligand-binding subunit IL-11Rα and the β-subunit gp130 (Figure 2). The α subunit of the receptor is expressed in lymphocytes, B cells, macrophages, endothelial cells, hematopoietic cells and osteoclasts [101]. Due to this abundant expression, IL-11 exerts pleiotropic effects such as the stimulation of hemopoiesis [102], thrombopoiesis [103] and the modulation of macrophage differentiation [104].
In several models of inflammation and infection, IL-11 was identified to be an immunomodulatory cytokine as it inhibits the release of pro-inflammatory cytokines [105,106,107,108]. However, also pro-inflammatory properties have been attributed to the cytokine. Thus, overexpression of IL-11 drives lymphocytic inflammation and fibrotic tissue remodeling in the murine airways [109] as well as heart and kidney fibrosis in mice [110]. Moreover, IL-11 is also involved in the recruitment of neutrophils to sites of inflammation or infection [111,112]. Most recently, IL-11 was demonstrated to promote the differentiation of TH17 cells in patients with multiple sclerosis (MS) and during relapsing-remitting experimental autoimmune encephalomyelitis (RREAE) [112]. Similar to IL-6, trans-signaling has also been described for IL-11 [113,114] (Figure 2). However, no biological function has been ascribed to IL-11 trans-signaling yet.
2.3. IL-27
In contrast to the IL-6 cytokine family members IL-6 and IL-11, the IL-12 cytokine family member IL-27 forms a heterodimeric complex [115] (Figure 2). The IL-12 family α subunit IL-27p28 resembles the unique up-up-down-down four-α-helix bundle structure of the IL-6 family made from a single polypeptide [12,116,117], which pairs with the β subunit Epstein-Barr-virus-induced gene 3 (EBI3) that is structurally related to sIL-6Rα [115,118]. The heterodimeric structure of IL-27 is analogous to complexes composed of soluble receptors such as sIL-6Rα that dimerize with their corresponding ligands [60]. IL-27 is produced by APCs in response to Toll-like receptor (TLR) activation [119,120]. It signals through a heterodimeric receptor complex composed of the private ligand-binding IL-27Rα and the common gp130 subunit [48] (Figure 2). IL-27R is expressed on various types of immune cells (e.g., T cells, macrophages, DC). Initially, IL-27 was shown to represent a key pro-inflammatory mediator of TH1 polarization by inducing the transcription factor T-bet and subsequently the expression of the IL-12 receptor β2 chain in naïve CD4+ cells. Hereby, IL-27 instructs TH0 cells to respond to IL-12 to differentiate into TH1 cells [119,121]. Hence, IL-27 instructs the initial TH1 response [122,123]. In contrast to this pro-inflammatory function during the initiation of a cell-mediated immune response, IL-27 induces a broad spectrum of suppressive mechanisms under inflammatory conditions. In general, it limits the intensity and duration of TH1, TH2 and TH17 cell activity and their cytokine production. Under strongly polarized TH1 immune responses, IL-27 suppresses the hyperactivity of CD4+ T cells and the development of inflammatory diseases [18,124]. Under TH2 conditions, IL-27 downregulates the TH2 transcription factor GATA3 [123,125]. Additionally, IL-27 also negatively regulates the differentiation of TH17 cells by several mechanisms, including the control of IL-6 production [126] (that, together with transforming growth factor-beta (TGF-β), supports the development of TH17 cells [32]) and of the TH17-specific transcription factor retinoid-related orphan receptor gamma t (RORγt) [127]. In line with this anti-inflammatory function just described, IL-27 also promotes the function of Treg under inflammatory conditions [128], which are central regulators of cellular immune responses and limit excessive inflammation. IL-27 upregulates in Treg-suppressive molecules such as LAG3, but the precise mechanisms by which IL-27 controls Treg functions remain elusive [128,129]. Additionally, IL-27 has been recognized as a differentiation factor for IL-10-producing Tr1 cells, which are crucial in controlling tissue inflammation [130] by inducing c-Maf, a transcription factor that transactivates IL-21 secretion which acts as an autocrine growth factor for Tr1 cells [131,132].
Furthermore, at the level of APCs, IL-27 is able to limit the release of pro-inflammatory cytokines and induce immunoregulatory molecules such as CD39 in activated macrophages and DCs, respectively. Therefore, IL-27 can also indirectly contribute to the modulation of TH1 and TH17 immune responses by suppressing the release of the TH1- and TH17-driving cytokines IL-12 and IL-23 in accessory cells, respectively [18,133,134].
3. gp130 Cytokines in Tuberculosis
This review aims to summarize the differential roles of gp130 cytokines in TB. Thereby, the main focus is on the presentation of mouse experimental data on the impact of the cytokines IL-6, IL-11 and IL-27, which are most explored in the context of TB. Moreover, the current study situation of these cytokines during human TB is delineated and potential therapeutic implementations are discussed. The animal experiments referred to here mainly deal with the impact of these mediators on TH1 and TH17 immune responses after infection with Mtb. Even if gp130 cytokines have pleiotropic effects, e.g., on TH2, cytotoxic T or B cells, these have only been little researched in connection with the role of IL-6, IL-11 and IL-27 in experimental TB. Although this review does not deal with these arms of the Mtb-induced immune response in the context of gp130 cytokines, future studies are certainly important.
The pleiotropic modes of action, along with the shared usage of both cytokine and receptor subunits within the gp130 cytokine family [52,116], imply a challenge for the experimental investigation of these cytokines during Mtb infection. Therefore, consideration of complementary mouse models, which interfere with the gp130-dependent signaling cascade at different levels, is of particular importance. Mice with a global cytokine deficiency can provide information on the overall influence of a certain cytokine on the outcome of Mtb infection, however, these mouse models carry the risk of concealing TB-related pleiotropic effector mechanisms. Mouse models, which cell-specifically interfere with gp130-mediated signaling, represent an interesting tool to highlight such effects. When interpreting findings obtained from these mouse models however, it has to be considered that the observable effects cannot necessarily be traced back to a specific cytokine. In addition to the choice of animal model, experimental parameters such as the bacterial dose and the route of exposure appear to have a considerable impact on the experimental outcome [135,136]. Likewise, the impact of specific cytokines on the outcome of experimental TB may also be influenced by the use of a more virulent Mtb strain or clinical isolate. Together, mouse experimental data on the impact of gp130 family cytokines during TB indeed provide important insights, however, the described limitations of these models may result in discrepancies in findings obtained from human studies.
3.1. IL-6
Although the cellular sources of IL-6 are known in principle, they have not been adequately investigated in either Mtb-infected mice or TB patients. Only in the context of murine type 2 diabetes was IL-6 described to be produced after infection by natural killer cells and CD11c DC [137]. Initially, IL-6 was considered a pro-inflammatory cytokine involved in protection against Mtb. However, during experimental TB, IL-6 plays a more complex role which differs between experimental models and, in addition, appears to be dependent on experimental conditions such as the infectious dose and the route of exposure. Accordingly, IL-6-/- mice are susceptible to a systemic infection with high doses of intravenously (i.v.) delivered Mtb [135]. The absence of IL-6 here leads to increased IL-4 levels and an attenuated expression of IFNγ. Moreover, the antibody-mediated depletion of IL-6 upon i.v. infection with Mycobacterium avium enhances mycobacterial growth [138]. In contrast, after aerosol infection with Mtb, IL-6-/- mice exhibit only an initial increase in bacterial burdens accompanied by a delayed IFNγ induction; however, these mice are eventually able to contain mycobacterial growth and to mount a protective memory response to secondary infection [136]. Moreover, frequencies of TH17 cells as well as of Treg are also only slightly modified in Mtb-infected IL-6-/- mice [139] (submitted). Together, different from the indispensable role of cytokines such as IFNγ and TNF during experimental TB, IL-6 may only be involved—either directly or indirectly—in early protective immune responses, but overall, effective anti-mycobacterial protection appears to be only marginally dependent on IL-6.
Due to the pleiotropy of IL-6, a more precise picture of its role during Mtb infection is presented by the cell type-specific analysis of IL-6/gp130-mediated effects. The CD4+ T cell-specific deficiency of gp130—and thus of IL-6-mediated signaling on T cells—leads to the abrogation of TH1 and TH17 immune responses in mouse models of autoimmunity [79] or extracellular parasitic infection [140]. Analysis of CD4+ T cell-specific gp130-deficient mice during Mtb infection, however, revealed that, here, not only the frequency of TH1 cells, but also the induction of TH17 cells and the overall expression of IL17a appear to be largely independent of gp130 expression on T cells [139] (submitted) (Figure 3A). In light of the repeatedly described impact of IL-6-mediated signaling on TH17 differentiation [32,79,140], this finding seems rather surprising. A compensatory effect of IL-27-mediated signaling on TH17 development, which is also abrogated in T cell-specific gp130-deficient mice, however, may partly explain this effect as the expansion of TH17 cells is only partly reduced in Mtb-infected IL-6-/- mice [139] (submitted). Hence, in contrast to IL-23 [33], IL-6 appears not to be required for a robust TH17 immune response during experimental TB. In accordance with the unaffected pro-inflammatory immune response, deficiency of gp130 on T cells results in only moderately decreased bacterial loads during the course of Mtb infection [139] (submitted).
Despite its pro-inflammatory properties, IL-6 appears to exert suppressive effects in macrophages [83,141]. Accordingly, IL-6 produced by Mtb-infected macrophages limits the responsiveness of uninfected macrophages to IFNγ [142], indicating that IL-6 also mediates anti-inflammatory mechanisms in TB (Table 2). In this context, IL-6 suppresses the transcription of selective IFNγ-responsive genes in infected macrophages [142]. An immunosuppressive role of IL-6 during mycobacterial infection was confirmed by VanHeyningen et al. [143] (Figure 3A; Table 2). Here, macrophages display a decreased T cell activation capacity in response to infection with Mycobacterium bovis BCG (Bacillus Calmette–Guérin), which is reversed by preincubation with neutralizing antibodies against IL-6. This cell type-specific effect has been further shown in vivo by using macrophage/neutrophil-specific gp130-deficient mice [82]. These mice exhibit elevated levels of pro-inflammatory cytokines and enhanced TH1 and TH17 immune responses, together with an increased expression of the anti-mycobacterial effector molecules inducible nitric oxide synthase (Nos2) and Lrg47 (EM in Figure 3A; Table 2). Nevertheless, the augmented inflammatory immune response in macrophage/neutrophil-specific gp130-deficient mice appears not to be critical for controlling mycobacterial growth. When assessing these data, it certainly has to be taken into account that the specific lack of gp130 in macrophages interferes not only with IL-6-, but also with IL-27-mediated signaling.
Notably, IL-6 also suppresses the expression of type I IFN-related genes in murine macrophages infected with Mtb strains of different virulence [144]. As it was demonstrated that type I IFNs–which are crucially important in the defense against viral infections—play a harmful role during mycobacterial infection [145,146,147], the inhibition of type I IFN-related genes by macrophage-produced IL-6 may contribute to the containment of TB progression [144] (Figure 3A).
A major physiological regulator of IL-6-mediated signaling is the Suppressor of cytokine signaling 3 (SOCS3) [148,149]. SOCS3 acts by inhibiting STAT3-mediated signaling by binding to gp130 [150]. Expression of Socs3 is induced in Mtb-infected macrophages through MyD88-dependent mechanisms [151] and, during experimental TB, in the lungs of Mtb-infected mice [152]. Mice with a T cell-specific lack of SOCS3 are highly susceptible to experimental TB [151]. Containment of Mtb infection is, however, also dramatically reduced in mice with SOCS3-deficient macrophages [151,153]. Both mouse models display increased levels of Il6-expression, which is, however, neither attributable to macrophages nor to T cells. In the absence of macrophage-derived SOCS3, IL-6 appears to contribute to susceptibility to Mtb infection by suppressing IL-12/23p40 secretion in macrophages and the subsequent impairment of TH1 immune responses [151]. However, IL-6 may also directly affect anti-mycobacterial effector mechanisms in Mtb-infected macrophages in the absence of SOCS3. Accordingly, in vitro data revealed that SOCS3 prevents the development of alternatively activated macrophages (AAM), while IL-6 induces the expression of the AAM marker arginase 1 (Arg1) in SOCS3-deficient macrophages [154,155]. During mycobacterial infection, Arg1 derived from AAM counteracts protective macrophage effector mechanisms [152,156,157]. In Mtb-infected mice with a macrophage-specific lack of SOCS3, depletion of IL-6 results in a reduced expression and activity of Arg1 rather than an impaired TH1 immune response accompanied by decreased bacterial loads and less lung pathology [153]. In total, SOCS3 appears to keep IL-6-dependent macrophage responses under control and therewith preserves protective macrophage effector mechanisms.
In humans, increased expression of IL-6 was found in the blood of patients with pulmonary TB when compared to a healthy control group [158,159]. Moreover, the diagnostic value of IL-6 was higher in tuberculous pleural effusion when compared to malignant effusion [160,161]. On the other hand, a preliminary multiplex analysis further revealed a significantly higher IL-6 secretion in latently Mtb-infected patients than in patients with active TB [162]. However, later studies did not confirm any difference in IL-6 secretion levels between latent and active TB patients [163] or between latently and active Mtb-infected groups and non-TB patients [164]. Conversely, patients with cavitary TB—a severe form of pulmonary disease—were found to exhibit a reduced content of IL-6 in the bronchoalveolar lavage (BAL) in comparison to TB patients without cavities, suggesting the cytokine as a potential biomarker for protection against tissue destruction during advanced TB [165]. Recently, the association of four single-nucleotide polymorphisms (SNPs) in the IL-6 gene with TB susceptibility was explored in the Western Chinese Han population [166]. However, a link between susceptibility to TB and IL-6 polymorphisms was not identified within the scope of this study.
Overall, the conflicting data regarding the role of IL-6 in the context of mycobacterial infection are not surprising, but rather reflect the pleiotropic and multifunctional nature of the cytokine [167,168]. During experimental TB, IL-6 appears to interfere with innate as well as adaptive immunity and to mediate pro-inflammatory as well as immunosuppressive effector responses [82,135,136,142,151,153]. Accordingly, depending on the experimental model and most importantly on the target cell, IL-6 has a protective or detrimental effect on the progression of TB [82,135,136,139,153]. The gp130-binding suppressor molecule SOCS3 may therefore be critical in specifically controlling the IL-6-mediated development of Mtb-permissive macrophages [153]. Together, counteraction of the IL-6-mediated pro- and anti-inflammatory effects along with the SOCS3-dependent regulation of IL-6 activity may account for the largely unaffected outcome of experimental TB in global IL-6-/- mice. Nevertheless, it can be stated that IL-6 plays a rather subordinate role in the protective immune response against Mtb (Table 2). Surprisingly, IL-6-mediated signaling also appears to be almost negligible for TH17 differentiation during TB [139] (submitted).
3.2. IL-6/sIL-6Rα
To fully consider the impact of IL-6 during Mtb infection, the specific role of IL-6/sIL-6Rα trans-signaling for the outcome of disease also needs to be addressed. As aforementioned, inhibition of the IL-6 trans-signaling mechanism—without interfering with the classical IL-6 cis-signaling pathway or with IL-6 trans-presentation—can be achieved by use of sgp130Fc [93,94,100]. With regard to experimental TB, administration of sgp130Fc to mice during the acute or chronic phase of Mtb infection has no significant effect on the expression of the effector cytokines TNF, IL-6, IFNγ and IL-17A, whereas it slightly hampers the release of IL-12/23p40 [169] (Figure 3A). At the same time, containment of Mtb infection is not impaired by treating infected mice with sgp130Fc. Permanent inhibition of IL-6/sIL-6Rα trans-signaling in sgp130Fc-overexpressing (sgp130Fctg) mice results in an inflammatory defect accompanied by reduced infiltration of neutrophils and monocytic cells during acute inflammation [95]. Accordingly, Mtb-infected sgp130Fctg mice exhibit a timely restricted increase in bacterial burdens in the acute phase of TB, however, overall, these mice also remain able to mount protective immune responses to infection [93]. In conclusion, these data clearly demonstrate that IL-6/sIL-6Rα trans-signaling is dispensable for anti-mycobacterial immunity and containment of Mtb (Table 2).
3.3. IL-11
Hitherto, the role of the IL-6 family member IL-11 during Mtb infection has barely been investigated. Research conducted by Alexander S. Apt and colleagues, however, suggests the cytokine as a potential host therapeutic target for immune modulatory treatment of TB [170,171,176]. They initially found that IL-11 is strongly expressed by interstitial lung macrophages [176]. Thereby, macrophages from I/St mice, which are susceptible to Mtb infection, exhibit substantially higher expression levels of IL-11 than those from resistant A/Sn mice. Importantly, because, after infection of the genetically heterogeneous F2 progeny of the I/St and A/Sn strains with Mtb, the individual expression levels of IL-11 mRNA in the lung tissue correlate with the degree of disease-associated body weight loss, a potential role of this cytokine as a risk factor for TB progression can reasonably be assumed [177]. Eventually, the antibody-mediated depletion of IL-11 in Mtb-infected susceptible I/St mice diminishes the extent of disease progression [170]. Thereby, in addition to reduced bacterial burdens, the blockade of IL-11 reduces histopathology and the infiltration of the lung tissue by neutrophils. Moreover, the antibody treatment decreases the protein levels of other inflammatory cytokines such as TNF and IL-6. As it also downregulates mRNA expression of Il11 itself, the authors presume a positive feedback loop at the transcriptional level. Together, these data imply that the self-reinforcing hyperproduction of IL-11 plays a causative role in lung pathogenesis during the early phase of experimental TB in genetically susceptible mice.
To investigate the potential of IL-11-directed therapy for immune modulatory treatment of TB, a recombinant mutated form of IL-11 was established and administered by aerosol in the lungs of Mtb-infected mice [171]. This recombinant IL-11 variant acts as a high-affinity antagonist of IL-11-mediated signaling by competitive disruption of the gp130/IL11R signaling complex formation [178]. Local administration of an IL-11 antagonist takes account for the involvement of IL-11 in several physiological pathways [171,179]. A systemic blockade of IL-11, on the contrary, bears the risk of serious adverse effects [171]. In response to therapy, Mtb-infected mice exhibit substantially reduced levels of TB-associated inflammation in the lung, which includes lower numbers of F4/80+ macrophages and Ly-6G+ neutrophils in the early phase of Mtb infection and a decreased pulmonary infiltration of lymphocytes during the further course of infection [171] (Table 2). Moreover, treatment decreases the expression of key inflammatory factors such as TNF and IFNγ at both mRNA and protein levels. Likewise, treated mice show lower expression levels of IL-6 and IL-11 itself. Eventually, IL-11-directed therapy prolongs the survival of Mtb-infected animals. Based on these data and the aforementioned findings obtained after systemic administration of anti–IL-11 antibodies, the authors claim that blockade of IL-11 may mediate protection during infection with Mtb particularly by attenuating pathogenic neutrophil accumulation in the affected lung tissue [170,171] (Figure 3B; Table 2). Hence, during experimental TB, gp130-mediated signaling through IL-11 appears to be involved in susceptibility (Table 2).
In contrast with the harmful effect of IL-11 expression during the early course of experimental TB, a human pilot proteome study revealed higher protein levels of IL-11 in fast responders to TB treatment when compared with slow treatment responders [180]. Future studies will therefore be necessary to obtain a more complete picture of the IL-11-mediated immune response and its consequences on the progression of experimental and human TB.
3.4. IL-27
In the context of human TB, elevated levels of the IL-12 family cytokine IL-27 are associated with active disease [173]. Both subunits of the heterodimeric cytokine are expressed in human TB granuloma [181] and in TB pleural effusion [182,183], the latter providing the potential to use IL-27 levels as a specific biomarker for the differential diagnosis of tuberculous pleurisy. IL-27 production in TB pleural effusion has been ascribed to various cellular sources including CD4+ and CD8+ T cells, B cells, NK/NKT cells, monocytes/macrophages and mesothelial cells [183]. IL-27-secreting CD4+ T cells were further demonstrated to constitute a terminally differentiated human T cell subpopulation with a distinct expression pattern of transcription factors and pro-inflammatory cytokines [184]. In contrast to these findings, however, gene polymorphisms associated with reduced secretion levels of IL-27 were identified in patients with pulmonary TB [185].
The increased levels of IL-27 during human TB indicate that IL-27 inhibits anti-mycobacterial immunity and thereby contributes to disease progression. In accordance with this hypothesis, IL-27 suppresses protective immune responses during Mtb infection of mice, leading to an impaired mycobacterial containment [17,18,172]. However, at the same time, it also dampens the pathological sequelae of a pathological systemic hyperinflammation [17,18]. Thus, Mtb-infected mice lacking the IL-27Rα chain (IL-27Rα-/-) show an elevated release of the pro-inflammatory cytokines TNF and IL-12, resulting in an increased activation of CD4+ T cells and amplified macrophage effector functions, which eventually results in significantly reduced bacterial loads [18]. On the other hand, IL-27Rα-/- mice exhibit a hyperinflammatory phenotype during Mtb infection associated with excessive systemic production of pro-inflammatory cytokines, splenomegaly, accelerated cachexia and a decreased survival.
Formation of granuloma represents a hallmark of host defense against Mtb infection [186]. Importantly, TB granuloma in IL-27Rα-/- mice are extremely well-organized, containing a core of activated macrophages surrounded by a layer of lymphocytes [17,18,172]. In addition, during the chronic phase of experimental TB, IL-27Rα-deficient CD4+ T cells were found to be superior in assessing the lung parenchyma and associating with an antigen within the lesions [173] (Figure 3C). These T cells exhibit an altered phenotype, as they maintain the expression of the surface molecules programmed death-1 (PD-1), CD69 and CD127, but simultaneously reduce killer cell lectin-like receptor-G1 (KLRG1) expression (Figure 3C). During experimental TB, PD1+ CD4+ T cells appear to represent a population of self-renewing effector cells, which contribute to anti-mycobacterial immunity, whereas KLRG1+ CD4+ T cells show all characteristics of terminally differentiated cytokine-secreting cells with a low proliferative potential and a shorter life span [187]. Furthermore, the loss of IL-27Rα-mediated signaling provokes the accumulation of antigen-specific multifunctional CD4+ T cells in the lungs of Mtb-infected mice [17]. Multifunctional CD4+ T cells are defined by their co-expression of the cytokines IFNγ, TNF and IL-2 and represent a high-quality effector T cell subpopulation [188,189,190] (Figure 3C). During vaccination against Mtb, the superiority of multifunctional T cells for protection has been ascribed, at least partially, to their high cytokine secretion levels on a per cell basis and to their enhanced longevity. Notably, contrary to multifunctional T cells, the overall frequencies of IFNγ-producing CD4+ T cells in the lungs of Mtb-infected mice are not affected in the absence of IL-27Rα [17]. Based on these data, it was hypothesized that IL-27 may limit the protective impact of Mtb-specific CD4+ T cells at different levels—intrinsically by directly altering the quality and fitness of the induced T cell immune responses and extrinsically by impeding optimal T cell localization within Mtb-containing granulomatous lesions [17,173].
As mentioned before, a further role ascribed to IL-27 comprises its promoting impact on the induction of IL-10-producing regulatory Tr1 cells [131,132] and on the functionality of Treg [128,129]. During experimental TB, Treg infiltrate the site of infection and delay effector T cell migration into the lung during the early course of infection with Mtb [36,37]. Moreover, the production of the immunomodulatory cytokine IL-10 dampens protective immune responses and thereby promotes TB progression in both C57BL/6 and CBA/J mice [191,192]. With regard to the impact of IL-27 on regulatory T cell populations during experimental TB, it was shown that IL-27 expression enhances the accumulation of Tr1 cells in the lung but does not appear to affect numbers of Treg [17] (Figure 3C; Table 2). IL-27-dependent Treg, however, have not yet been functionally investigated in the context of TB.
IL-27 is known to suppress the development of IL-17A-producing TH17 cells in mouse models of chronic inflammation [126,127,193,194]. During experimental TB, IL-27Rα-/- mice also exhibit a significantly increased production of IL-17A by TH17 cells [17]. Furthermore, the impact of IL-27 on anti-mycobacterial immunity and TB progression has been connected, at least partially, with the IL-27-mediated suppression of Il-17A production (Figure 3C; Table 2). Thus, in Mtb-infected IL-27Rα-/- mice that additionally lack IL-17A expression, both the increased protection and the exacerbated immunopathology observed in the absence of IL-27Rα are abrogated. While IL-17A appears to mediate neither the suppression of Tr1 cells nor the expression of KLRG1 on CD4+ T cells in Mtb-infected IL-27Rα-/- mice, it regulates the accumulation of multifunctional CD4+ T cells in the lung. Moreover, the formation of highly structured TB granuloma in the absence of IL-27Rα clearly depends on the production of IL-17A—a function that has been attributed to the cytokine before [30]. In this context, IL-17A also triggers the expression of T cell-attracting chemokines in the lungs of Mtb-infected IL-27Rα-/- mice.
Beyond the capacity of IL-27 to indirectly limit protective cell-mediated immune responses to Mtb through the inhibition of IL-17A production, IL-27-mediated signaling may also directly impair the antimicrobial activity of infected macrophages [126,174] (Figure 3C; Table 2). In human macrophages, IL-27 suppresses phagosomal acidification and phagosome–lysosome fusion by inhibition of vacuolar ATPase (V-ATPase) and lysosomal integrated membrane protein-1 (CD63) and impairment of cathepsin D maturation [126] (Figure 3C). Furthermore, IL-27 induces the autophagy-regulating molecules mTOR and Mcl-1 to suppress IFNγ-mediated autophagy and eventually elimination of intracellular mycobacteria in Mtb-infected human macrophages [174] (Figure 3C; Table 2). In addition to these direct effects on macrophage anti-mycobacterial activity, IL-27 was demonstrated to differentially affect the production of pro- and anti-inflammatory cytokines in macrophages (Figure 3C). Accordingly, IL-27Rα-mediated signaling in murine activated peritoneal macrophages limits the release of IL-12/23p40 and TNF possibly by induction of STAT3 phosphorylation [18] (Table 2). Incubation of Mtb-infected human macrophages with a soluble receptor to neutralize IL-27 (sIL27RA) revealed that IL-27 also antagonizes the activity of IL-18 [195]. Along with IFNγ and TNF, IL-18 plays a pivotal role in reducing mycobacterial growth in human macrophages when IL-12 is supplied and signaling via IL-27 is blocked [175] (Figure 3C; Table 2). On the other hand, in mouse macrophages, IL-27 promotes the transcription of IL-10 through activation of STAT1 and STAT3, which are subsequently recruited to the IL-10 promoter [196] (Figure 3C). Overall, IL-27 can undermine protective immune responses in Mtb-infected macrophages directly by suppressing effector mechanisms and indirectly by restricting the production of pro-inflammatory cytokines. The impaired release of TH1- and TH17-driving cytokines diminishes the subsequent development of a protective T cell response. Future in vivo studies may investigate the overall impact of IL-27Rα-mediated signaling in macrophages on the outcome of Mtb infection.
In summary, during TB, IL-27 represents a “double-edged sword”, as it regulates both protective and immunopathological immune responses [17,18] (Table 2). Both effects can be ascribed to the IL-27-mediated inhibition of IL-17A production [17]. IL-27-dependent immunoregulation appears to impair both the quality and optimal localization of protective T cell responses against Mtb [17,18,173]; however, it may also directly affect the antimicrobial activity of macrophages [174,197].
3.5. Other gp130 Cytokines
While, in the context of experimental TB, there are currently only available data in regard to the gp130 cytokines IL-6, IL-11 and IL-27, different human studies indicate the additional contribution of further gp130 cytokines to the TB-associated immunopathology. Secretion levels of the IL-6 cytokine LIF were found to be upregulated in the blood plasma of patients with active and latent TB when compared to non-TB patients [164]. Furthermore, secretion of OSM—which can alternatively signal through the receptor complexes OSMR:gp130 and LIFR:gp130 [55]—appears to be increased in Mtb-infected human monocytes [198]. Together with TNF, the cytokine thereby stimulates the production of the matrix metalloproteinases (MMP)-1 and -3 from human pulmonary fibroblasts. MPPs are involved in the degradation of the extracellular matrix (ECM) [199] and were demonstrated to drive the development of TB cavities in rabbits [200]. Therewith, OSM might contribute to tissue destruction in TB. Finally, the gp130 family-related cytokine IL-31 has been identified as a novel sensitive and specific biomarker for the diagnosis of tuberculous pleurisy [201]. Another recent study further suggests a potential role for IL-31 as a biomarker for the discrimination between patients with latent TB and healthy individuals [202].
Future studies may bring new insights into the possible impact of other gp130 cytokine family members during experimental and human Mtb infection.
4. Therapeutical Aspects
4.1. Prevention of TB during Therapeutic Targeting of gp130 Cytokines in Autoimmune and Chronic Inflammatory Diseases
The aforementioned specific IL-6/sIL-6Rα trans-signaling inhibitor sgp130Fc represents a promising drug candidate for the management of chronic inflammatory disorders [71]. Under the name olamkicept, the inhibitor is undergoing phase II clinical trials for the treatment of inflammatory bowel disease (IBD) [203] and ulcerative colitis [204]. Investigation of the IL-6/sIL-6Rα trans-signaling pathway in the context of TB therefore provides an important contribution to the prevention of TB.
Cytokine-targeting therapies have been successfully used in the management of numerous chronic inflammatory diseases ever since TNF-neutralizing antibodies were deployed to treat rheumatoid arthritis (RA) in the 1990s [205]. However, continuous therapy with such immunomodulatory agents carries the threat of promoting susceptibility to severe bacterial infections. Accordingly, during experimental TB, TNF antagonist-treated mice are highly susceptible to Mtb infection, accompanied by impaired macrophage activation, an unorganized granulomatous response and necrosis development [169,206,207]. In individuals with latent TB, treatment of autoimmune and chronic inflammatory diseases with TNF antagonists is linked to an increased risk of reactivated Mtb infection [6,208,209,210]. Depending on the clinical setting, under TNF antagonist therapy, the relative risk of TB can be increased up to 40 times [6].
In accordance with the pathogenic role of IL-6 in autoimmune and chronic inflammatory diseases such as RA and juvenile idiopathic arthritis [211,212], the IL-6 signaling cascade has been targeted in numerous inflammatory diseases [213]. Several of those anti-IL-6 therapeutics have already been approved or are currently being evaluated in clinical trials. Most accurately described is the mIL-6Rα-neutralizing antibody tocilizumab, which is, inter alia, approved for the treatment of RA and juvenile inflammatory arthritis in multiple countries [71]. Although it appears that, in contrast to inhibition of TNF, blockade of mIL-6Rα does not account for TB progression in mice [214], human studies indicate that treatment with tocilizumab increases the risk of serious infections to a similar extent as TNF antagonists [215]. A meta-analysis published by Schiff and colleagues in 2011 reported enhanced rates of opportunistic infections including TB as well as infections with nontuberculous mycobacteria in patients receiving tocilizumab when compared to a placebo-treated control group [216]. In line with this study, real-world data gathered in Japan suggest that the risk for development of TB during treatment with tocilizumab is comparable to the infection risk during TNF antagonist therapy [217,218]. Even though the discrepancy between these findings and the aforementioned data obtained in mice [214] might result from the choice of mouse model as well as the experimental design, future clinical studies may be necessary to further elucidate the role of the drug in the predisposition to serious infections such as TB.
Tocilizumab, similar to the other IL-6- or mIL-6Rα-neutralizing drugs, blocks both IL-6 cis- and IL-6/sIL-6Rα trans-signaling [213,219]. Therewith, host-protective activities of IL-6 are suppressed during therapy in the same manner as chronic inflammation. In contrast, the specific blockade of the IL-6/sIL-6Rα trans-signaling pathway, which is achieved by treatment with sgp130Fc, still allows non-blocked IL-6 to support productive immune responses in defense against bacterial infections. This may especially include the hepatic acute phase response at the level of innate immunity but also TH1 and TH17 adaptive immune responses [169,213]. Therapeutic targeting of IL-6/sIL-6Rα trans-signaling during the treatment of chronic inflammatory diseases hence offers the potential to imply a reduced risk for opportunistic infections. With regard to TB, the previously described mouse experimental data demonstrating that the specific blockade of IL-6/sIL-6Rα trans-signaling does not impair protective immunity during Mtb infection support this hypothesis [169]. Moreover, in other animal models of disease, blockade of the trans-signaling pathway also appears to be superior when compared to a global inhibition of IL-6-mediated signaling. These include, inter alia, murine models for polymicrobial sepsis [99], bone fracture healing [220] and experimental infection with Listeria monocytogenes [221]. However, it still remains to be seen whether the clinical use of the IL-6/sIL-6Rα trans-signaling inhibitor olamkicept may be connected with any increase in the risk of TB reactivation. Together, the introduction of sgp130Fc therapeutics for the treatment of IL-6-driven chronic inflammatory diseases may contribute to TB prevention by providing a lower risk of disease reactivation as one of the major adverse effects of cytokine-directed immunosuppressive therapy.
4.2. Potential of Targeting gp130 Cytokines during Treatment of TB
As outlined in several review articles, in the context of novel therapeutic approaches for the treatment of TB, HDTs play an increasingly important role [8,222,223,224,225]. During this type of therapy, administration of antibiotics is combined with immunomodulatory drugs, particularly to reduce the duration of treatment. A number of different candidate host-directed therapeutics against TB are currently undergoing preclinical or clinical trials [222]. These therapeutics target different host pathways to optimize autophagy and phagosomal killing of Mtb within macrophages, immunometabolism, granuloma structure and T cell immunity, but they also aim to dampen exacerbated inflammation [222,224]. One important part of the present HDT approaches comprises the modulation of cytokine signaling either to activate anti-mycobacterial effector mechanisms in macrophages and shape protective T cell immunity or to modulate excessive inflammation.
Antagonists of the gp130 family cytokines, as the present review demonstrates, may also constitute promising candidates for HDT against TB, although no clinical data are yet available on this matter. Unfortunately, in this context, the only currently clinically available drugs are the mIL-6Rα-neutralizing antibodies tocilizumab and siltuximab. Although, in regard to their immunosuppressive properties, both therapeutics are mentioned as potential host-directed anti-TB drugs [222], the aforementioned findings on the risk of treatment with tocilizumab for the development of TB [216,217,218] might argue against this application of mIL-6Rα-neutralizing drugs. However, it should be considered that in connection with a simultaneous antibiotic treatment during adjunct therapy, the administration of cytokine-directed anti-inflammatory drugs may nonetheless have a beneficial effect, as demonstrated by usage of the soluble TNF receptor etanercept during the treatment of HIV-associated TB [226]. In this context, adjunct treatment has a positive therapeutic impact, possibly by disrupting mycobacterial containment within granuloma and eventually promoting the elimination of metabolically active bacteria [222,227]. Importantly, IL-11 as well as IL-27 antagonist treatment may also represent exciting future perspectives for improving therapy for TB. In this regard, IL-11 with a W147A substitution [171] appears to be a candidate antagonist for blocking IL-11 signaling during antibiotic treatment. Accordingly, in Mtb–infected I/St mice, as described above, administration of W147A indeed results in attenuated inflammation in the lung along with an increased survival time of the animals [171]. Since a soluble form of IL-27Rα—sIL-27RA—has been described to naturally control IL-27 receptor binding [228] and treatment with recombinant sIL-27RA has a beneficial effect in septic peritonitis [229], adjunct treatment with sIL-27RA also constitutes an interesting future option to improve the therapy for TB. The inhibitor, however, has not yet been investigated in the context of experimental TB. Nevertheless, it has to be remarked that, whereas the overall knowledge of the IL-11-driven immune mechanisms during Mtb infection is still at the beginning, IL-27 apparently provides a broad spectrum of regulatory functions in the context of TB, underlining the therapeutic potential of interference with IL-27-mediated signaling. However, as IL-27 also prevents immunopathology induced by excessive production of IL-17A [17], inhibition of IL-27-mediated signaling during adjunct TB therapy would have to be strictly controlled.
5. Conclusions
The gp130 cytokines IL-6, IL-11 and IL-27 differentially affect the outcome of infection with Mtb. The cytokines thereby exert pleiotropic effects on myeloid and lymphoid cells and modulate pro- and anti-inflammatory immune responses. In particular, the impact of the respective cytokines on IL-17A-producing TH17 cells appears to correlate with disease outcome—whereas, in the absence of IL-6-mediated signaling, both bacterial growth and TH17 immune response are largely unaffected [139] (submitted), IL-27Rα deficiency leads to improved anti-mycobacterial protection by enhanced production of IL-17A [17]. To further revise this connection, however, a potential role of IL-11 in the induction of TH17 cells during TB would also need to be examined. The differential roles of the gp130 cytokines in TB provoke varying potential therapeutic implementations.
Author Contributions
Conceptualization, K.R., J.R. and C.H.; writing—original draft preparation, K.R., J.R. and C.H.; writing—review and editing, K.R., J.R. and C.H.; funding acquisition, C.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the DFG International Research Training Group (IRTG 1911), the German Centre for Infection Research (DZIF TTU 02.810.) and the Cluster of Excellence Inflammation-at-Interfaces (EXC306).
Conflicts of Interest
The authors declare no conflict of interest.
References
- WHO. Global Tuberculosis Report 2019; WHO: Geneva, Switzerland, 2019. [Google Scholar]
- Kaufmann, S.H.E. Protection Against Tuberculosis: Cytokines, T Cells, and Macrophages. Ann. Rheum. Dis. 2002, 6, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, S.; Schaible, U.E. The Granuloma in Tuberculosis: Dynamics of a Host-Pathogen Collusion. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, J. TNF-blocking agents and tuberculosis: New drugs illuminate an old topic. Rheumatology 2005, 44, 714–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, O.N.; Noiseux, C.R.; Martin, J.A.C.; Lara, V.G. Reactivation tuberculosis in a patient with anti-TNF-alpha treatment. Am. J. Gastroenterol. 2001, 96, 1665–1666. [Google Scholar] [CrossRef] [PubMed]
- Sartori, N.S.; de Andrade, N.P.B.; da Silva Chakr, R.M. Incidence of tuberculosis in patients receiving anti-TNF therapy for rheumatic diseases: A systematic review. Clin. Rheumatol. 2020, 39, 1439–1447. [Google Scholar] [CrossRef]
- Zhang, Z.; Fan, W.; Yang, G.; Xu, Z.; Wang, J.; Cheng, Q.; Yu, M. Risk of Tuberculosis in Patients Treated with TNF-α Antagonists: A Systematic Review and Meta-Analysis of Randomised Controlled Trials. BMJ Open 2017, 7, e012567. [Google Scholar] [CrossRef] [Green Version]
- Tobin, D.M. Host-Directed Therapies for Tuberculosis. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef]
- Hoft, D.; Abate, G. Immunotherapy for tuberculosis: Future prospects. ImmunoTargets Ther. 2016, 5, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotgiu, G.; Centis, R.; D’Ambrosio, L.; Battista Migliori, G. Tuberculosis treatment and drug regimens. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Dara, Y.; Volcani, D.; Shah, K.; Shin, K.; Venketaraman, V. Potentials of host-directed therapies in tuberculosis management. J. Clin. Med. 2019, 8, 1166. [Google Scholar] [CrossRef] [Green Version]
- Domingo-Gonzalez, R.; Prince, O.; Cooper, A.; Khader, S.A. Cytokines and chemokines in mycobacterium tuberculosis infection. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cicchese, J.M.; Evans, S.; Hult, C.; Joslyn, L.R.; Wessler, T.; Millar, J.A.; Marino, S.; Cilfone, N.A.; Mattila, J.T.; Linderman, J.J.; et al. Dynamic balance of pro- and anti-inflammatory signals controls disease and limits pathology. Immunol. Rev. 2018, 285, 147–167. [Google Scholar] [CrossRef]
- Torrado, E.; Cooper, A.M. Cytokines in the balance of protection and pathology during mycobacterial infections. Adv. Exp. Med. Biol. 2013. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.M.; Mayer-Barber, K.D.; Sher, A. Role of Innate Cytokines in Mycobacterial Infection. Mucosal Immunol. 2011, 4, 252–260. [Google Scholar] [CrossRef]
- Sharma, S.; Bose, M. Role of Cytokines in Immune Response to Pulmonary Tuberculosis. Asian Pac. J. Allergy Immunol. 2001, 19, 213–219. [Google Scholar]
- Erdmann, H.; Behrends, J.; Ritter, K.; Holscher, A.; Volz, J.; Rosenkrands, I.; Holscher, C. The increased protection and pathology in Mycobacterium tuberculosis-infected IL-27R-alpha-deficient mice is supported by IL-17A and is associated with the IL-17A-induced expansion of multifunctional T cells. Mucosal Immunol. 2018, 11, 1168–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hölscher, C.; Hölscher, A.; Rückerl, D.; Yoshimoto, T.; Yoshida, H.; Mak, T.; Saris, C.; Ehlers, S. The IL-27 receptor chain WSX-1 differentially regulates antibacterial immunity and survival during experimental tuberculosis. J. Immunol. 2005, 174, 3534–3544. [Google Scholar] [CrossRef] [PubMed]
- Van Crevel, R.; Ottenhoff, T.H.M.; Van der Meer, J.W.M. Innate immunity to Mycobacterium tuberculosis. Am. Soc. Microbiol. J. 2002, 15, 294–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, J.A.L.; Goldstein, M.M.; Chan, J.; Triebold, K.J.; Pfeffer, K.; Lowenstein, C.J.; Schrelber, R.; Mak, T.W.; Bloom, B.R. Tumor necrosis factor-α is required in the protective immune response against mycobacterium tuberculosis in mice. Immunity 1995, 2, 561–572. [Google Scholar] [CrossRef] [Green Version]
- Zeng, G.; Zhang, G.; Chen, X. Th1 cytokines, true functional signatures for protective immunity against TB? Chin. Soc. Immunol. 2018, 15, 206–215. [Google Scholar] [CrossRef] [Green Version]
- Giacomini, E.; Iona, E.; Ferroni, L.; Miettinen, M.; Fattorini, L.; Orefici, G.; Julkunen, I.; Coccia, E.M. Infection of human macrophages and dendritic cells with mycobacterium tuberculosis induces a differential cytokine gene expression that modulates T Cell response. J. Immunol. 2001, 166, 7033–7041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guirado, E.; Schlesinger, L.S.; Kaplan, G. Macrophages in Tuberculosis: Friend or Foe. Semin Immunopathol. 2013, 35, 563–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, J.L.; Chan, J.; Triebold, K.J.; Dalton, D.K.; Stewart, T.A.; Bloom, B.R. An essential role for interferon 7 in resistance to mycobacterium tuberculosis infection. J. Exp. Med. 1993, 178, 2249–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, A.M.; Dalton, D.K.; Stewart, T.A.; Griffin, J.P.; Russell, D.G.; Orme, I.M. Disseminated tuberculosis in interferon gamma gene-distrupted mice. J. Exp. Med. 1993, 178, 2243–2247. [Google Scholar] [CrossRef] [Green Version]
- Newport, M.J.; Huxley, C.M.; Huston, S.; Hawrylowicz, C.M.; Oostra, B.A.; Williamson, R.; Levin, M. A mutation in the interferon-γ–receptor gene and susceptibility to mycobacterial infection. N. Engl. J. Med. 1996, 335, 1941–1949. [Google Scholar] [CrossRef]
- Gallegos, A.M.; van Heijst, J.W.J.; Samstein, M.; Su, X.; Pamer, E.G.; Glickman, M.S. A gamma interferon independent mechanism of CD4 T cell mediated control of M. tuberculosis infection In Vivo. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef] [Green Version]
- Lyadova, I.V.; Panteleev, A.V. Th1 and Th17 Cells in tuberculosis: Protection, pathology, and biomarkers. Mediat. Inflamm. 2015. [Google Scholar] [CrossRef] [Green Version]
- Nikitina, I.Y.; Panteleev, A.V.; Sosunova, E.V.; Karpina, N.L.; Bagdasarian, T.R.; Burmistrova, I.A.; Andreevskaya, S.N.; Chernousova, L.N.; Vasilyeva, I.A.; Lyadova, I.V. Antigen-Specific IFN-γ Responses correlate with the activity of m. tuberculosis infection but are not associated with the severity of tuberculosis disease. J. Immunol. Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Okamoto Yoshida, Y.; Umemura, M.; Yahagi, A.; O’Brien, R.L.; Ikuta, K.; Kishihara, K.; Hara, H.; Nakae, S.; Iwakura, Y.; Matsuzaki, G. Essential role of IL-17A in the formation of a mycobacterial infection-induced granuloma in the lung. J. Immunol. 2010, 184, 4414–4422. [Google Scholar] [CrossRef] [Green Version]
- Torrado, E.; Cooper, A.M. IL-17 and Th17 cells in tuberculosis. Cytokine Growth Factor Rev. 2010, 21, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khader, S.A.; Pearl, J.E.; Sakamoto, K.; Gilmartin, L.; Bell, G.K.; Jelley-Gibbs, D.M.; Ghilardi, N.; de Sauvage, F.; Cooper, A.M. IL-23 compensates for the absence of IL-12p70 and is essential for the IL-17 response during tuberculosis but is dispensable for protection and antigen-specific IFN-gamma responses if IL-12p70 is available. J. Immunol. 2005, 175, 788–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umemura, M.; Yahagi, A.; Hamada, S.; Begum, M.D.; Watanabe, H.; Kawakami, K.; Suda, T.; Sudo, K.; Nakae, S.; Iwakura, Y.; et al. IL-17-mediated regulation of innate and acquired immune response against pulmonary Mycobacterium bovis bacille Calmette-Guerin infection. J. Immunol. 2007, 178, 3786–3796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lázár-Molnár, E.; Chen, B.; Sweeney, K.A.; Wang, E.J.; Liu, W.; Lin, J.; Porcelli, S.A.; Almo, S.C.; Nathenson, S.G.; Jacobs, W.R. Programmed death-1 (PD-1)-deficient mice are extraordinarily sensitive to tuberculosis. Proc. Natl. Acad. Sci. USA 2010, 107, 13402–13407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott-Browne, J.P.; Shafiani, S.; Tucker-Heard, G.; Ishida-Tsubota, K.; Fontenot, J.D.; Rudensky, A.Y.; Bevan, M.J.; Urdahl, K.B. Expansion and function of Foxp3-expressing T regulatory cells during tuberculosis. J. Exp. Med. 2007, 204, 2159–2169. [Google Scholar] [CrossRef] [Green Version]
- Shafiani, S.; Tucker-Heard, G.; Kariyone, A.; Takatsu, K.; Urdahl, K.B. Pathogen-specific regulatory T cells delay the arrival of effector T cells in the lung during early tuberculosis. J. Exp. Med. 2010, 207, 1409–1420. [Google Scholar] [CrossRef] [Green Version]
- Dorhoi, A.; Yeremeev, V.; Nouailles, G.; Weiner, J., 3rd; Jorg, S.; Heinemann, E.; Oberbeck-Muller, D.; Knaul, J.K.; Vogelzang, A.; Reece, S.T.; et al. Type I IFN signaling triggers immunopathology in tuberculosis-susceptible mice by modulating lung phagocyte dynamics. Eur. J. Immunol. 2014, 44, 2380–2393. [Google Scholar] [CrossRef]
- Lin, P.L.; Plessner, H.L.; Voitenok, N.N.; Flynn, J.A.L. Tumor necrosis factor and tuberculosis. J. Investig. Dermatol. Symp. Proc. 2007, 12, 22–25. [Google Scholar] [CrossRef] [Green Version]
- Collison, L.W.; Delgoffe, G.M.; Guy, C.S.; Vignali, K.M.; Chaturvedi, V.; Fairweather, D.; Satoskar, A.R.; Garcia, K.C.; Hunter, C.A.; Drake, C.G.; et al. The composition and signaling of the IL-35 receptor are unconventional. Nat. Immunol. 2012, 13, 290–299. [Google Scholar] [CrossRef] [Green Version]
- Derouet, D.; Rousseau, F.; Alfonsi, F.; Froger, J.; Hermann, J.; Barbier, F.; Perret, D.; Diveu, C.; Guillet, C.; Preisser, L.; et al. Neuropoietin, a new IL-6-related cytokine signaling through the ciliary neurotrophic factor receptor. Proc. Natl. Acad Sci. USA 2004, 101, 4827–4832. [Google Scholar] [CrossRef] [Green Version]
- Huyton, T.; Zhang, J.-G.; Luo, C.S.; Lou, M.-Z.; Hilton, D.J.; Nicola, N.A.; Garrett, T.P.J. An unusual cytokine:Ig-domain interaction revealed in the crystal structure of leukemia inhibitory factor (LIF) in complex with the LIF receptor. Proc. Natl. Acad Sci. USA 2007, 104, 12737–12742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lelievre, E.; Plun-Favreau, H.; Chevalier, S.; Froger, J.; Guillet, C.; Elson, G.C.; Gauchat, J.F.; Gascan, H. Signaling pathways recruited by the cardiotrophin-like cytokine/cytokine-like factor-1 composite cytokine: Specific requirement of the membrane-bound form of ciliary neurotrophic factor receptor alpha component. J. Biol. Chem. 2001, 276, 22476–22484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Man, D.; He, W.; Sze, K.H.; Gong, K.; Smith, D.K.; Zhu, G.; Ip, N.Y. Solution Structure of the C-terminal Domain of the Ciliary Neurotrophic Factor (CNTF) Receptor and Ligand Free Associations among Components of the CNTF Receptor Complex. J. Biol. Chem. 2003, 278, 23285–23294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metcalfe, R.D.; Aizel, K.; Zlatic, C.O.; Nguyen, P.M.; Morton, C.J.; Lio, D.S.S.; Cheng, H.-C.; Dobson, R.C.J.; Parker, M.W.; Gooley, P.R.; et al. The structure of the extracellular domains of human interleukin 11 α-receptor reveals mechanisms of cytokine engagement. J. Biol. Chem. 2020. [Google Scholar] [CrossRef] [Green Version]
- Mosley, B.; De Imus, C.; Friend, D.; Boiani, N.; Thoma, B.; Park, L.S.; Cosman, D. Dual oncostatin M (OSM) receptors. Cloning and characterization of an alternative signaling subunit conferring OSM-specific receptor activation. J. Biol. Chem. 1996, 271, 32635–32643. [Google Scholar] [CrossRef] [Green Version]
- Pennica, D.; Shaw, K.J.; Swanson, T.A.; Moore, M.W.; Shelton, D.L.; Zioncheck, K.A.; Rosenthal, A.; Taga, T.; Paoni, N.F.; Wood, W.I. Cardiotrophin-1. Biological activities and binding to the leukemia inhibitory factor receptor/gp130 signaling complex. J. Biol. Chem. 1995, 270, 10915–10922. [Google Scholar] [CrossRef] [Green Version]
- Pflanz, S.; Hibbert, L.; Mattson, J.; Rosales, R.; Vaisberg, E.; Bazan, J.F.; Phillips, J.H.; McClanahan, T.K.; de Waal Malefyt, R.; Kastelein, R.A. WSX-1 and Glycoprotein 130 Constitute a Signal-Transducing Receptor for IL-27. J. Immunol. 2004, 172, 2225. [Google Scholar] [CrossRef]
- Robledo, O.; Fourcin, M.; Chevalier, S.; Guillet, C.; Auguste, P.; Pouplard-Barthelaix, A.; Pennica, D.; Gascan, H. Signaling of the cardiotrophin-1 receptor. Evidence for a third receptor component. J. Biol. Chem. 1997, 272, 4855–4863. [Google Scholar] [CrossRef] [Green Version]
- Simpson, R.J.; Hammacher, A.; Smith, D.K.; Matthews, J.M.; Ward, L.D. Interleukin-6: Structure-function relationships. Protein Sci. 1997, 6, 929–955. [Google Scholar] [CrossRef]
- Somers, W.; Stahl, M.; Seehra, J.S. 1.9 A crystal structure of interleukin 6: Implications for a novel mode of receptor dimerization and signaling. EMBO J. 1997, 16, 989–997. [Google Scholar] [CrossRef] [Green Version]
- Taga, T.; Kishimoto, T. Gp130 and the interleukin-6 family of cytokines. Annu. Rev. Immunol. 1997, 15, 797–819. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wei, Y.; Xiao, H.; Liu, X.; Zhang, Y.; Han, G.; Chen, G.; Hou, C.; Ma, N.; Shen, B.; et al. A novel IL-23p19/Ebi3 (IL-39) cytokine mediates inflammation in Lupus-like mice. Eur. J. Immunol. 2016, 46, 1343–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dillon, S.R.; Sprecher, C.; Hammond, A.; Bilsborough, J.; Rosenfeld-Franklin, M.; Presnell, S.R.; Haugen, H.S.; Maurer, M.; Harder, B.; Johnston, J.; et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat. Immunol. 2004, 5, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Putheti, P.; Zhou, Q.; Liu, Q.; Gao, W. Structures and biological functions of IL-31 and IL-31 receptors. Cytokine Growth Factor Rev. 2008, 19, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Ma, D.; Wang, Y.; Zhou, G.; Wang, Y.; Li, X. Review: The Roles and Mechanisms of Glycoprotein 130 Cytokines in the Regulation of Adipocyte Biological Function. Inflammation 2019, 42, 790–798. [Google Scholar] [CrossRef]
- Reeh, H.; Rudolph, N.; Billing, U.; Christen, H.; Streif, S.; Bullinger, E.; Schliemann-Bullinger, M.; Findeisen, R.; Schaper, F.; Huber, H.J.; et al. Response to IL-6 trans- and IL-6 classic signalling is determined by the ratio of the IL-6 receptor α to gp130 expression: Fusing experimental insights and dynamic modelling. Cell Commun. Signal. 2019, 17, 46. [Google Scholar] [CrossRef] [Green Version]
- West, N.R. Coordination of Immune-Stroma Crosstalk by IL-6 Family Cytokines. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Silver, J.S.; Hunter, C.A. gp130 at the nexus of inflammation, autoimmunity, and cancer. J. Leukoc. Biol. 2010, 88, 1145–1156. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Y.; Clemens, J.C.; Schubert, H.L.; Stuckey, J.A.; Fischer, M.W.; Hume, D.M.; Saper, M.A.; Dixon, J.E. Expression, purification, and physicochemical characterization of a recombinant Yersinia protein tyrosine phosphatase. J. Biol. Chem. 1992, 267, 23759–23766. [Google Scholar]
- Hirano, T.; Taga, T.; Nakano, N.; Yasukawa, K.; Kashiwamura, S.; Shimizu, K.; Nakajima, K.; Pyun, K.H.; Kishimoto, T. Purification to homogeneity and characterization of human B-cell differentiation factor (BCDF or BSFp-2). Proc. Natl. Acad. Sci. USA 1985, 82, 5490–5494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Snick, J.; Cayphas, S.; Vink, A.; Uyttenhove, C.; Coulie, P.G.; Rubira, M.R.; Simpson, R.J. Purification and NH2-terminal amino acid sequence of a T-cell-derived lymphokine with growth factor activity for B-cell hybridomas. Proc. Natl. Acad. Sci. USA 1986, 83, 9679–9683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordan, R.P.; Pumphrey, J.G.; Rudikoff, S. Purification and NH2-terminal sequence of a plasmacytoma growth factor derived from the murine macrophage cell line P388D1. J. Immunol. 1987, 139, 813–817. [Google Scholar] [PubMed]
- Gauldie, J.; Richards, C.; Harnish, D.; Lansdorp, P.; Baumann, H. Interferon beta 2/B-cell stimulatory factor type 2 shares identity with monocyte-derived hepatocyte-stimulating factor and regulates the major acute phase protein response in liver cells. Proc. Natl. Acad. Sci. USA 1987, 84, 7251. [Google Scholar] [CrossRef] [Green Version]
- Takai, Y.; Wong, G.G.; Clark, S.C.; Burakoff, S.J.; Herrmann, S.H. B cell stimulatory factor-2 is involved in the differentiation of cytotoxic T lymphocytes. J. Immunol. 1988, 140, 508. [Google Scholar]
- Kishimoto, T. Factors affecting B-cell growth and differentiation. Annu. Rev. Immunol. 1985, 3, 133–157. [Google Scholar] [CrossRef]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect. Biol. 2014, 6, a016295. [Google Scholar] [CrossRef]
- Akdis, M.; Aab, A.; Altunbulakli, C.; Azkur, K.; Costa, R.A.; Crameri, R.; Duan, S.; Eiwegger, T.; Eljaszewicz, A.; Ferstl, R.; et al. Interleukins (from IL-1 to IL-38), interferons, transforming growth factor β and TNF-α: Receptors, functions, and roles in diseases. J. Allergy Clin. Immunol. 2016, 138, 984–1010. [Google Scholar] [CrossRef] [Green Version]
- Gabay, C. Interleukin-6 and chronic inflammation. Arthritis Res. Ther. 2006, 8 (Suppl. 2), S3. [Google Scholar] [CrossRef] [Green Version]
- Rose-John, S.; Winthrop, K.; Calabrese, L. The role of IL-6 in host defence against infections: Immunobiology and clinical implications. Nat. Rev. Rheumatol. 2017, 13, 399–409. [Google Scholar] [CrossRef]
- Schaper, F.; Rose-John, S. Interleukin-6: Biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. 2015, 26, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Narazaki, M.; Yasukawa, K.; Saito, T.; Ohsugi, Y.; Fukui, H.; Koishihara, Y.; Yancopoulos, G.D.; Taga, T.; Kishimoto, T. Soluble forms of the interleukin-6 signal-transducing receptor component gp130 in human serum possessing a potential to inhibit signals through membrane-anchored gp130. Blood 1993, 82, 1120–1126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopf, M.; Baumann, H.; Freer, G.; Freudenberg, M.; Lamers, M.; Kishimoto, T.; Zinkernagel, R.; Bluethmann, H.; Kohler, G. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 1994, 368, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Simpson, R.J.; Cheers, C. Role of IL-6 in activation of T cells for acquired cellular resistance to Listeria monocytogenes. J. Immunol. 1994, 152, 5375–5380. [Google Scholar]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef]
- Hagenstein, J.; Melderis, S.; Nosko, A.; Warkotsch, M.T.; Richter, J.V.; Ramcke, T.; Herrnstadt, G.R.; Scheller, J.; Yan, I.; Mittrücker, H.-W.; et al. A Novel Role for IL-6 Receptor Classic Signaling: Induction of RORγt(+)Foxp3(+) Tregs with Enhanced Suppressive Capacity. J. Am. Soc. Nephrol. 2019, 30, 1439–1453. [Google Scholar] [CrossRef]
- Harbour, S.N.; DiToro, D.F.; Witte, S.J.; Zindl, C.L.; Gao, M.; Schoeb, T.R.; Jones, G.W.; Jones, S.A.; Hatton, R.D.; Weaver, C.T. TH17 cells require ongoing classic IL-6 receptor signaling to retain transcriptional and functional identity. Sci. Immunol. 2020, 5, eaaw2262. [Google Scholar] [CrossRef]
- Korn, T.; Mitsdoerffer, M.; Croxford, A.L.; Awasthi, A.; Dardalhon, V.A.; Galileos, G.; Vollmar, P.; Stritesky, G.L.; Kaplan, M.H.; Waisman, A.; et al. IL-6 controls Th17 immunity In Vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2008, 105, 18460–18465. [Google Scholar] [CrossRef] [Green Version]
- Kimura, A.; Kishimoto, T. IL-6: Regulator of Treg/Th17 balance. Eur. J. Immunol. 2010, 40, 1830–1835. [Google Scholar] [CrossRef]
- Noack, M.; Miossec, P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev. 2014, 13, 668–677. [Google Scholar] [CrossRef]
- Sodenkamp, J.; Behrends, J.; Forster, I.; Muller, W.; Ehlers, S.; Holscher, C. gp130 on macrophages/granulocytes modulates inflammation during experimental tuberculosis. Eur. J. Cell Biol. 2011, 90, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Aderka, D.; Le, J.M.; Vilcek, J. IL-6 inhibits lipopolysaccharide-induced tumor necrosis factor production in cultured human monocytes, U937 cells, and in mice. J. Immunol. 1989, 143, 3517–3523. [Google Scholar] [PubMed]
- Mauer, J.; Chaurasia, B.; Goldau, J.; Vogt, M.C.; Ruud, J.; Nguyen, K.D.; Theurich, S.; Hausen, A.C.; Schmitz, J.; Brönneke, H.S.; et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat. Immunol. 2014, 15, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S.; Heinrich, P.C. Soluble receptors for cytokines and growth factors: Generation and biological function. Biochem. J. 1994, 300 (Pt 2), 281–290. [Google Scholar] [CrossRef]
- Mülberg, J.; Schooltink, H.; Stoyan, T.; Günther, M.; Graeve, L.; Buse, G.; Mackiewicz, A.; Heinrich, P.C.; Rose-John, S. The soluble interleukin-6 receptor is generated by shedding. Eur. J. Immunol. 1993, 23, 473–480. [Google Scholar] [CrossRef]
- Mackiewicz, A.; Schooltink, H.; Heinrich, P.C.; Rose-John, S. Complex of soluble human IL-6-receptor/IL-6 up-regulates expression of acute-phase proteins. J. Immunol. 1992, 149, 2021–2027. [Google Scholar]
- Fischer, M.; Goldschmitt, J.; Peschel, C.; Brakenhoff, J.P.; Kallen, K.J.; Wollmer, A.; Grötzinger, J.; Rose-John, S.I. A bioactive designer cytokine for human hematopoietic progenitor cell expansion. Nat. Biotechnol. 1997, 15, 142–145. [Google Scholar] [CrossRef]
- Romano, M.; Sironi, M.; Toniatti, C.; Polentarutti, N.; Fruscella, P.; Ghezzi, P.; Faggioni, R.; Luini, W.; van Hinsbergh, V.; Sozzani, S.; et al. Role of IL-6 and Its Soluble Receptor in Induction of Chemokines and Leukocyte Recruitment. Immunity 1997, 6, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Ebihara, N.; Matsuda, A.; Nakamura, S.; Matsuda, H.; Murakami, A. Role of the IL-6 Classic- and Trans-Signaling Pathways in Corneal Sterile Inflammation and Wound Healing. Investig. Ophthalmol. Vis. Sci. 2011, 52, 8549–8557. [Google Scholar] [CrossRef]
- Atreya, R.; Mudter, J.; Finotto, S.; Müllberg, J.; Jostock, T.; Wirtz, S.; Schütz, M.; Bartsch, B.; Holtmann, M.; Becker, C.; et al. Blockade of interleukin 6 trans signaling suppresses T-cell resistance against apoptosis in chronic intestinal inflammation: Evidence in Crohn disease and experimental colitis In Vivo. Nat. Med. 2000, 6, 583–588. [Google Scholar] [CrossRef]
- Dominitzki, S.; Fantini, M.C.; Neufert, C.; Nikolaev, A.; Galle, P.R.; Scheller, J.; Monteleone, G.; Rose-John, S.; Neurath, M.F.; Becker, C. Cutting edge: Trans-signaling via the soluble IL-6R abrogates the induction of FoxP3 in naive CD4+CD25 T cells. J. Immunol. 2007, 179, 2041–2045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jostock, T.; Müllberg, J.; Özbek, S.; Atreya, R.; Blinn, G.; Voltz, N.; Fischer, M.; Neurath, M.F.; Rose-John, S. Soluble gp130 is the natural inhibitor of soluble interleukin-6 receptor transsignaling responses. Eur. J. Biochem. 2001, 268, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S.; Waetzig, G.H.; Scheller, J.; Grötzinger, J.; Seegert, D. The IL-6/sIL-6R complex as a novel target for therapeutic approaches. Expert Opin. Ther. Targets 2007, 11, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Rabe, B.; Chalaris, A.; May, U.; Waetzig, G.H.; Seegert, D.; Williams, A.S.; Jones, S.A.; Rose-John, S.; Scheller, J. Transgenic blockade of interleukin 6 transsignaling abrogates inflammation. Blood 2008, 111, 1021–1028. [Google Scholar] [CrossRef] [PubMed]
- Hurst, S.M.; Wilkinson, T.S.; McLoughlin, R.M.; Jones, S.; Horiuchi, S.; Yamamoto, N.; Rose-John, S.; Fuller, G.M.; Topley, N.; Jones, S.A. Il-6 and its soluble receptor orchestrate a temporal switch in the pattern of leukocyte recruitment seen during acute inflammation. Immunity 2001, 14, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Greenhill, C.J.; Rose-John, S.; Lissilaa, R.; Ferlin, W.; Ernst, M.; Hertzog, P.J.; Mansell, A.; Jenkins, B.J. IL-6 trans-signaling modulates TLR4-dependent inflammatory responses via STAT3. J. Immunol. 2011, 186, 1199–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doganci, A.; Sauer, K.; Karwot, R.; Finotto, S. Pathological role of IL-6 in the experimental allergic bronchial asthma in mice. Clin. Rev. Allergy Immunol. 2005, 28, 257–270. [Google Scholar] [CrossRef]
- Barkhausen, T.; Tschernig, T.; Rosenstiel, P.; van Griensven, M.; Vonberg, R.P.; Dorsch, M.; Mueller-Heine, A.; Chalaris, A.; Scheller, J.; Rose-John, S.; et al. Selective blockade of interleukin-6 trans-signaling improves survival in a murine polymicrobial sepsis model. Crit. Care Med. 2011, 39, 1407–1413. [Google Scholar] [CrossRef]
- Heink, S.; Yogev, N.; Garbers, C.; Herwerth, M.; Aly, L.; Gasperi, C.; Husterer, V.; Croxford, A.L.; Möller-Hackbarth, K.; Bartsch, H.S.; et al. Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic T(H)17 cells. Nat. Immunol. 2017, 18, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Putoczki, T.; Ernst, M. More than a sidekick: The IL-6 family cytokine IL-11 links inflammation to cancer. J. Leukoc. Biol. 2010, 88, 1109–1117. [Google Scholar] [CrossRef]
- Du, X.X.; Williams, D.A. Interleukin-11: A multifunctional growth factor derived from the hematopoietic microenvironment. Blood 1994, 83, 2023–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yonemura, Y.; Kawakita, M.; Masuda, T.; Fujimoto, K.; Kato, K.; Takatsuki, K. Synergistic effects of interleukin 3 and interleukin 11 on murine megakaryopoiesis in serum-free culture. Exp. Hematol. 1992, 20, 1011–1016. [Google Scholar]
- Jenkins, B.J.; Grail, D.; Inglese, M.; Quilici, C.; Bozinovski, S.; Wong, P.; Ernst, M. Imbalanced gp130-Dependent Signaling in Macrophages Alters Macrophage Colony-Stimulating Factor Responsiveness via Regulation of c-fms Expression. Mol. Cell. Biol. 2004, 24, 1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redlich, C.A.; Gao, X.; Rockwell, S.; Kelley, M.; Elias, J.A. IL-11 enhances survival and decreases TNF production after radiation-induced thoracic injury. J. Immunol. 1996, 157, 1705–1710. [Google Scholar] [PubMed]
- Trepicchio, W.L.; Bozza, M.; Pedneault, G.; Dorner, A.J. Recombinant human IL-11 attenuates the inflammatory response through down-regulation of proinflammatory cytokine release and nitric oxide production. J. Immunol. 1996, 157, 3627–3634. [Google Scholar] [PubMed]
- Opal, S.M.; Keith, J.C.; Palardy, J.E.; Parejo, N. Recombinant human interleukin-11 has anti-inflammatory actions yet does not exacerbate systemic Listeria infection. J. Infect. Dis. 2000, 181, 754–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Opal, S.M.; Jhung, J.W.; Keith, J.C., Jr.; Palardy, J.E.; Parejo, N.A.; Young, L.D.; Bhattacharjee, A. Recombinant human interleukin-11 in experimental Pseudomonas aeruginosa sepsis in immunocompromised animals. J. Infect. Dis. 1998, 178, 1205–1208. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.; Geba, G.P.; Zheng, T.; Ray, P.; Homer, R.J.; Kuhn, C., 3rd; Flavell, R.A.; Elias, J.A. Targeted expression of IL-11 in the murine airway causes lymphocytic inflammation, bronchial remodeling, and airways obstruction. J. Clin. Investig. 1996, 98, 2845–2853. [Google Scholar] [CrossRef]
- Schafer, S.; Viswanathan, S.; Widjaja, A.A.; Lim, W.W.; Moreno-Moral, A.; DeLaughter, D.M.; Ng, B.; Patone, G.; Chow, K.; Khin, E.; et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature 2017, 552, 110–115. [Google Scholar] [CrossRef]
- Traber, K.E.; Dimbo, E.L.; Symer, E.M.; Korkmaz, F.T.; Jones, M.R.; Mizgerd, J.P.; Quinton, L.J. Roles of interleukin-11 during acute bacterial pneumonia. PLoS ONE 2019, 14, e0221029. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Kiapour, N.; Kapoor, S.; Khan, T.; Thamilarasan, M.; Tao, Y.; Cohen, S.; Miller, R.; Sobel, R.A.; Markovic-Plese, S. IL-11 Induces Encephalitogenic Th17 Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. J. Immunol. 2019, 203, 1142–1150. [Google Scholar] [CrossRef]
- Lokau, J.; Nitz, R.; Agthe, M.; Monhasery, N.; Aparicio-Siegmund, S.; Schumacher, N.; Wolf, J.; Möller-Hackbarth, K.; Waetzig, G.H.; Grötzinger, J.; et al. Proteolytic Cleavage Governs Interleukin-11 Trans-signaling. Cell Rep. 2016, 14, 1761–1773. [Google Scholar] [CrossRef] [Green Version]
- Lamertz, L.; Rummel, F.; Polz, R.; Baran, P.; Hansen, S.; Waetzig, G.H.; Moll, J.M.; Floss, D.M.; Scheller, J. Soluble gp130 prevents interleukin-6 and interleukin-11 cluster signaling but not intracellular autocrine responses. Sci. Signal. 2018, 11, eaar7388. [Google Scholar] [CrossRef] [Green Version]
- Jones, L.L.; Vignali, D.A.A. Molecular interactions within the IL-6/IL-12 cytokine/receptor superfamily. Immunol. Res. 2011, 51, 5–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tait Wojno, E.D.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lupardus, P.; LaPorte, S.L.; Garcia, K.C. Structural Biology of Shared Cytokine Receptors. Annu. Rev. Immunol. 2009, 27, 29–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gearing, D.P.; Cosman, D. Homology of the p40 subunit of natural killer cell stimulatory factor (NKSF) with the extracellular domain of the interleukin-6 receptor. Cell 1991, 66, 9–10. [Google Scholar] [CrossRef]
- Pflanz, S.; Timans, J.C.; Cheung, J.; Rosales, R.; Kanzler, H.; Gilbert, J.; Hibbert, L.; Churakova, T.; Travis, M.; Vaisberg, E.; et al. IL-27, a Heterodimeric Cytokine Composed of EBI3 and p28 Protein, Induces Proliferation of Naive CD4+ T Cells. Immunity 2002, 16, 779–790. [Google Scholar] [CrossRef] [Green Version]
- Abdalla, A.E.; Li, Q.; Xie, L.; Xie, J. Biology of IL-27 and its Role in the Host Immunity against Mycobacterium Tuberculosis. Int. J. Biol. Sci. 2015, 11, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Lucas, S.; Ghilardi, N.; Li, J.; de Sauvage, F.J. IL-27 regulates IL-12 responsiveness of naive CD4+ T cells through Stat1-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2003, 100, 15047–15052. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, H.; Hamano, S.; Senaldi, G.; Covey, T.; Faggioni, R.; Mu, S.; Xia, M.; Wakeham, A.C.; Nishina, H.; Potter, J.; et al. WSX-1 Is Required for the Initiation of Th1 Responses and Resistance to L. major Infection. Immunity 2001, 15, 569–578. [Google Scholar] [CrossRef] [Green Version]
- Artis, D.; Johnson, L.M.; Joyce, K.; Saris, C.; Villarino, A.; Hunter, C.A.; Scott, P. Cutting Edge: Early IL-4 Production Governs the Requirement for IL-27-WSX-1 Signaling in the Development of Protective Th1 Cytokine Responses following Leishmania major Infection. J. Immunol. 2004, 172, 4672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarino, A.; Hibbert, L.; Lieberman, L.; Wilson, E.; Mak, T.; Yoshida, H.; Kastelein, R.A.; Saris, C.; Hunter, C.A. The IL-27R (WSX-1) Is Required to Suppress T Cell Hyperactivity during Infection. Immunity 2003, 19, 645–655. [Google Scholar] [CrossRef] [Green Version]
- Yoshimoto, T.; Yoshimoto, T.; Yasuda, K.; Mizuguchi, J.; Nakanishi, K. IL-27 Suppresses Th2 Cell Development and Th2 Cytokines Production from Polarized Th2 Cells: A Novel Therapeutic Way for Th2-Mediated Allergic Inflammation. J. Immunol. 2007, 179, 4415. [Google Scholar] [CrossRef] [Green Version]
- Batten, M.; Li, J.; Yi, S.; Kljavin, N.M.; Danilenko, D.M.; Lucas, S.; Lee, J.; de Sauvage, F.J.; Ghilardi, N. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17–producing T cells. Nat. Immunol. 2006, 7, 929–936. [Google Scholar] [CrossRef]
- Diveu, C.; McGeachy, M.J.; Boniface, K.; Stumhofer, J.S.; Sathe, M.; Joyce-Shaikh, B.; Chen, Y.; Tato, C.M.; McClanahan, T.K.; de Waal Malefyt, R.; et al. IL-27 blocks RORc expression to inhibit lineage commitment of Th17 cells. J. Immunol. 2009, 182, 5748–5756. [Google Scholar] [CrossRef] [Green Version]
- Do, J.; Kim, D.; Kim, S.; Valentin-Torres, A.; Dvorina, N.; Jang, E.; Nagarajavel, V.; DeSilva, T.M.; Li, X.; Ting, A.H.; et al. Treg-specific IL-27Rα deletion uncovers a key role for IL-27 in Treg function to control autoimmunity. Proc. Natl. Acad. Sci. USA 2017, 114, 10190–10195. [Google Scholar] [CrossRef] [Green Version]
- Do, J.S.; Visperas, A.; Sanogo, Y.O.; Bechtel, J.J.; Dvorina, N.; Kim, S.; Jang, E.; Stohlman, S.A.; Shen, B.; Fairchild, R.L.; et al. An IL-27/Lag3 axis enhances Foxp3+ regulatory T cell-suppressive function and therapeutic efficacy. Mucosal Immunol. 2016, 9, 137–145. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, A.; Carrier, Y.; Peron, J.P.; Bettelli, E.; Kamanaka, M.; Flavell, R.A.; Kuchroo, V.K.; Oukka, M.; Weiner, H.L. A dominant function for interleukin 27 in generating interleukin 10-producing anti-inflammatory T cells. Nat. Immunol. 2007, 8, 1380–1389. [Google Scholar] [CrossRef]
- Apetoh, L.; Quintana, F.J.; Pot, C.; Joller, N.; Xiao, S.; Kumar, D.; Burns, E.J.; Sherr, D.H.; Weiner, H.L.; Kuchroo, V.K. The aryl hydrocarbon receptor interacts with c-Maf to promote the differentiation of type 1 regulatory T cells induced by IL-27. Nat. Immunol. 2010, 11, 854–861. [Google Scholar] [CrossRef] [Green Version]
- Pot, C.; Jin, H.; Awasthi, A.; Liu, S.M.; Lai, C.Y.; Madan, R.; Sharpe, A.H.; Karp, C.L.; Miaw, S.C.; Ho, I.C.; et al. Cutting edge: IL-27 induces the transcription factor c-Maf, cytokine IL-21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL-10-producing Tr1 cells. J. Immunol. 2009, 183, 797–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascanfroni, I.D.; Yeste, A.; Vieira, S.M.; Burns, E.J.; Patel, B.; Sloma, I.; Wu, Y.; Mayo, L.; Ben-Hamo, R.; Efroni, S.; et al. IL-27 acts on DCs to suppress the T cell response and autoimmunity by inducing expression of the immunoregulatory molecule CD39. Nat. Immunol. 2013, 14, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Ruckerl, D.; Hessmann, M.; Yoshimoto, T.; Ehlers, S.; Holscher, C. Alternatively activated macrophages express the IL-27 receptor alpha chain WSX-1. Immunobiology 2006, 211, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Ladel, C.H.; Blum, C.; Dreher, A.; Reifenberg, K.; Kopf, M.; Kaufmann, S.H. Lethal tuberculosis in interleukin-6-deficient mutant mice. Infect. Immun. 1997, 65, 4843–4849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, B.M.; Frank, A.A.; Orme, I.M.; Cooper, A.M. Interleukin-6 induces early gamma interferon production in the infected lung but is not required for generation of specific immunity to Mycobacterium tuberculosis infection. Infect. Immun. 2000, 68, 3322–3326. [Google Scholar] [CrossRef] [Green Version]
- Cheekatla, S.S.; Tripathi, D.; Venkatasubramanian, S.; Nathella, P.K.; Paidipally, P.; Ishibashi, M.; Welch, E.; Tvinnereim, A.R.; Ikebe, M.; Valluri, V.L.; et al. NK-CD11c+ Cell Crosstalk in Diabetes Enhances IL-6-Mediated Inflammation during Mycobacterium tuberculosis Infection. PLoS Pathog. 2016, 12, e1005972. [Google Scholar] [CrossRef]
- Appelberg, R.; Castro, A.G.; Pedrosa, J.; Minoprio, P. Role of interleukin-6 in the induction of protective T cells during mycobacterial infections in mice. Immunology 1994, 82, 361–364. [Google Scholar]
- Ritter, K.; Sodenkamp, J.; Hölscher, A.; Behrends, J.; Holscher, C. IL-6 is not absolutely essential for the development of a TH17 immune response after an aerosol infection with Mycobacterium tuberculosis H37rv. Cells 2020. under review. [Google Scholar]
- Fasnacht, N.; Greweling, M.C.; Bollati-Fogolín, M.; Schippers, A.; Müller, W. T-cell-specific deletion of gp130 renders the highly susceptible IL-10-deficient mouse resistant to intestinal nematode infection. Eur. J. Immunol. 2009, 39, 2173–2183. [Google Scholar] [CrossRef]
- Hatzigeorgiou, D.E.; He, S.; Sobel, J.; Grabstein, K.H.; Hafner, A.; Ho, J.L. IL-6 down-modulates the cytokine-enhanced antileishmanial activity in human macrophages. J. Immunol. 1993, 151, 3682–3692. [Google Scholar]
- Nagabhushanam, V.; Solache, A.; Ting, L.M.; Escaron, C.J.; Zhang, J.Y.; Ernst, J.D. Innate inhibition of adaptive immunity: Mycobacterium tuberculosis-induced IL-6 inhibits macrophage responses to IFN-gamma. J. Immunol. 2003, 171, 4750–4757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- VanHeyningen, T.K.; Collins, H.L.; Russell, D.G. IL-6 produced by macrophages infected with Mycobacterium species suppresses T cell responses. J. Immunol. 1997, 158, 330–337. [Google Scholar] [PubMed]
- Martinez, A.N.; Mehra, S.; Kaushal, D. Role of interleukin 6 in innate immunity to Mycobacterium tuberculosis infection. J. Infect. Dis. 2013, 207, 1253–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manca, C.; Tsenova, L.; Freeman, S.; Barczak, A.K.; Tovey, M.; Murray, P.J.; Barry, C.; Kaplan, G. Hypervirulent M. tuberculosis W/Beijing strains upregulate type I IFNs and increase expression of negative regulators of the Jak-Stat pathway. J. Interferon Cytokine Res. 2005, 25, 694–701. [Google Scholar] [CrossRef]
- Ordway, D.; Henao-Tamayo, M.; Harton, M.; Palanisamy, G.; Troudt, J.; Shanley, C.; Basaraba, R.J.; Orme, I.M. The hypervirulent Mycobacterium tuberculosis strain HN878 induces a potent TH1 response followed by rapid down-regulation. J. Immunol. 2007, 179, 522–531. [Google Scholar] [CrossRef] [Green Version]
- Telesca, C.; Angelico, M.; Piccolo, P.; Nosotti, L.; Morrone, A.; Longhi, C.; Carbone, M.; Baiocchi, L. Interferon-alpha treatment of hepatitis D induces tuberculosis exacerbation in an immigrant. J. Infect. 2007, 54, e223–e226. [Google Scholar] [CrossRef]
- Croker, B.A.; Krebs, D.L.; Zhang, J.G.; Wormald, S.; Willson, T.A.; Stanley, E.G.; Robb, L.; Greenhalgh, C.J.; Forster, I.; Clausen, B.E.; et al. SOCS3 negatively regulates IL-6 signaling In Vivo. Nat. Immunol. 2003, 4, 540–545. [Google Scholar] [CrossRef]
- Lang, R.; Pauleau, A.L.; Parganas, E.; Takahashi, Y.; Mages, J.; Ihle, J.N.; Rutschman, R.; Murray, P.J. SOCS3 regulates the plasticity of gp130 signaling. Nat. Immunol. 2003, 4, 546–550. [Google Scholar] [CrossRef]
- Carow, B.; Rottenberg, M.E. SOCS3, a Major Regulator of Infection and Inflammation. Front. Immunol. 2014, 5, 58. [Google Scholar] [CrossRef] [Green Version]
- Carow, B.; Reuschl, A.K.; Gavier-Widen, D.; Jenkins, B.J.; Ernst, M.; Yoshimura, A.; Chambers, B.J.; Rottenberg, M.E. Critical and independent role for SOCS3 in either myeloid or T cells in resistance to Mycobacterium tuberculosis. PLoS Pathog. 2013, 9, e1003442. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, T.; Ehlers, S.; Heitmann, L.; Rausch, A.; Mages, J.; Murray, P.J.; Lang, R.; Holscher, C. Autocrine IL-10 induces hallmarks of alternative activation in macrophages and suppresses antituberculosis effector mechanisms without compromising T cell immunity. J. Immunol. 2009, 183, 1301–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmok, E.; Abad Dar, M.; Behrends, J.; Erdmann, H.; Ruckerl, D.; Endermann, T.; Heitmann, L.; Hessmann, M.; Yoshimura, A.; Rose-John, S.; et al. Suppressor of Cytokine Signaling 3 in Macrophages Prevents Exacerbated Interleukin-6-Dependent Arginase-1 Activity and Early Permissiveness to Experimental Tuberculosis. Front. Immunol. 2017, 8, 1537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Stewart, K.N.; Bishop, E.; Marek, C.J.; Kluth, D.C.; Rees, A.J.; Wilson, H.M. Unique expression of suppressor of cytokine signaling 3 is essential for classical macrophage activation in rodents in vitro and In Vivo. J. Immunol. 2008, 180, 6270–6278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qualls, J.E.; Neale, G.; Smith, A.M.; Koo, M.S.; DeFreitas, A.A.; Zhang, H.; Kaplan, G.; Watowich, S.S.; Murray, P.J. Arginine usage in mycobacteria-infected macrophages depends on autocrine-paracrine cytokine signaling. Sci. Signal. 2010, 3, ra62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Kasmi, K.C.; Qualls, J.E.; Pesce, J.T.; Smith, A.M.; Thompson, R.W.; Henao-Tamayo, M.; Basaraba, R.J.; Konig, T.; Schleicher, U.; Koo, M.S.; et al. Toll-like receptor-induced arginase 1 in macrophages thwarts effective immunity against intracellular pathogens. Nat. Immunol. 2008, 9, 1399–1406. [Google Scholar] [CrossRef] [Green Version]
- Heitmann, L.; Abad Dar, M.; Schreiber, T.; Erdmann, H.; Behrends, J.; McKenzie, A.N.; Brombacher, F.; Ehlers, S.; Holscher, C. The IL-13/IL-4Ralpha axis is involved in tuberculosis-associated pathology. J. Pathol. 2014, 234, 338–350. [Google Scholar] [CrossRef] [Green Version]
- el-Ahmady, O.; Mansour, M.; Zoeir, H.; Mansour, O. Elevated concentrations of interleukins and leukotriene in response to Mycobacterium tuberculosis infection. Ann. Clin. Biochem. 1997, 34 Pt 2, 160–164. [Google Scholar] [CrossRef] [Green Version]
- Buha, I.; Skodric-Trifunovic, V.; Adzic-Vukicevic, T.; Ilic, A.; Blanka-Protic, A.; Stjepanovic, M.; Andelkovic, M.; Vreca, M.; Milin-Lazovic, J.; Spasovski, V.; et al. Relevance of TNF-alpha, IL-6 and IRAK1 gene expression for assessing disease severity and therapy effects in tuberculosis patients. J. Infect. Dev. Ctries 2019, 13, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Hu, Y.J.; Li, F.G.; Chang, X.J.; Zhang, T.H.; Wang, Z.T. Analysis of Cytokine Levers in Pleural Effusions of Tuberculous Pleurisy and Tuberculous Empyema. Mediat. Inflamm. 2016, 2016, 3068103. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Chen, Y.; He, G.; Jiang, X.; Chen, P.; Ouyang, J. Differential diagnosis of tuberculous and malignant pleural effusions: Comparison of the Th1/Th2 cytokine panel, tumor marker panel and chemistry panel. Scand. J. Clin. Lab. Investig. 2020, 80, 265–270. [Google Scholar] [CrossRef]
- Anbarasu, D.; Raja, C.P.; Raja, A. Multiplex analysis of cytokines/chemokines as biomarkers that differentiate healthy contacts from tuberculosis patients in high endemic settings. Cytokine 2013, 61, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Suzukawa, M.; Akashi, S.; Nagai, H.; Nagase, H.; Nakamura, H.; Matsui, H.; Hebisawa, A.; Ohta, K. Combined Analysis of IFN-γ, IL-2, IL-5, IL-10, IL-1RA and MCP-1 in QFT Supernatant Is Useful for Distinguishing Active Tuberculosis from Latent Infection. PLoS ONE 2016, 11, e0152483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Manna, M.P.; Orlando, V.; Li Donni, P.; Sireci, G.; Di Carlo, P.; Cascio, A.; Dieli, F.; Caccamo, N. Identification of plasma biomarkers for discrimination between tuberculosis infection/disease and pulmonary non tuberculosis disease. PLoS ONE 2018, 13, e0192664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nolan, A.; Condos, R.; Huie, M.L.; Dawson, R.; Dheda, K.; Bateman, E.; Rom, W.N.; Weiden, M.D. Elevated IP-10 and IL-6 from bronchoalveolar lavage cells are biomarkers of non-cavitary tuberculosis. Int. J. Tuberc. Lung Dis. 2013, 17, 922–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Jiao, L.; Liu, T.; Song, J.; Wang, M.; Liang, L.; Wen, C.; Hu, L.; Qu, W.; Ying, B. No Significant Effects of IL-6 and IL-13 Gene Variants on Tuberculosis Susceptibility in the Chinese Population. DNA Cell Biol. 2020, 39, 1356–1367. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T. Interleukin-6: Discovery of a pleiotropic cytokine. Arthritis Res. Ther. 2006, 8 (Suppl. 2), S2. [Google Scholar] [CrossRef] [Green Version]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [Green Version]
- Sodenkamp, J.; Waetzig, G.H.; Scheller, J.; Seegert, D.; Grötzinger, J.; Rose-John, S.; Ehlers, S.; Hölscher, C. Therapeutic targeting of interleukin-6 trans-signaling does not affect the outcome of experimental tuberculosis. Immunobiology 2012, 217, 996–1004. [Google Scholar] [CrossRef]
- Kapina, M.A.; Shepelkova, G.S.; Avdeenko, V.G.; Guseva, A.N.; Kondratieva, T.K.; Evstifeev, V.V.; Apt, A.S. Interleukin-11 drives early lung inflammation during Mycobacterium tuberculosis infection in genetically susceptible mice. PLoS ONE 2011, 6, e21878. [Google Scholar] [CrossRef] [Green Version]
- Shepelkova, G.; Evstifeev, V.; Majorov, K.; Bocharova, I.; Apt, A. Therapeutic Effect of Recombinant Mutated Interleukin 11 in the Mouse Model of Tuberculosis. J. Infect. Dis. 2016, 214, 496–501. [Google Scholar] [CrossRef] [Green Version]
- Pearl, J.E.; Khader, S.A.; Solache, A.; Gilmartin, L.; Ghilardi, N.; de Sauvage, F.; Cooper, A.M. IL-27 signaling compromises control of bacterial growth in mycobacteria-infected mice. J. Immunol. 2004, 173, 7490–7496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrado, E.; Fountain, J.J.; Liao, M.; Tighe, M.; Reiley, W.W.; Lai, R.P.; Meintjes, G.; Pearl, J.E.; Chen, X.; Zak, D.E.; et al. Interleukin 27R regulates CD4+ T cell phenotype and impacts protective immunity during Mycobacterium tuberculosis infection. J. Exp. Med. 2015, 212, 1449–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, G.; Dutta, R.K.; Khan, M.A.; Ishaq, M.; Sharma, K.; Malhotra, H.; Majumdar, S. IL-27 inhibits IFN-γ induced autophagy by concomitant induction of JAK/PI3 K/Akt/mTOR cascade and up-regulation of Mcl-1 in Mycobacterium tuberculosis H37Rv infected macrophages. Int. J. Biochem. Cell Biol. 2014, 55, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.M.; Jung, J.Y.; Nau, G.J. Interferon-γ, tumor necrosis factor, and interleukin-18 cooperate to control growth of Mycobacterium tuberculosis in human macrophages. Cytokine 2012, 60, 233–241. [Google Scholar] [CrossRef] [Green Version]
- Orlova, M.O.; Majorov, K.B.; Lyadova, I.V.; Eruslanov, E.B.; M’Lan, C.E.; Greenwood, C.M.; Schurr, E.; Apt, A.S. Constitutive differences in gene expression profiles parallel genetic patterns of susceptibility to tuberculosis in mice. Infect. Immun. 2006, 74, 3668–3672. [Google Scholar] [CrossRef] [Green Version]
- Lyadova, I.V.; Tsiganov, E.N.; Kapina, M.A.; Shepelkova, G.S.; Sosunov, V.V.; Radaeva, T.V.; Majorov, K.B.; Shmitova, N.S.; van den Ham, H.J.; Ganusov, V.V.; et al. In mice, tuberculosis progression is associated with intensive inflammatory response and the accumulation of Gr-1 cells in the lungs. PLoS ONE 2010, 5, e10469. [Google Scholar] [CrossRef]
- Underhill-Day, N.; McGovern, L.A.; Karpovich, N.; Mardon, H.J.; Barton, V.A.; Heath, J.K. Functional characterization of W147A: A high-affinity interleukin-11 antagonist. Endocrinology 2003, 144, 3406–3414. [Google Scholar] [CrossRef] [Green Version]
- Ernst, M.; Thiem, S.; Nguyen, P.M.; Eissmann, M.; Putoczki, T.L. Epithelial gp130/Stat3 functions: An intestinal signaling node in health and disease. Semin. Immunol. 2014, 26, 29–37. [Google Scholar] [CrossRef]
- Nahid, P.; Bliven-Sizemore, E.; Jarlsberg, L.G.; De Groote, M.A.; Johnson, J.L.; Muzanyi, G.; Engle, M.; Weiner, M.; Janjic, N.; Sterling, D.G.; et al. Aptamer-based proteomic signature of intensive phase treatment response in pulmonary tuberculosis. Tuberculosis (Edinb.) 2014, 94, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Larousserie, F.; Pflanz, S.; Coulomb-L’Herminé, A.; Brousse, N.; Kastelein, R.; Devergne, O. Expression of IL-27 in human Th1-associated granulomatous diseases. J. Pathol. 2004, 202, 164–171. [Google Scholar] [CrossRef]
- Wu, Y.B.; Ye, Z.J.; Qin, S.M.; Wu, C.; Chen, Y.Q.; Shi, H.Z. Combined detections of interleukin 27, interferon-γ, and adenosine deaminase in pleural effusion for diagnosis of tuberculous pleurisy. Chin. Med. J. (Engl.) 2013, 126, 3215–3221. [Google Scholar] [PubMed]
- Yang, W.B.; Liang, Q.L.; Ye, Z.J.; Niu, C.M.; Ma, W.L.; Xiong, X.Z.; Du, R.H.; Zhou, Q.; Zhang, J.C.; Shi, H.Z. Cell origins and diagnostic accuracy of interleukin 27 in pleural effusions. PLoS ONE 2012, 7, e40450. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Ye, Z.J.; Zhou, Q.; You, W.J.; Cui, A.; Wang, X.J.; Zhai, K.; Jin, X.G.; Tong, Z.H.; Shi, H.Z. IL-27 and IL-27-producing CD4+ T cells in human tuberculous pleural effusion. Tuberculosis (Edinb.) 2014, 94, 579–588. [Google Scholar] [CrossRef]
- Urazova, O.I.; Churina, E.G.; Hasanova, R.R.; Novitskiy, V.V.; Poletika, V.S. Association between polymorphisms of cytokine genes and secretion of IL-12p70, IL-18, and IL-27 by dendritic cells in patients with pulmonary tuberculosis. Tuberculosis (Edinb.) 2019, 115, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Saunders, B.M.; Britton, W.J. Life and death in the granuloma: Immunopathology of tuberculosis. Immunol. Cell Biol. 2007, 85, 103–111. [Google Scholar] [CrossRef]
- Reiley, W.W.; Shafiani, S.; Wittmer, S.T.; Tucker-Heard, G.; Moon, J.J.; Jenkins, M.K.; Urdahl, K.B.; Winslow, G.M.; Woodland, D.L. Distinct functions of antigen-specific CD4 T cells during murine Mycobacterium tuberculosis infection. Proc. Natl. Acad. Sci. USA 2010, 107, 19408–19413. [Google Scholar] [CrossRef] [Green Version]
- Aagaard, C.; Hoang, T.T.; Izzo, A.; Billeskov, R.; Troudt, J.; Arnett, K.; Keyser, A.; Elvang, T.; Andersen, P.; Dietrich, J. Protection and polyfunctional T cells induced by Ag85B-TB10.4/IC31 against Mycobacterium tuberculosis is highly dependent on the antigen dose. PLoS ONE 2009, 4, e5930. [Google Scholar] [CrossRef]
- Derrick, S.C.; Yabe, I.M.; Yang, A.; Morris, S.L. Vaccine-induced anti-tuberculosis protective immunity in mice correlates with the magnitude and quality of multifunctional CD4 T cells. Vaccine 2011, 29, 2902–2909. [Google Scholar] [CrossRef]
- Lindenstrøm, T.; Agger, E.M.; Korsholm, K.S.; Darrah, P.A.; Aagaard, C.; Seder, R.A.; Rosenkrands, I.; Andersen, P. Tuberculosis subunit vaccination provides long-term protective immunity characterized by multifunctional CD4 memory T cells. J. Immunol. 2009, 182, 8047–8055. [Google Scholar] [CrossRef]
- Beamer, G.L.; Flaherty, D.K.; Assogba, B.D.; Stromberg, P.; Gonzalez-Juarrero, M.; de Waal Malefyt, R.; Vesosky, B.; Turner, J. Interleukin-10 promotes Mycobacterium tuberculosis disease progression in CBA/J mice. J. Immunol. 2008, 181, 5545–5550. [Google Scholar] [CrossRef] [Green Version]
- Turner, J.; Gonzalez-Juarrero, M.; Ellis, D.L.; Basaraba, R.J.; Kipnis, A.; Orme, I.M.; Cooper, A.M. In Vivo IL-10 production reactivates chronic pulmonary tuberculosis in C57BL/6 mice. J. Immunol. 2002, 169, 6343–6351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzgerald, D.C.; Ciric, B.; Touil, T.; Harle, H.; Grammatikopolou, J.; Das Sarma, J.; Gran, B.; Zhang, G.X.; Rostami, A. Suppressive effect of IL-27 on encephalitogenic Th17 cells and the effector phase of experimental autoimmune encephalomyelitis. J. Immunol. 2007, 179, 3268–3275. [Google Scholar] [CrossRef] [Green Version]
- Hirahara, K.; Ghoreschi, K.; Yang, X.P.; Takahashi, H.; Laurence, A.; Vahedi, G.; Sciumè, G.; Hall, A.O.; Dupont, C.D.; Francisco, L.M.; et al. Interleukin-27 priming of T cells controls IL-17 production in trans via induction of the ligand PD-L1. Immunity 2012, 36, 1017–1030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, C.M.; O’Dee, D.; Hamilton, T.; Nau, G.J. Cytokines involved in interferon-gamma production by human macrophages. J. Innate Immun. 2010, 2, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.S.; Ghaffari, A.A.; Cheng, G. Lipopolysaccharide-mediated IL-10 transcriptional regulation requires sequential induction of type I IFNs and IL-27 in macrophages. J. Immunol. 2010, 185, 6599–6607. [Google Scholar] [CrossRef] [Green Version]
- Jung, J.Y.; Robinson, C.M. IL-12 and IL-27 regulate the phagolysosomal pathway in mycobacteria-infected human macrophages. Cell Commun. Signal. 2014, 12, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Kane, C.M.; Elkington, P.T.; Friedland, J.S. Monocyte-dependent oncostatin M and TNF-alpha synergize to stimulate unopposed matrix metalloproteinase-1/3 secretion from human lung fibroblasts in tuberculosis. Eur. J. Immunol. 2008, 38, 1321–1330. [Google Scholar] [CrossRef]
- Visse, R.; Nagase, H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: Structure, function, and biochemistry. Circ. Res. 2003, 92, 827–839. [Google Scholar] [CrossRef] [Green Version]
- Kübler, A.; Luna, B.; Larsson, C.; Ammerman, N.C.; Andrade, B.B.; Orandle, M.; Bock, K.W.; Xu, Z.; Bagci, U.; Mollura, D.J.; et al. Mycobacterium tuberculosis dysregulates MMP/TIMP balance to drive rapid cavitation and unrestrained bacterial proliferation. J. Pathol. 2015, 235, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Ou, Q.; Wu, J.; Zhang, B.; Shen, L.; Chen, S.; Weng, X.; Zhang, Y.; Zhang, W.; Shao, L. Potential diagnostic value of serum/pleural fluid IL-31 levels for tuberculous pleural effusion. Sci. Rep. 2016, 6, 20607. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Fan, Y.; Shen, D.; Yu, M.; Shi, L.; Ding, S.; Li, L. Characterization of cytokine profile to distinguish latent tuberculosis from active tuberculosis and healthy controls. Cytokine 2020, 135, 155218. [Google Scholar] [CrossRef] [PubMed]
- German Clinical Trials Register. A multi-centre, exploratory trial to assess the mechanisms of molecular activity, safety and tolerability of one dose level of FE 999301 by intravenous infusions in patients with active inflammatory bowel disease (IBD). German Clinical Trials Register 2016. Available online: http://www.drks.de/drks_web/navigate.do?navigationId=trial.HTML&TRIAL_ID=DRKS00010101 (accessed on 15 December 2020).
- I-Mab Biopharma HongKong Limited. Safety and efficacy of TJ301 IV in participants with active ulcerative colitis. U.S. Natl. Libary Med. ClinicalTrials.gov. 2020. Available online: https://www.clinicaltrials.gov/ct2/show/NCT03235752 (accessed on 15 December 2020).
- Elliott, M.J.; Maini, R.N.; Feldmann, M.; Long-Fox, A.; Charles, P.; Bijl, H.; Woody, J.N. Repeated therapy with monoclonal antibody to tumour necrosis factor alpha (cA2) in patients with rheumatoid arthritis. Lancet 1994, 344, 1125–1127. [Google Scholar] [CrossRef]
- Mohan, V.P.; Scanga, C.A.; Yu, K.; Scott, H.M.; Tanaka, K.E.; Tsang, E.; Tsai, M.M.; Flynn, J.L.; Chan, J. Effects of tumor necrosis factor alpha on host immune response in chronic persistent tuberculosis: Possible role for limiting pathology. Infect. Immun. 2001, 69, 1847–1855. [Google Scholar] [CrossRef] [Green Version]
- Scanga, C.A.; Mohan, V.P.; Joseph, H.; Yu, K.; Chan, J.; Flynn, J.L. Reactivation of latent tuberculosis: Variations on the Cornell murine model. Infect. Immun. 1999, 67, 4531–4538. [Google Scholar] [CrossRef] [Green Version]
- Afif, W.; Loftus, E.V., Jr. Safety profile of IBD therapeutics: Infectious risks. Gastroenterol. Clin. N. Am. 2009, 38, 691–709. [Google Scholar] [CrossRef]
- Solovic, I.; Sester, M.; Gomez-Reino, J.J.; Rieder, H.L.; Ehlers, S.; Milburn, H.J.; Kampmann, B.; Hellmich, B.; Groves, R.; Schreiber, S.; et al. The risk of tuberculosis related to tumour necrosis factor antagonist therapies: A TBNET consensus statement. Eur. Respir. J. 2010, 36, 1185–1206. [Google Scholar] [CrossRef] [Green Version]
- Stallmach, A.; Hagel, S.; Bruns, T. Adverse effects of biologics used for treating IBD. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 167–182. [Google Scholar] [CrossRef]
- Narazaki, M.; Tanaka, T.; Kishimoto, T. The role and therapeutic targeting of IL-6 in rheumatoid arthritis. Expert. Rev. Clin. Immunol. 2017, 13, 535–551. [Google Scholar] [CrossRef]
- Akioka, S. Interleukin-6 in juvenile idiopathic arthritis. Mod. Rheumatol. 2019, 29, 275–286. [Google Scholar] [CrossRef] [Green Version]
- Garbers, C.; Heink, S.; Korn, T.; Rose-John, S. Interleukin-6: Designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018, 17, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Kita, Y.; Kanamaru, N.; Hashimoto, S.; Uchiyama, Y.; Mihara, M.; Inoue, Y.; Ohsugi, Y.; Kishimoto, T.; Sakatani, M. Anti-IL-6 receptor antibody causes less promotion of tuberculosis infection than anti-TNF-alpha antibody in mice. Clin. Dev. Immunol. 2011, 2011, 404929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strand, V.; Ahadieh, S.; French, J.; Geier, J.; Krishnaswami, S.; Menon, S.; Checchio, T.; Tensfeldt, T.G.; Hoffman, E.; Riese, R.; et al. Systematic review and meta-analysis of serious infections with tofacitinib and biologic disease-modifying antirheumatic drug treatment in rheumatoid arthritis clinical trials. Arthritis Res. Ther. 2015, 17, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiff, M.H.; Kremer, J.M.; Jahreis, A.; Vernon, E.; Isaacs, J.D.; van Vollenhoven, R.F. Integrated safety in tocilizumab clinical trials. Arthritis Res. Ther. 2011, 13, R141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, T.; Harigai, M.; Inokuma, S.; Ishiguro, N.; Ryu, J.; Takeuchi, T.; Takei, S.; Tanaka, Y.; Ito, K.; Yamanaka, H. Postmarketing surveillance of tocilizumab for rheumatoid arthritis in Japan: Interim analysis of 3881 patients. Ann. Rheum. Dis. 2011, 70, 2148–2151. [Google Scholar] [CrossRef] [PubMed]
- Koike, T.; Harigai, M.; Inokuma, S.; Ishiguro, N.; Ryu, J.; Takeuchi, T.; Takei, S.; Tanaka, Y.; Sano, Y.; Yaguramaki, H.; et al. Effectiveness and safety of tocilizumab: Postmarketing surveillance of 7901 patients with rheumatoid arthritis in Japan. J. Rheumatol. 2014, 41, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Rose-John, S. IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the pro-inflammatory activities of IL-6. Int. J. Biol. Sci. 2012, 8, 1237–1247. [Google Scholar] [CrossRef]
- Prystaz, K.; Kaiser, K.; Kovtun, A.; Haffner-Luntzer, M.; Fischer, V.; Rapp, A.E.; Liedert, A.; Strauss, G.; Waetzig, G.H.; Rose-John, S.; et al. Distinct Effects of IL-6 Classic and Trans-Signaling in Bone Fracture Healing. Am. J. Pathol. 2018, 188, 474–490. [Google Scholar] [CrossRef] [Green Version]
- Hoge, J.; Yan, I.; Jänner, N.; Schumacher, V.; Chalaris, A.; Steinmetz, O.M.; Engel, D.R.; Scheller, J.; Rose-John, S.; Mittrücker, H.W. IL-6 controls the innate immune response against Listeria monocytogenes via classical IL-6 signaling. J. Immunol. 2013, 190, 703–711. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, S.H.E.; Dorhoi, A.; Hotchkiss, R.S.; Bartenschlager, R. Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug. Discov. 2018, 17, 35–56. [Google Scholar] [CrossRef]
- Ndlovu, H.; Marakalala, M.J. Granulomas and Inflammation: Host-Directed Therapies for Tuberculosis. Front. Immunol. 2016, 7, 434. [Google Scholar] [CrossRef] [Green Version]
- Palucci, I.; Delogu, G. Host Directed Therapies for Tuberculosis: Futures Strategies for an Ancient Disease. Chemotherapy 2018, 63, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Rao, M.; Parida, S.K.; Keshavjee, S.; Cassell, G.; Wallis, R.; Axelsson-Robertsson, R.; Doherty, M.; Andersson, J.; Maeurer, M. Inflammation and tuberculosis: Host-directed therapies. J. Intern. Med. 2015, 277, 373–387. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.S.; Kyambadde, P.; Johnson, J.L.; Horter, L.; Kittle, R.; Pohle, M.; Ducar, C.; Millard, M.; Mayanja-Kizza, H.; Whalen, C.; et al. A study of the safety, immunology, virology, and microbiology of adjunctive etanercept in HIV-1-associated tuberculosis. AIDS 2004, 18, 257–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aige, C.; Bishai, W.R. Penitentiary or penthouse condo: The tuberculous granuloma from the microbe’s point of view. Cell Microbiol. 2010, 12, 301–309. [Google Scholar] [CrossRef]
- Dietrich, C.; Candon, S.; Ruemmele, F.M.; Devergne, O. A soluble form of IL-27Ralpha is a natural IL-27 antagonist. J. Immunol. 2014, 192, 5382–5389. [Google Scholar] [CrossRef] [Green Version]
- Wirtz, S.; Tubbe, I.; Galle, P.R.; Schild, H.J.; Birkenbach, M.; Blumberg, R.S.; Neurath, M.F. Protection from lethal septic peritonitis by neutralizing the biological function of interleukin 27. J. Exp. Med. 2006, 203, 1875–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
The immune response to Mtb infection. After phagocytosis of Mtb (pink rods) by macrophages (Mϕ), cytokines such as TNF, IL-12 and IL-23 are released. IL-12 is critically important for the induction of TH1 cells, whereas IL-23 mediates the differentiation of IL-17A-producing TH17 cells. By activating various chemokines, IL-17A indirectly contributes to granuloma formation and the recruitment of IFNγ/TNF/IL-2-producing multifunctional T cells to the site of Mtb infection. IFNγ and TNF in turn synergistically activate effector mechanisms (EM) in infected Mϕ. Through this activation cascade, TH17 and TH1 cells mediate protection against Mtb infection. However, an elevated TH17 immune response can also have pathological consequences. Susceptibility to and the subsequent pathology of tuberculosis (TB) are mediated by the activity of neutrophils dependent on type 1 interferons (IFNs). Treg and Tr1 cells accumulate at the site of infection and restrict protective T cell responses.
Figure 1.
The immune response to Mtb infection. After phagocytosis of Mtb (pink rods) by macrophages (Mϕ), cytokines such as TNF, IL-12 and IL-23 are released. IL-12 is critically important for the induction of TH1 cells, whereas IL-23 mediates the differentiation of IL-17A-producing TH17 cells. By activating various chemokines, IL-17A indirectly contributes to granuloma formation and the recruitment of IFNγ/TNF/IL-2-producing multifunctional T cells to the site of Mtb infection. IFNγ and TNF in turn synergistically activate effector mechanisms (EM) in infected Mϕ. Through this activation cascade, TH17 and TH1 cells mediate protection against Mtb infection. However, an elevated TH17 immune response can also have pathological consequences. Susceptibility to and the subsequent pathology of tuberculosis (TB) are mediated by the activity of neutrophils dependent on type 1 interferons (IFNs). Treg and Tr1 cells accumulate at the site of infection and restrict protective T cell responses.

Figure 2.
Ligands and receptor complexes of the gp130 cytokines IL-6, sIL-6R/IL-6, IL-11 and IL-27. The gp130 cytokines IL-6 and IL-11 represent monomeric four-helix bundle cytokines. In contrast, sIL-6R/IL-6 and IL-27 are heterodimeric cytokines. Both comprise a secreted receptor α subunit and a four-helix bundle protein (sIL-6R/IL-6 and EBI3/IL-27p28, respectively). These cytokines all signal through receptor complexes that contain the gp130. The receptor complexes for IL-6 and IL-11 comprise a non-signaling receptor α subunit and a signaling gp130 homodimer. IL-6 signals through this receptor complex in cis. In contrast, a gp130 homodimer is required for sIL-6R/IL-6-mediated signaling. Accordingly, the receptor complex for IL-27 contains gp130 and the IL-27R subunit-α, which is structurally similar to gp130.
Figure 2.
Ligands and receptor complexes of the gp130 cytokines IL-6, sIL-6R/IL-6, IL-11 and IL-27. The gp130 cytokines IL-6 and IL-11 represent monomeric four-helix bundle cytokines. In contrast, sIL-6R/IL-6 and IL-27 are heterodimeric cytokines. Both comprise a secreted receptor α subunit and a four-helix bundle protein (sIL-6R/IL-6 and EBI3/IL-27p28, respectively). These cytokines all signal through receptor complexes that contain the gp130. The receptor complexes for IL-6 and IL-11 comprise a non-signaling receptor α subunit and a signaling gp130 homodimer. IL-6 signals through this receptor complex in cis. In contrast, a gp130 homodimer is required for sIL-6R/IL-6-mediated signaling. Accordingly, the receptor complex for IL-27 contains gp130 and the IL-27R subunit-α, which is structurally similar to gp130.

Figure 3.
The effect of gp130 cytokines on the immune response to an infection with Mtb. (A) While IL-6 and sIL-6R/IL-6 do not have any effect on TH1, TH17 and Treg, IL-6 is able to restrict the production of IL-12, IL-23 and type 1 interferons (IFNs) in activated macrophages. This can indirectly influence the activation of neutrophils and T cells but has no influence on the outcome of the infection. (B) IL-11 promotes the accumulation of pathogenic neutrophils and could thereby contribute to TB pathology. (C) IL-27 suppresses the parenchymal accumulation of PD-1-, CD69- and CD127-expressing self-renewing TH1 cells and, by limiting the expansion of IL-17A-producing TH17 cells, the recruitment of IFNγ/TNF/IL-2-producing multifunctional TH1 cells. TH1 and TH17 immune responses may also be indirectly diminished by IL-27 through the activation of Tr1 cells rather than of Treg. Importantly, IL-27 also has a suppressive effect on macrophage effector functions (EM) and on the release of pro-inflammatory cytokines by these cells. Even if IL-27 suppresses protective functions, it also prevents immunopathological consequences of an infection with Mtb by controlling an excessively strong TH17 immune response.
Figure 3.
The effect of gp130 cytokines on the immune response to an infection with Mtb. (A) While IL-6 and sIL-6R/IL-6 do not have any effect on TH1, TH17 and Treg, IL-6 is able to restrict the production of IL-12, IL-23 and type 1 interferons (IFNs) in activated macrophages. This can indirectly influence the activation of neutrophils and T cells but has no influence on the outcome of the infection. (B) IL-11 promotes the accumulation of pathogenic neutrophils and could thereby contribute to TB pathology. (C) IL-27 suppresses the parenchymal accumulation of PD-1-, CD69- and CD127-expressing self-renewing TH1 cells and, by limiting the expansion of IL-17A-producing TH17 cells, the recruitment of IFNγ/TNF/IL-2-producing multifunctional TH1 cells. TH1 and TH17 immune responses may also be indirectly diminished by IL-27 through the activation of Tr1 cells rather than of Treg. Importantly, IL-27 also has a suppressive effect on macrophage effector functions (EM) and on the release of pro-inflammatory cytokines by these cells. Even if IL-27 suppresses protective functions, it also prevents immunopathological consequences of an infection with Mtb by controlling an excessively strong TH17 immune response.


{kind=link}
{kind=link}
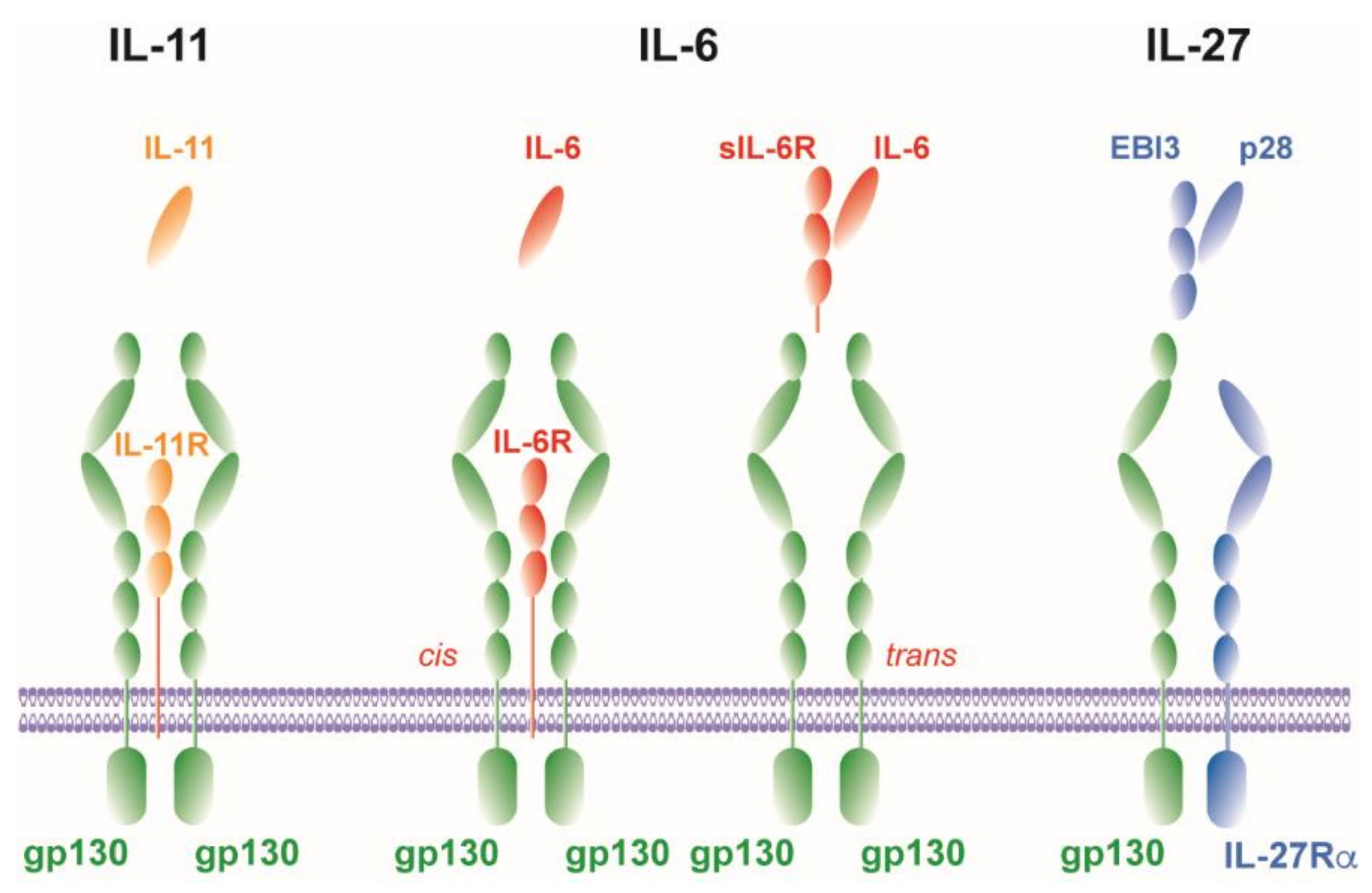{kind=link}
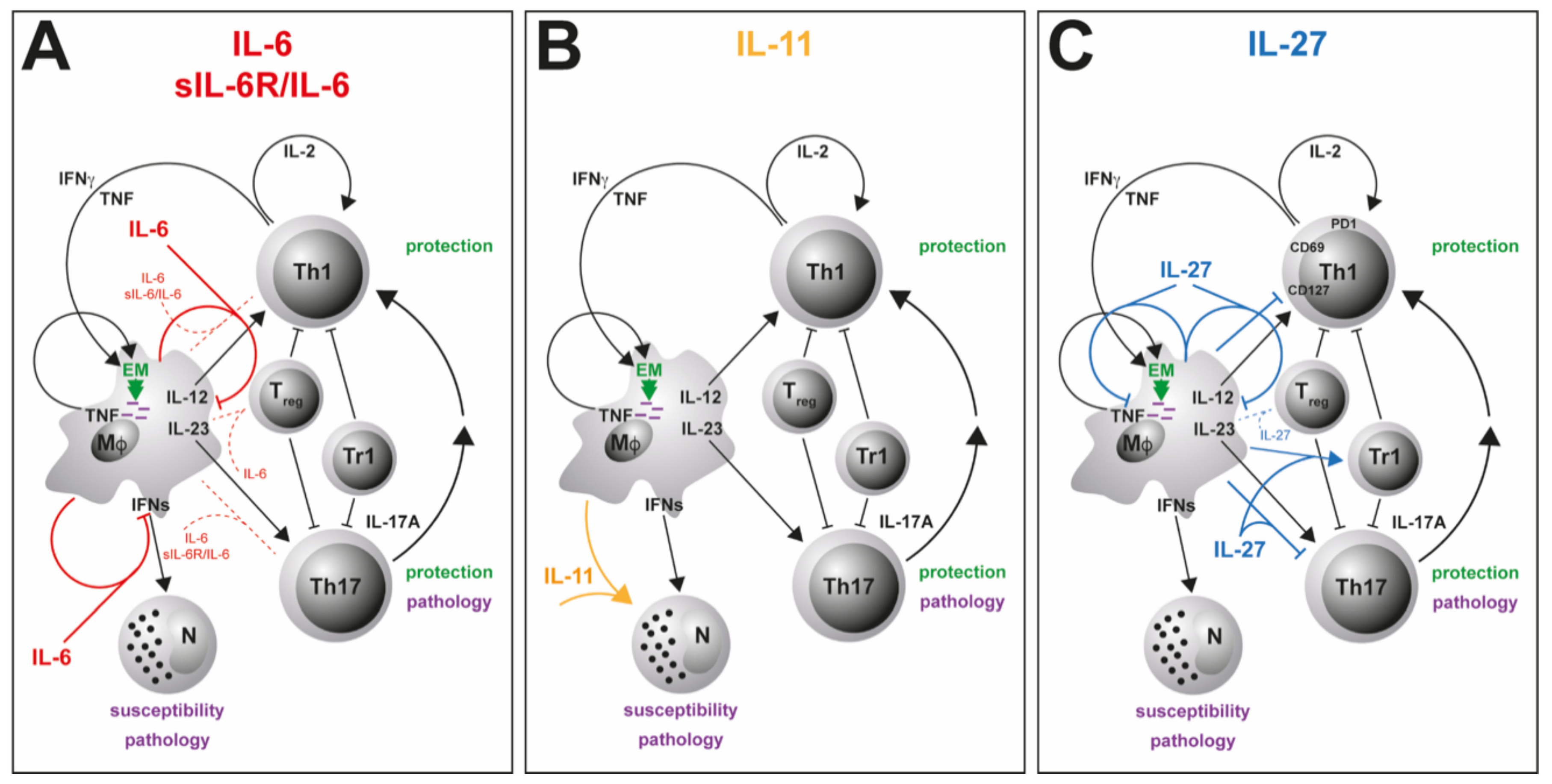{kind=link}
Table 1.
Cytokines and receptor complexes of the gp130 family.
| Cytokine | Receptor Complex | Reference 1 |
|---|---|---|
| IL-6 | IL-6Rα:gp130:gp130 | [50,51] |
| IL-11 | IL11Rα:gp130:gp130 | [45] |
| IL-27 (p28/EBI3) | IL27Rα (WSX-1):gp130 | [48] |
| IL-35 (p35/EBI3) | IL-12Rβ2:gp130 | [40] |
| gp130:gp130 | ||
| IL-39 (p19/EBI3) | IL-23R:gp130 | [53] |
| LIF | LIFR:gp130 | [42] |
| OSM | OSMR:gp130 | [46] |
| LIFR:gp130 | ||
| CNTF | LIFR:CNTFRα:gp130 | [44] |
| LIFR:IL-6Rα:gp130 | ||
| LIFR:sIL-6Rα:gp130 | ||
| CT-1 | LIFR:gp130 | [47,49] |
| LIFR:gp190:gp130 | ||
| CLC | LIFR:mCNTFRα:gp130 | [43] |
| NP | LIFR:CNTFRα:gp130 | [41] |
1 determined by different methodologies.
Table 2.
The role of gp130 in experimental TB 1.
| Effect 2 | Protection 3 | Pathology 4 | |
|---|---|---|---|
| IL-6 | → inflammation [139] | → [139] | → [139] |
| ↓ macrophages [82,142,143] | |||
| IL-6/sIL-6Rα | → inflammation [169] | → [93] | → [93] |
| IL-11 | ↑ inflammation [170,171] | ↓ [170,171] | ↑ [170,171] |
| ↑ macrophages [171] | |||
| ↑ neutrophils [170,171] | |||
| IL-27 | ↓ inflammation [17,18,172] | ↓ [17,18,172] | ↓ [17,18,172] |
| ↓ CD4 [17,173] | |||
| ↑ Tr1 [17] | |||
| ↓ TH17 [17] | |||
| ↓ macrophages [18,174,175] |
1 determined after aerosol infection of experimental mice; 2 pro- and anti-inflammatory functions measured by the overall cytokine response or the cell type-specific effect in lungs; 3 protection as determined by counts of colony-forming units in lungs; 4 pathology as determined by survival or body weight changes; ↑, increasing effect; ↓, decreasing effect; →, no effect.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ritter, K.; Rousseau, J.; Hölscher, C. The Role of gp130 Cytokines in Tuberculosis. Cells 2020, 9, 2695. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9122695
AMA Style
Ritter K, Rousseau J, Hölscher C. The Role of gp130 Cytokines in Tuberculosis. Cells. 2020; 9(12):2695. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9122695
Chicago/Turabian StyleRitter, Kristina, Jasmin Rousseau, and Christoph Hölscher. 2020. "The Role of gp130 Cytokines in Tuberculosis" Cells 9, no. 12: 2695. https://0-doi-org.brum.beds.ac.uk/10.3390/cells9122695
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.