The Application of Next Generation Sequencing in DNA Methylation Analysis


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
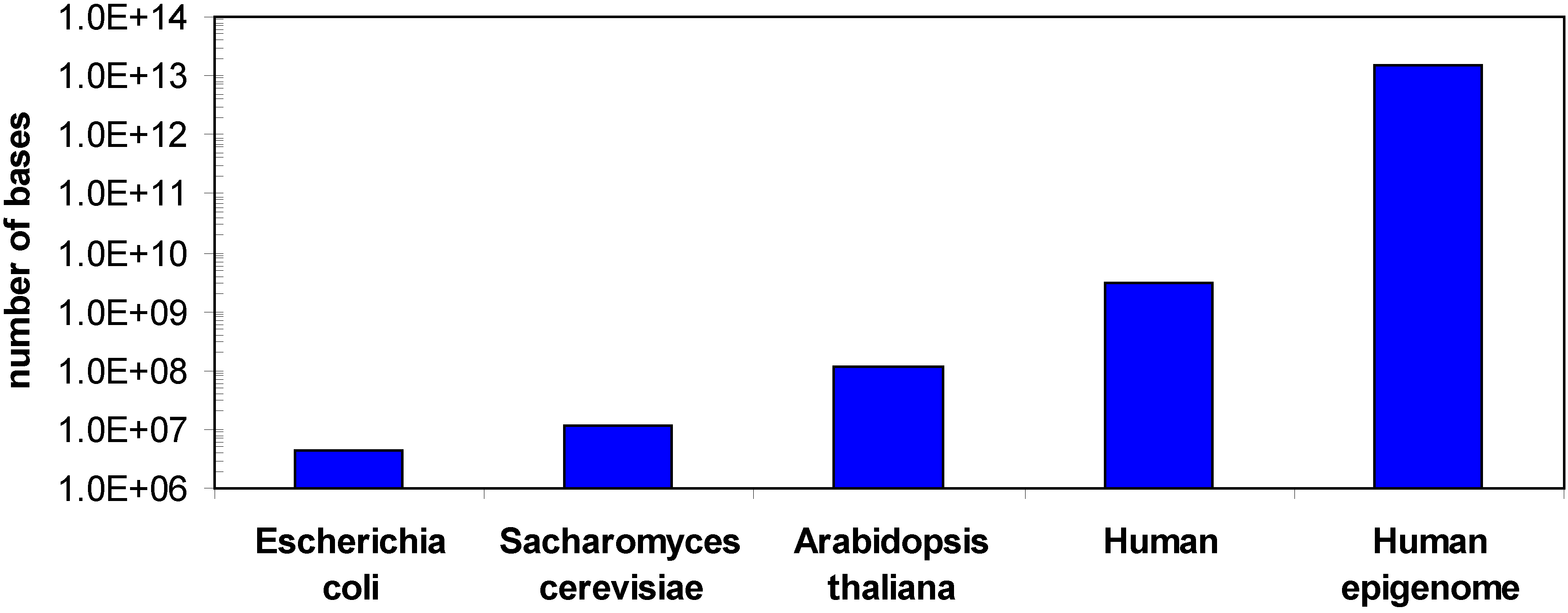
1.1. DNA methylation
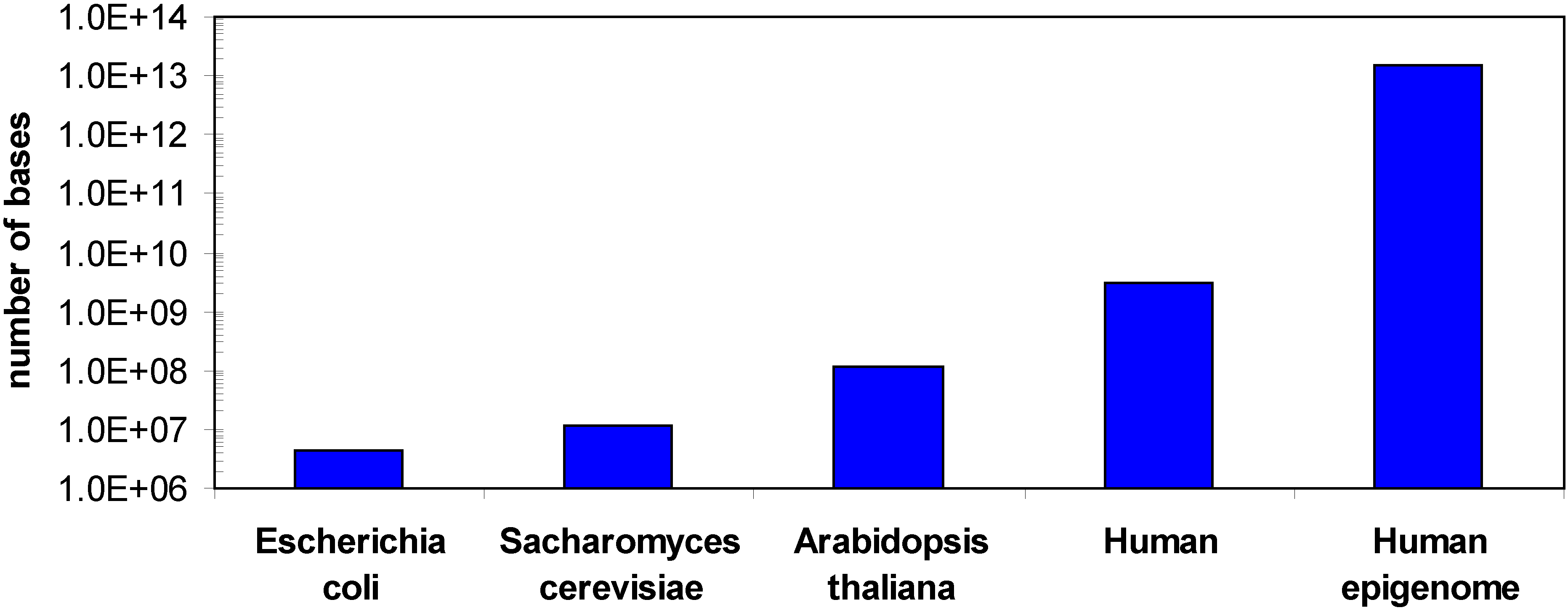
1.2. Methods for DNA methylation analysis
- 1)
- Methylation sensitive restriction enzyme digestion. The genomic DNA can be digested by methylation sensitive restriction enzymes like HpaII and McrBC to discriminate and/or enrich methylated or unmethylated DNA. The methods based on this approach are limited by providing methylation data only at the restriction enzyme recognition sites or adjacent regions.
- 2)
- Affinity purification. The methylated or unmethylated fractions of genomic DNA can be immunoprecipitated by using antibodies against methylated cytosine, methyl-CpG binding domains or other protein domains [32,33,34,35,36]. Using this method, the genome coverage is limited by the composition of the array for hybridization, and the distribution of the potential affinity targets in the genome, e.g. the density of methylated cytosines or CpG sites, which are unevenly distributed in the genome. The exact methylation state of individual CpG sites cannot be determined using this approach.
- 3)
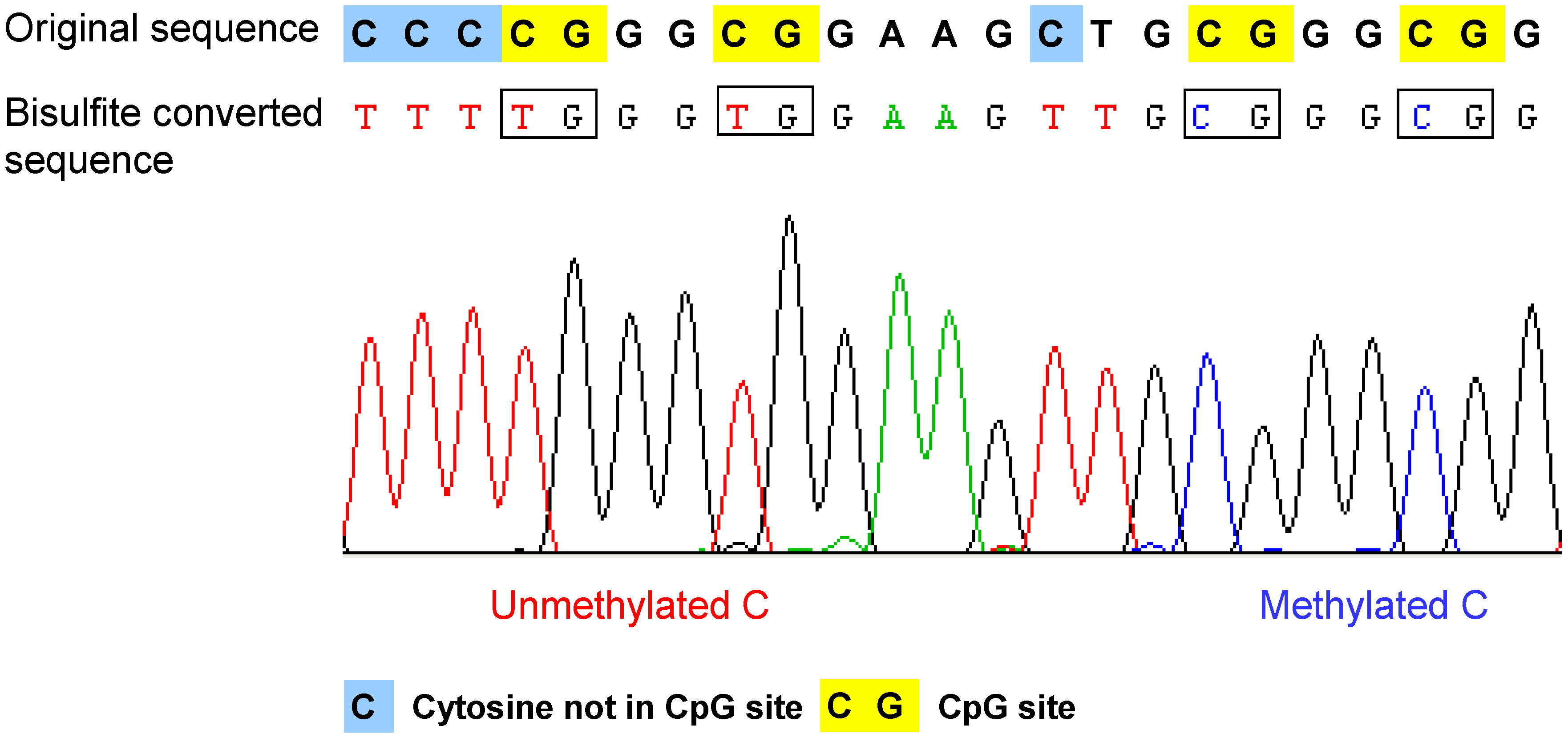
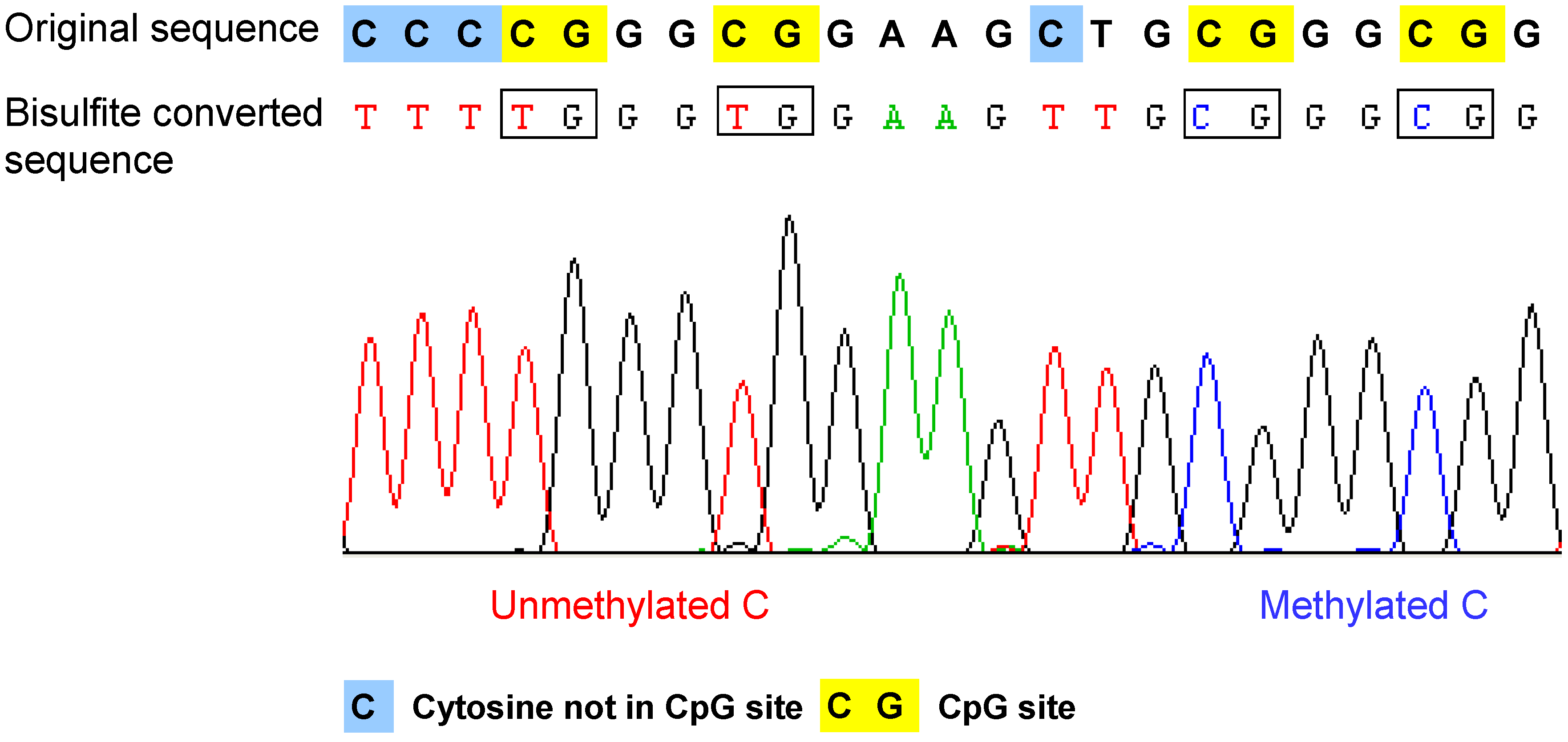
- Bisulfite conversion of DNA. The method is based on the selective deamination of cytosine but not 5-methylcytosine by treatment with sodium bisulfite [37,38]. Briefly, in the presence of sodium bisulfite, all the unmethylated cytosines are chemically converted to uracil, which is amplified as thymine during PCR. In contrast, the methylated cytosines are not converted, such that in the final sequencing result, the 5-methylcytosine will be still detected as cytosine. Therefore, after bisulfite conversion, the methylated and unmethylated cytosines can be distinguished according to the sequence changes (Figure 2). The bisulfite conversion efficiency is critical for the accuracy and the reliability of the results, especially for non-CpG methylation analysis. The incomplete conversion of unmethylated cytosine to uracil or inappropriate conversion of methylcytosine to thymine can cause over- or underestimination of the methylation level [39,40]. It is also noteworthy that the bisulfite conversion technique cannot be used to discriminate the methylated cytosine from 5-hydroxymethylcytosine (5hmC), which has been recently detected in the Purkinje neurons and embryonic stem cells [41,42]. The underlying reason is that after bisulfite conversion, 5hmC is not deaminated to thymine, but converted to cytosine 5-methylenesulfonate, which is read as cytosine during Sanger sequencing [43,44].
2. Application of next generation sequencing methods for DNA methylation analyses
2.1. 454 sequencing based DNA methylation analysis
2.2. Illumina Solexa sequencing based DNA methylation analysis
- 1)
- The reduced representation bisulfite sequencing (RRBS) method was developed and employed to map the methylation status of the murine genome in different cell lines [21]. The principle of the method is to reduce the complexity of genomic DNA by digesting the genomic DNA into small fragments using methylation insensitive restrictive enzymes like MspI, which recognizes CCGG sequence that are enriched in CGIs. After size selection for short fragments, the digested DNA was bisulfite converted and sequenced by Illumina Genome Analyzer. The library generated from MspI digestion was predicted to contain nearly 90% of CGIs in the mouse genome [58]. Recently, this method has been adapted for the DNA methylation profiling in the human genome, especially for the identification of the methylation changes in human clinical samples based on small amounts (30 ng) of genomic DNA [59]. The genomic coverage is not limited to the CGI (50%) and gene core promoters (65%), but other regions e.g. exons, 3’untranslated regions and repetitive elements are included as well [59].
- 2)
- Two similar approaches based on restriction enzymes digestion and Illumina sequencing, but without bisulfite conversion, have been developed for human methylome analysis. One is called Methyl-sensitive cut counting (MSCC) [60]. Here, the methylation sensitive restriction enzyme HpaII, which cuts unmethylated CCGG sequence, is used to digest the genomic DNA and the generated library is sequenced by Illumina sequencing to reveal unmethylated sites [60]. Another similar approach is called methyl-sequencing [61]. Here, the isoschizomers HpaII and MspI are both used to digest the genomic DNA. After adaptor ligation and size selection, Illumina sequencing is used to sequence the library. As HpaII only digest the unmethylated sites and MspI can digest the sites regardless of methylation, the methylation state of single CpG site can be determined by comparing the different reads number from the two libraries [61]. These methods are useful in reducing the complexity of genomic DNA by focusing on the CpG sites in specific sequence context, but on the other hand they cannot provide a methylation map at single base pair resolution.
- 3)
- Based on Illumina sequencing and bisulfite conversion, array capture [62] and Padlock capture [60,63] were developed for the target specific DNA methylation analysis. Hodges et al. developed a method called bisulfite capture based on hybrid selection techniques. An array containing the probes designed to be complementary to the sequence of interest is used to enrich the target sequences from bisulfite converted genomic DNA. Illumina sequencing was used to sequence the fragments eluted from the arrays. The padlock capture strategy was developed by different groups for DNA methylation analysis in target regions [60,63]. Padlock probes were designed to capture the bisulfite converted targeted sequences. Then, they are ligated to form a circularized single strand of DNA in the target region, which can be further amplified and sequenced. Thousands of probes can be designed for the targeted regions like CGIs and the captured fragments can be sequenced in a single run by Illumina Genome Analyzer. For both above mentioned technologies, the capture efficiency of the designed probes, can potentially affect the measurement of DNA methylation state.
- 4)
- Illumina sequencing of bisulfite converted DNA was also used to quantify the DNA methylation level in mouse primordial germ cells at lower coverage that allowed to analyse some global methylation properties like a strong global reduction of DNA methylation in primordial germ cells [26].
2.3. Other NGS sequencing based DNA methylation analysis
3. Discussion
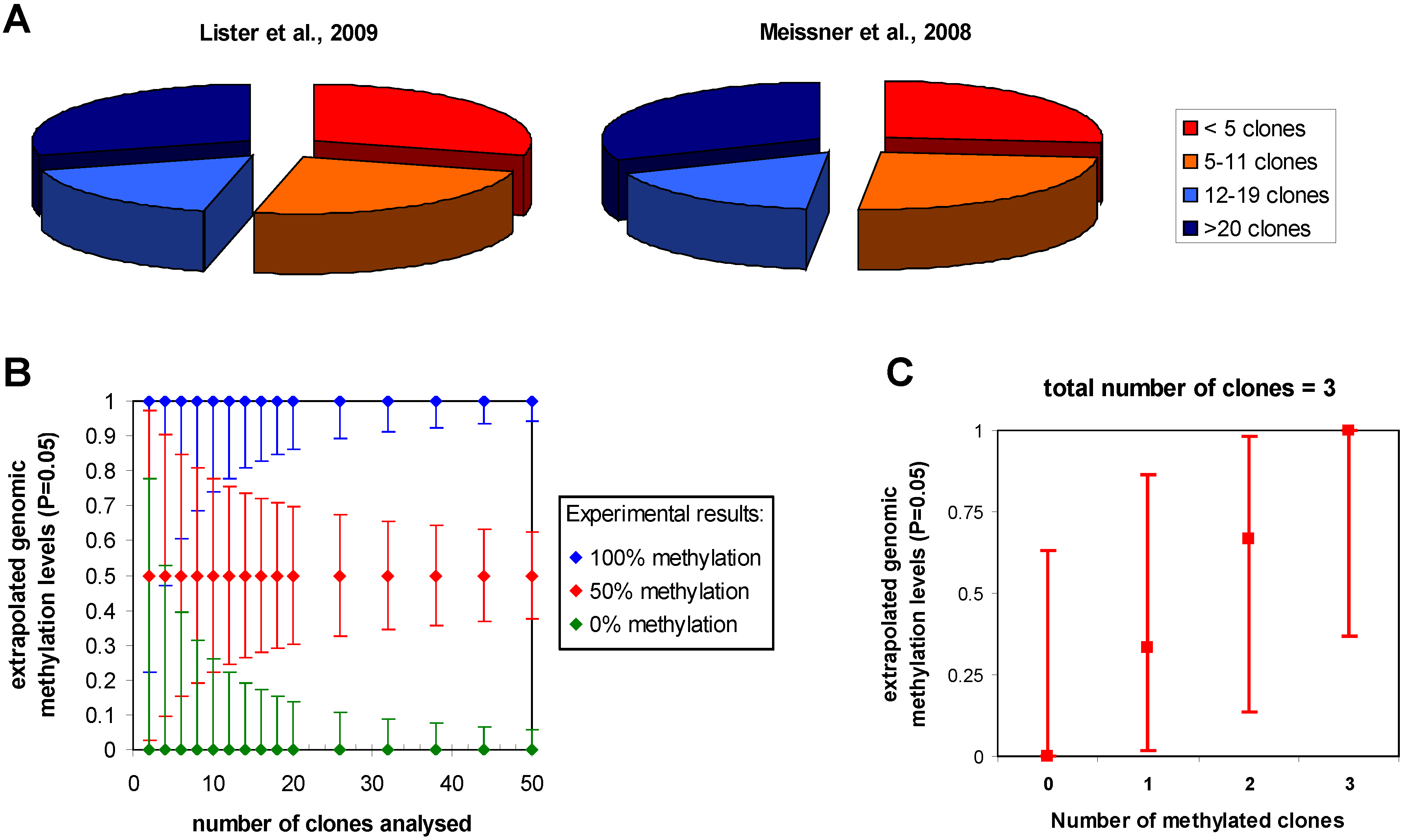
3.1. Statistical issues in bisulfite sequencing DNA methylation analysis
3.2. Methylation analysis in repeats
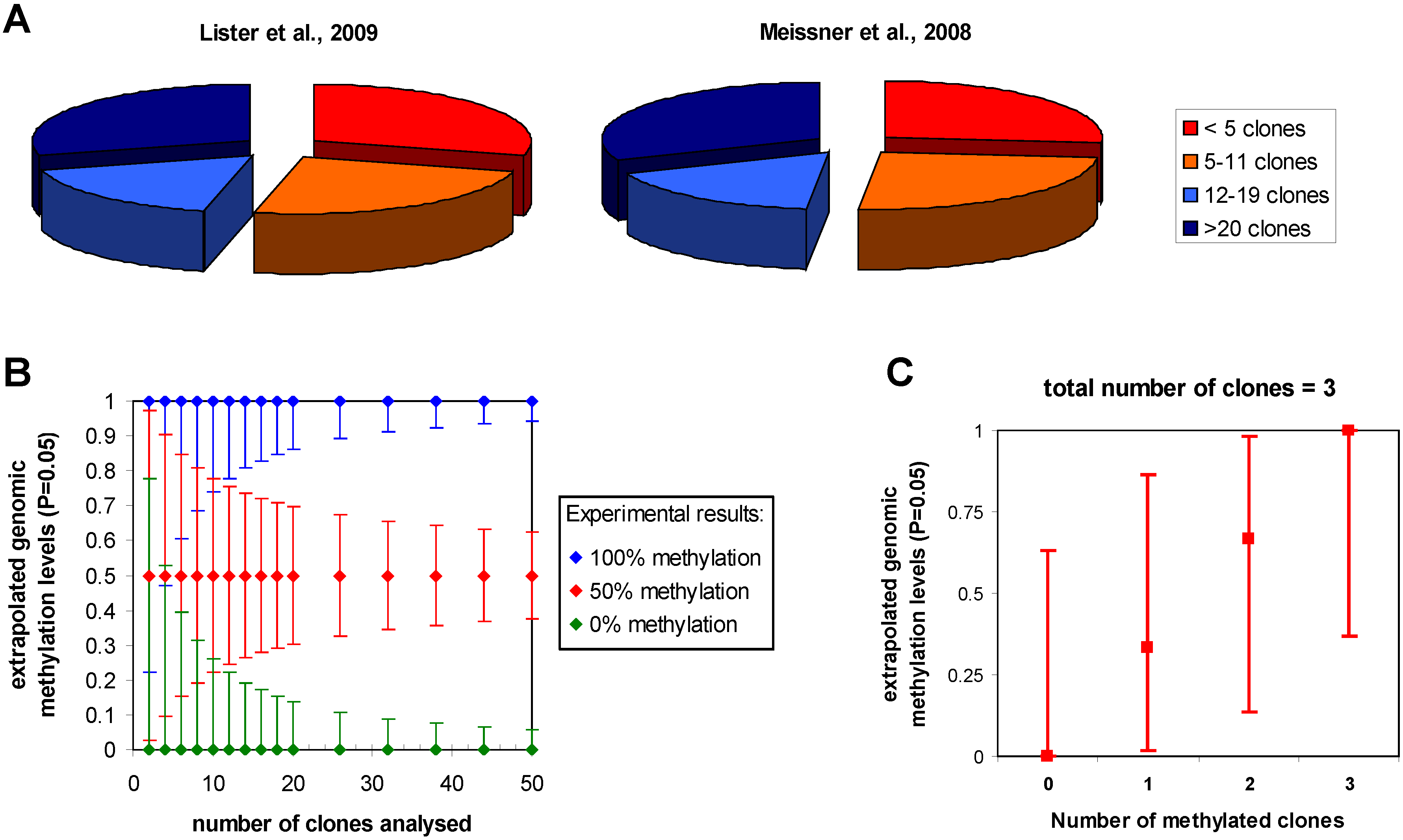
3.3. Clonal DNA amplification after bisulfite conversion
3.4. Detection of non-CpG methylation
- Analysis of mitochondrial DNA which is not methylated can be included for conversion control. However, this is not bound to chromatin and the DNA sequence is different so it may not be sufficient as control.
- Sequencing depths can be increased. If non-CpG methylation is happening and biologically relevant, it should be observed in several reads at the same cytosine residue. It is essential at the same time to use barcoded adaptors to exclude clonal amplification of the DNA templates giving rise to the independent reads with same non-CpG methylation patterns.
- Results can be reproduced with independent DNA preparations.
- In order to avoid conversion problems related to the primary sequence, recombinant DNA with same sequence can be added to the genomic DNA and analyzed.
- At key positions, methylation may be confirmed by methods not based on bisulfite conversion.
3.5. The challenge of data analysis
Acknowledgements
References and Notes
- Allis, C.D.; Jenuwein, T.; Reinberg, D. Epigenetics. 2007. [Google Scholar]
- Watson, J.D.; Baker, T.A.; Bell, S.P.; Gann, A.; Levine, M.; Losick, R. Molecular Biology of the Gene. 2008. [Google Scholar]
- Robertson, K.D. DNA methylation, methyltransferases, and cancer. Oncogene 2001, 20, 3139–3155. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef]
- Esteller, M. The necessity of a human epigenome project. Carcinogenesis 2006, 27, 1121–1125. [Google Scholar] [CrossRef]
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Klose, R.J.; Bird, A.P. Genomic DNA methylation: the mark and its mediators. Trends Biochem. Sci. 2006, 31, 89–97. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Laird, P.W. Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef]
- Illingworth, R.S.; Bird, A.P. CpG islands--'a rough guide'. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef]
- Rollins, R.A.; Haghighi, F.; Edwards, J.R.; Das, R.; Zhang, M. Q.; Ju, J.; Bestor, T.H. Large-scale structure of genomic methylation patterns. Genome Res. 2006, 16, 157–163. [Google Scholar]
- Saxonov, S.; Berg, P.; Brutlag, D.L. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 1412–1417. [Google Scholar] [CrossRef]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; Edsall, L.; Antosiewicz-Bourget, J.; Stewart, R.; Ruotti, V.; Millar, A.H.; Thomson, J.A.; Ren, B.; Ecker, J.R. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef]
- Laurent, L.; Wong, E.; Li, G.; Huynh, T.; Tsirigos, A.; Ong, C.T.; Low, H.M.; Kin Sung, K.W.; Rigoutsos, I.; Loring, J.; Wei, C.L. Dynamic changes in the human methylome during differentiation. Genome Res. 2010, 20, 320–331. [Google Scholar] [CrossRef]
- Goll, M.G.; Bestor, T.H. Eukaryotic cytosine methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Feinberg, A.P. Phenotypic plasticity and the epigenetics of human disease. Nature 2007, 447, 433–440. [Google Scholar] [CrossRef]
- Egger, G.; Liang, G.; Aparicio, A.; Jones, P.A. Epigenetics in human disease and prospects for epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; Gnirke, A.; Jaenisch, R.; Lander, E.S. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar]
- Zhang, Y.; Rohde, C.; Tierling, S.; Jurkowski, T.P.; Bock, C.; Santacruz, D.; Ragozin, S.; Reinhardt, R.; Groth, M.; Walter, J.; Jeltsch, A. DNA methylation analysis of chromosome 21 gene promoters at single base pair and single allele resolution. PLoS Genet. 2009, 5, e1000438. [Google Scholar] [CrossRef]
- Maegawa, S.; Hinkal, G.; Kim, H.S.; Shen, L.; Zhang, L.; Zhang, J.; Zhang, N.; Liang, S.; Donehower, L.A.; Issa, J.P. Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 2010, 20, 332–340. [Google Scholar] [CrossRef]
- Irizarry, R.A.; Ladd-Acosta, C.; Wen, B.; Wu, Z.; Montano, C.; Onyango, P.; Cui, H.; Gabo, K.; Rongione, M.; Webster, M.; Ji, H.; Potash, J.B.; Sabunciyan, S.; Feinberg, A. P. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009, 41, 178–186. [Google Scholar] [CrossRef]
- Straussman, R.; Nejman, D.; Roberts, D.; Steinfeld, I.; Blum, B.; Benvenisty, N.; Simon, I.; Yakhini, Z.; Cedar, H. Developmental programming of CpG island methylation profiles in the human genome. Nat. Struct. Mol. Biol. 2009, 16, 564–571. [Google Scholar] [CrossRef]
- Popp, C.; Dean, W.; Feng, S.; Cokus, S. J.; Andrews, S.; Pellegrini, M.; Jacobsen, S.E.; Reik, W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 2010, 463, 1101–1105. [Google Scholar] [CrossRef]
- Kerkel, K.; Spadola, A.; Yuan, E.; Kosek, J.; Jiang, L.; Hod, E.; Li, K.; Murty, V.V.; Schupf, N.; Vilain, E.; Morris, M.; Haghighi, F.; Tycko, B. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nat. Genet. 2008, 40, 904–908. [Google Scholar] [CrossRef]
- Zhang, Y.; Rohde, C.; Reinhardt, R.; Voelcker-Rehage, C.; Jeltsch, A. Non-imprinted allele-specific DNA methylation on human autosomes. Genome Biol. 2009, 10, R138. [Google Scholar] [CrossRef]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; Boix-Chornet, M.; Sanchez-Aguilera, A.; Ling, C.; Carlsson, E.; Poulsen, P.; Vaag, A.; Stephan, Z.; Spector, T.D.; Wu, Y.Z.; Plass, C.; Esteller, M. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 10604–10609. [Google Scholar] [CrossRef]
- Kaminsky, Z.A.; Tang, T.; Wang, S.C.; Ptak, C.; Oh, G.H.; Wong, A.H.; Feldcamp, L.A.; Virtanen, C.; Halfvarson, J.; Tysk, C.; McRae, A.F.; Visscher, P.M.; Montgomery, G.W.; Gottesman, II; Martin, N.G.; Petronis, A. DNA methylation profiles in monozygotic and dizygotic twins. Nat. Genet. 2009, 41, 240–245. [Google Scholar] [CrossRef]
- Beck, S.; Rakyan, V.K. The methylome: approaches for global DNA methylation profiling. Trends Genet. 2008, 24, 231–237. [Google Scholar] [CrossRef]
- Keshet, I.; Schlesinger, Y.; Farkash, S.; Rand, E.; Hecht, M.; Segal, E.; Pikarski, E.; Young, R.A.; Niveleau, A.; Cedar, H.; Simon, I. Evidence for an instructive mechanism of de novo methylation in cancer cells. Nat. Genet. 2006, 38, 149–153. [Google Scholar] [CrossRef]
- Rakyan, V.K.; Down, T.A.; Thorne, N.P.; Flicek, P.; Kulesha, E.; Graf, S.; Tomazou, E.M.; Backdahl, L.; Johnson, N.; Herberth, M.; Howe, K.L.; Jackson, D.K.; Miretti, M.M.; Fiegler, H.; Marioni, J.C.; Birney, E.; Hubbard, T.J.; Carter, N.P.; Tavare, S.; Beck, S. An integrated resource for genome-wide identification and analysis of human tissue-specific differentially methylated regions (tDMRs). Genome Res. 2008, 18, 1518–1529. [Google Scholar] [CrossRef]
- Weber, M.; Davies, J.J.; Wittig, D.; Oakeley, E.J.; Haase, M.; Lam, W.L.; Schubeler, D. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat. Genet. 2005, 37, 853–862. [Google Scholar] [CrossRef]
- Weber, M.; Hellmann, I.; Stadler, M.B.; Ramos, L.; Paabo, S.; Rebhan, M.; Schubeler, D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007, 39, 457–466. [Google Scholar] [CrossRef]
- Illingworth, R.; Kerr, A.; Desousa, D.; Jorgensen, H.; Ellis, P.; Stalker, J.; Jackson, D.; Clee, C.; Plumb, R.; Rogers, J.; Humphray, S.; Cox, T.; Langford, C.; Bird, A. A novel CpG island set identifies tissue-specific methylation at developmental gene loci. PLoS Biol. 2008, 6, e22. [Google Scholar] [CrossRef]
- Clark, S.J.; Harrison, J.; Paul, C.L.; Frommer, M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994, 22, 2990–2997. [Google Scholar] [CrossRef]
- Frommer, M.; McDonald, L.; Millar, D.; Collis, C.; Watt, F.; Grigg, G.; Molloy, P.; Paul, C. A Genomic Sequencing Protocol that Yields a Positive Display of 5-Methylcytosine Residues in Individual DNA Strands. Proc. Natl. Acad. Sci. U.S.A. 1992, 89, 1827–1831. [Google Scholar]
- Grunau, C.; Clark, S.J.; Rosenthal, A. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001, 29, E65–65. [Google Scholar] [CrossRef]
- Genereux, D.P.; Johnson, W.C.; Burden, A.F.; Stoger, R.; Laird, C.D. Errors in the bisulfite conversion of DNA: modulating inappropriate- and failed-conversion frequencies. Nucleic Acids Res. 2008, 36, e150. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; Rao, A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Huang, Y.; Pastor, W.A.; Shen, Y.; Tahiliani, M.; Liu, D.R.; Rao, A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One 2010, 5, e8888. [Google Scholar]
- Jin, S.G.; Kadam, S.; Pfeifer, G.P. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res. 2010. [Google Scholar]
- Costello, J.F.; Fruhwald, M.C.; Smiraglia, D.J.; Rush, L.J.; Robertson, G.P.; Gao, X.; Wright, F.A.; Feramisco, J.D.; Peltomaki, P.; Lang, J.C.; Schuller, D.E.; Yu, L.; Bloomfield, C.D.; Caligiuri, M.A.; Yates, A.; Nishikawa, R.; Su Huang, H.; Petrelli, N. J.; Zhang, X.; O'Dorisio, M.S.; Held, W.A.; Cavenee, W.K.; Plass, C. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. Nat. Genet. 2000, 24, 132–138. [Google Scholar] [CrossRef]
- Zhang, D.; Bai, Y.; Ge, Q.; Qiao, Y.; Wang, Y.; Chen, Z.; Lu, Z. Microarray-based molecular margin methylation pattern analysis in colorectal carcinoma. Anal Biochem 2006, 355, 117–124. [Google Scholar] [CrossRef]
- Hurd, P.J.; Nelson, C.J. Advantages of next-generation sequencing versus the microarray in epigenetic research. Brief Funct. Genomic Proteomic 2009, 8, 174–183. [Google Scholar] [CrossRef]
- Herman, J.G.; Graff, J.R.; Myohanen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 9821–9826. [Google Scholar] [CrossRef]
- Xiong, Z.; Laird, P.W. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997, 25, 2532–2534. [Google Scholar] [CrossRef]
- Voelkerding, K.V.; Dames, S.A.; Durtschi, J. D. Next-generation sequencing: from basic research to diagnostics. Clin. Chem. 2009, 55, 641–658. [Google Scholar] [CrossRef]
- Metzker, M.L. Sequencing technologies - the next generation. Nat Rev Genet 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Schilling, E.; El Chartouni, C.; Rehli, M. Allele-specific DNA methylation in mouse strains is mainly determined by cis-acting sequences. Genome Res. 2009, 19, 2028–2035. [Google Scholar] [CrossRef]
- Taylor, K.H.; Kramer, R.S.; Davis, J.W.; Guo, J.; Duff, D.J.; Xu, D.; Caldwell, C.W.; Shi, H. Ultradeep bisulfite sequencing analysis of DNA methylation patterns in multiple gene promoters by 454 sequencing. Cancer Res. 2007, 67, 8511–8518. [Google Scholar] [CrossRef]
- Zeschnigk, M.; Martin, M.; Betzl, G.; Kalbe, A.; Sirsch, C.; Buiting, K.; Gross, S.; Fritzilas, E.; Frey, B.; Rahmann, S.; Horsthemke, B. Massive parallel bisulfite sequencing of CG-rich DNA fragments reveals that methylation of many X-chromosomal CpG islands in female blood DNA is incomplete. Hum. Mol. Genet. 2009, 18, 1439–1448. [Google Scholar] [CrossRef]
- Korshunova, Y.; Maloney, R.K.; Lakey, N.; Citek, R.W.; Bacher, B.; Budiman, A.; Ordway, J.M.; McCombie, W.R.; Leon, J.; Jeddeloh, J.A.; McPherson, J.D. Massively parallel bisulphite pyrosequencing reveals the molecular complexity of breast cancer-associated cytosine-methylation patterns obtained from tissue and serum DNA. Genome Res. 2008, 18, 19–29. [Google Scholar]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef]
- Lister, R.; O'Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef]
- Smith, Z.D.; Gu, H.; Bock, C.; Gnirke, A.; Meissner, A. High-throughput bisulfite sequencing in mammalian genomes. Methods 2009, 48, 226–232. [Google Scholar] [CrossRef]
- Gu, H.; Bock, C.; Mikkelsen, T.S.; Jager, N.; Smith, Z.D.; Tomazou, E.; Gnirke, A.; Lander, E.S.; Meissner, A. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat. Methods 2010, 7, 133–136. [Google Scholar] [CrossRef] [Green Version]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.H.; LeProust, E.M.; Park, I.H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef]
- Brunner, A.L.; Johnson, D.S.; Kim, S.W.; Valouev, A.; Reddy, T.E.; Neff, N.F.; Anton, E.; Medina, C.; Nguyen, L.; Chiao, E.; Oyolu, C.B.; Schroth, G.P.; Absher, D.M.; Baker, J.C.; Myers, R.M. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res. 2009, 19, 1044–1056. [Google Scholar] [CrossRef]
- Hodges, E.; Smith, A.D.; Kendall, J.; Xuan, Z.; Ravi, K.; Rooks, M.; Zhang, M.Q.; Ye, K.; Bhattacharjee, A.; Brizuela, L.; McCombie, W.R.; Wigler, M.; Hannon, G.J.; Hicks, J.B. High definition profiling of mammalian DNA methylation by array capture and single molecule bisulfite sequencing. Genome Res. 2009, 19, 1593–1605. [Google Scholar] [CrossRef]
- Deng, J.; Shoemaker, R.; Xie, B.; Gore, A.; LeProust, E.M.; Antosiewicz-Bourget, J.; Egli, D.; Maherali, N.; Park, I.H.; Yu, J.; Daley, G.Q.; Eggan, K.; Hochedlinger, K.; Thomson, J.; Wang, W.; Gao, Y.; Zhang, K. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat. Biotechnol. 2009, 27, 353–360. [Google Scholar] [CrossRef]
- Bormann Chung, C.A.; Boyd, V.L.; McKernan, K.J.; Fu, Y.; Monighetti, C.; Peckham, H.E.; Barker, M. Whole methylome analysis by ultra-deep sequencing using two-base encoding. PLoS One 2010, 5, e9320. [Google Scholar]
- Miner, B.E.; Stoger, R.J.; Burden, A.F.; Laird, C.D.; Hansen, R.S. Molecular barcodes detect redundancy and contamination in hairpin-bisulfite PCR. Nucleic Acids Res. 2004, 32, e135. [Google Scholar] [CrossRef]
- Lefrancois, P.; Euskirchen, G.M.; Auerbach, R.K.; Rozowsky, J.; Gibson, T.; Yellman, C.M.; Gerstein, M.; Snyder, M. Efficient yeast ChIP-Seq using multiplex short-read DNA sequencing. BMC Genomics 2009, 10, 37. [Google Scholar] [CrossRef]
- Baird, N.A.; Etter, P.D.; Atwood, T.S.; Currey, M.C.; Shiver, A.L.; Lewis, Z.A.; Selker, E.U.; Cresko, W.A.; Johnson, E.A. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 2008, 3, e3376. [Google Scholar] [CrossRef]
- Zhang, Y.; Rohde, C.; Tierling, S.; Stamerjohanns, H.; Reinhardt, R.; Walter, J.; Jeltsch, A. DNA methylation analysis by bisulfite conversion, cloning, and sequencing of individual clones. Methods Mol. Biol. 2009, 507, 177–187. [Google Scholar] [CrossRef]
- Rohde, C.; Zhang, Y.; Reinhardt, R.; Jeltsch, A. BISMA - Fast and accurate bisulfite sequencing data analysis of individual clones from unique and repetitive sequences. BMC Bioinformatics 2010, 11, 230. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Clarke, J.; Wu, H.C.; Jayasinghe, L.; Patel, A.; Reid, S.; Bayley, H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009, 4, 265–270. [Google Scholar] [CrossRef]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 2010, 7, 461–465. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, Y.; Jeltsch, A. The Application of Next Generation Sequencing in DNA Methylation Analysis. Genes 2010, 1, 85-101. https://0-doi-org.brum.beds.ac.uk/10.3390/genes1010085
Zhang Y, Jeltsch A. The Application of Next Generation Sequencing in DNA Methylation Analysis. Genes. 2010; 1(1):85-101. https://0-doi-org.brum.beds.ac.uk/10.3390/genes1010085
Chicago/Turabian StyleZhang, Yingying, and Albert Jeltsch. 2010. "The Application of Next Generation Sequencing in DNA Methylation Analysis" Genes 1, no. 1: 85-101. https://0-doi-org.brum.beds.ac.uk/10.3390/genes1010085