454-Pyrosequencing: A Molecular Battiscope for Freshwater Viral Ecology

Abstract
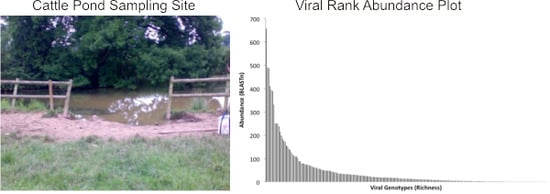
:
1. Introduction
2. Results and Discussion
2.1. Preparation of DNA

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2.2. Metagenomic library output
2.3. Post-pyrosequencing analyses
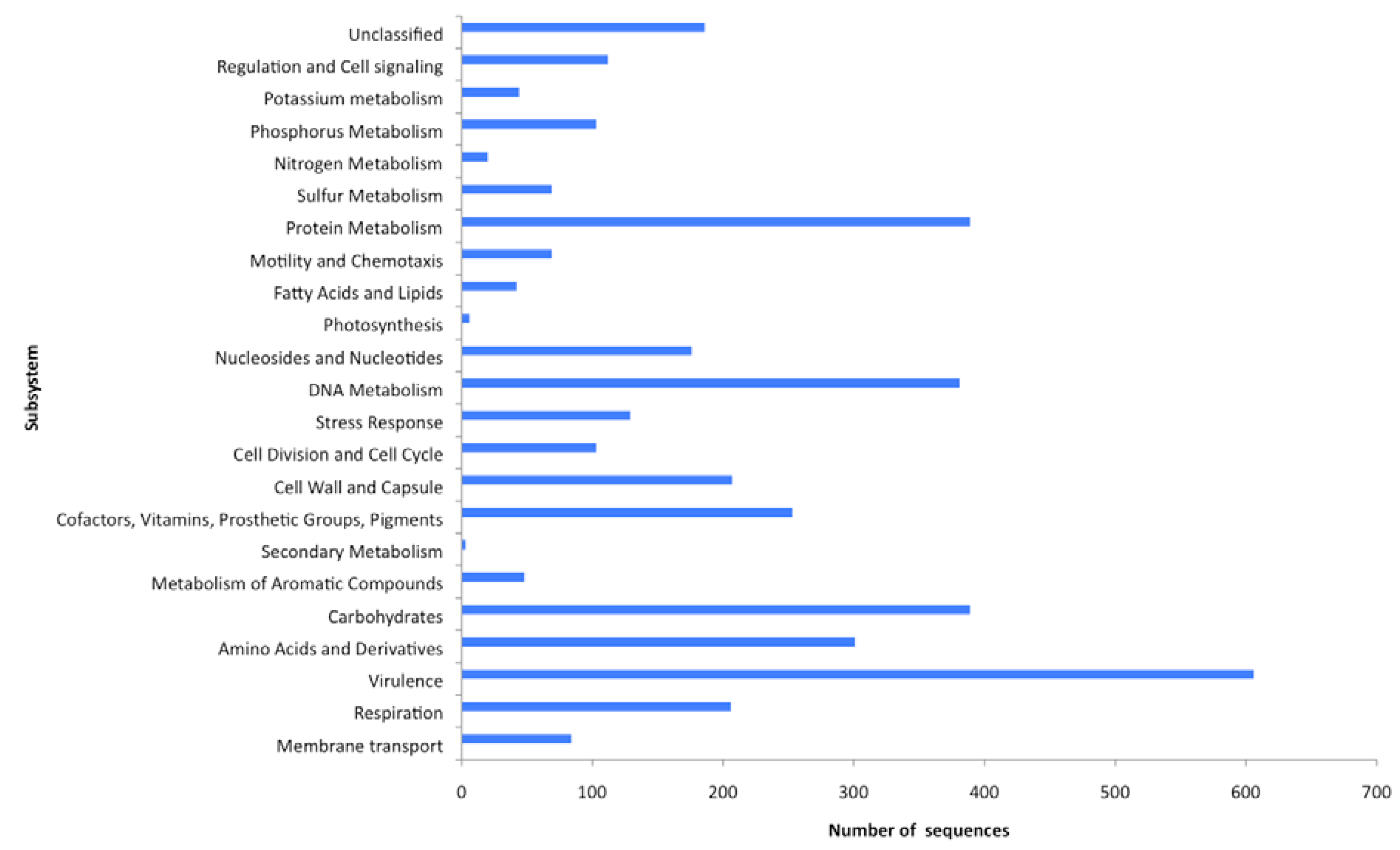
2.4. Functional analyses of the virome
| SSU rRNA homology (%) RDP/ERDP/GREENGENES | Protein Based (%)(SEED database*) | |
|---|---|---|
| Archaea | 0 | 1.99 |
| Bacteria | 0.02 | 41.71 |
| Eukaryota | 0 | 3.10 |
| Virus | 0 | 51.25 |
| Other | 0 | 0.02 |


2.5. Population Diversity and Structure


3. Experimental Section
3.1. Viral DNA Extraction
3.2. End-Point polymerase chain reaction
3.3. pyrosequencing
3.4. Bioinformatical analyses
3.5. MG-RAST
4. Conclusions
Acknowledgments
Supplementary Files
{kind=link}
References and Notes
- Edwards, R.A.; Rohwer, F. Viral metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Weinbauer, M.G.; Rassoulzadegan, F. Are viruses driving microbial diversification and diversity? Environ. Microbiol. 2004, 6, 1–11. [Google Scholar] [CrossRef]
- Thingstad, E.V. Elements of a theory for the mechanisms controlling abundance, diversity and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol. Oceanogr. 2000, 45, 1320–1328. [Google Scholar] [CrossRef]
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Angly, F.E.; Felts, B.; Breitbart, M.; Salamon, P.; Edwards, R.A.; Carlson, C.; Chan, A.M.; Haynes, M.; Kelley, S.; Liu, H.; et al. The marine viromes of four oceanic regions. PLoS Biol. 2006, 4, 368. [Google Scholar] [CrossRef]
- Brussow, H.; Hendrix, R.W. Phage genomics: small is beautiful. Cell 2002, 108, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.-W.; DuBow, M.S. Viruses of prokaryotes, Volume 1: General properties of bacteriophages; 1987; CRC Press: Boca Raton, FL, USA. [Google Scholar]
- Rohwer, F. Global phage diversity. Cell 2003, 113, 141. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Edwards, R. The Phage Proteomic Tree: a genome-based taxonomy for phage. J. Bacteriol. 2002, 184, 4529–4535. [Google Scholar] [CrossRef] [PubMed]
- Thurber, R.V. Current insights into phage biodiversity and biogeography. Curr. Opin. Microbiol. 2009, 12, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Rooks, D.J.; Yan, Y.; McDonald, J.E.; Woodward, M.J.; McCarthy, A.J.; Allison, H.E. Development and validation of a qPCR-based method for quantifying Shiga toxin-encoding and other lambdoid bacteriophages. Environ. Microbiol. 2010, 12, 1194–1204. [Google Scholar] [CrossRef] [PubMed]
- Short, C.M.; Suttle, C.A. Nearly identical bacteriophage structural gene sequences are widely distributed in both marine and freshwater environments. Appl. Environ. Microbiol. 2005, 71, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Hall, N. Advanced sequencing technologies and their wider impact in microbiology. J. Exp. Biol. 2007, 210, 1518–1525. [Google Scholar] [CrossRef] [PubMed]
- Beja, O.; Spudich, E.N.; Spudich, J.L.; Leclerc, M.; DeLong, E.F. Proteorhodopsin phototrophy in the ocean. Nature 2001, 411, 786–789. [Google Scholar] [CrossRef] [PubMed]
- Beja, O.; Suzuki, M.T.; Koonin, E.V.; Aravind, L.; Hadd, A.; Nguyen, L.P.; Villacorta, R.; Amjadi, M.; Garrigues, C.; Jovanovich, S.B.; et al. Construction and analysis of bacterial artificial chromosome libraries from a marine microbial assemblage. Environ. Microbiol. 2000, 2, 516–529. [Google Scholar] [CrossRef]
- Martiny, J.B.; Bohannan, B.J.; Brown, J.H.; Colwell, R.K.; Fuhrman, J.A.; Green, J.L.; Horner-Devine, M.C.; Kane, M.; Krumins, J.A.; Kuske, C.R.; et al. Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 2006, 4, 102–112. [Google Scholar] [CrossRef]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Steward, G.F.; Montiel, J.L.; Azam, F. Genome size distributions indicate variability and similarities among marine viral assemblages from diverse environments. Limnol. Oceanogr. 2000, 45, 1697–1706. [Google Scholar] [CrossRef]
- Warren, R.A. Modified bases in bacteriophage DNAs. Annu Rev Microbiol 1980, 34, 137–158. [Google Scholar] [PubMed]
- Wang, I.N.; Smith, D.L.; Young, R. Holins: the protein clocks of bacteriophage infections. Annu. Rev. Microbiol. 2000, 54, 799–825. [Google Scholar] [CrossRef] [PubMed]
- Delwart, E.L. Viral metagenomics. Rev. Med. Virol. 2007, 17, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Vega Thurber, R.; Willner-Hall, D.; Rodriguez-Mueller, B.; Desnues, C.; Edwards, R.A.; Angly, F.; Dinsdale, E.; Kelly, L.; Rohwer, F. Metagenomic analysis of stressed coral holobionts. Environ. Microbiol. 2009, 11, 2148–2163. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, T.; Patterson, M.; Richardson, P.M.; Wommack, K.E.; Young, M.; Mead, D. Assembly of viral metagenomes from yellowstone hot springs. Appl. Environ. Microbiol. 2008, 74, 4164–4174. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bueno, A.; Tamames, J.; Velazquez, D.; Moya, A.; Quesada, A.; Alcami, A. High diversity of the viral community from an Antarctic lake. Science 2009, 326, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, F.; Prangishvili, D.; Lindell, D. Roles of viruses in the environment. Environ. Microbiol. 2009, 11, 2771–2774. [Google Scholar] [CrossRef] [PubMed]
- Dinsdale, E.A.; Edwards, R.A.; Hall, D.; Angly, F.; Breitbart, M.; Brulc, J.M.; Furlan, M.; Desnues, C.; Haynes, M.; Li, L.; et al. Functional metagenomic profiling of nine biomes. Nature 2008, 452, 629–632. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Nilsson, C.; Lim, Y.W.; Ruan, Y.; Breitbart, M. Metagenomic analysis of viruses in reclaimed water. Environ. Microbiol. 2009, 11, 2806–2820. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Felts, B.; Kelley, S.; Mahaffy, J.M.; Nulton, J.; Salamon, P.; Rohwer, F. Diversity and population structure of a near-shore marine-sediment viral community. Proc. Biol. Sci. 2004, 271, 565–574. [Google Scholar] [CrossRef]
- Breitbart, M.; Salamon, P.; Andresen, B.; Mahaffy, J.M.; Segall, A.M.; Mead, D.; Azam, F.; Rohwer, F. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 14250–14255. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Hewson, I.; Felts, B.; Mahaffy, J.M.; Nulton, J.; Salamon, P.; Rohwer, F. Metagenomic analyses of an uncultured viral community from human feces. J. Bacteriol. 2003, 185, 6220–6223. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.; Wilson, W. Aquatic virus diversity accessed through omic techniques: a route map to function. Curr. Opin. Microbiol. 2008, 11, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Thurber, R.V.; Haynes, M.; Breitbart, M.; Wegley, L.; Rohwer, F. Laboratory procedures to generate viral metagenomes. Nat. Protoc. 2009, 4, 470–483. [Google Scholar] [CrossRef] [PubMed]
- McDonald, J.E.; Smith, D.L.; Fogg, P.C.; McCarthy, A.J.; Allison, H.E. High-throughput method for rapid induction of prophages from lysogens and its application in the study of Shiga toxin-encoding Escherichia coli strains. Appl. Environ. Microbiol. 2010, 76, 2360–2365. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.E.; Wright, E.J.; Hart, C.A.; Bennett, M.; French, N.P. Intermittent and persistent shedding of Escherichia coli O157 in cohorts of naturally infected calves. J. Appl. Microbiol. 2004, 97, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.L.; Wareing, B.M.; Fogg, P.C.; Riley, L.M.; Spencer, M.; Cox, M.J.; Saunders, J.R.; McCarthy, A.J.; Allison, H.E. Multilocus characterization scheme for Shiga toxin-encoding bacteriophages. Appl. Environ. Microbiol. 2007, 73, 8032–8040. [Google Scholar] [CrossRef] [PubMed]
- Edwards, U.; Rogall, T.; Blocker, H.; Emde, M.; Bottger, E.C. Isolation and direct complete nucleotide determination of entire genes. Characterization of a gene coding for 16S ribosomal RNA. Nucleic Acids Res. 1989, 17, 7843–7853. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Burns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics . In PCR protocols: a guide to methods and applications; Innis, M.A., Gelfand, D.H., Sninsky, J.I., White, T.J., Eds.; 1990; Academic Press: San Diego, CA, USA. [Google Scholar]
- Martinsohn, J.T.; Radman, M.; Petit, M.A. The lambda red proteins promote efficient recombination between diverged sequences: implications for bacteriophage genome mosaicism. PLoS Genet. 2008, 4, 1000065. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Auch, A.F.; Qi, J.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Paarmann, D.; D'Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.Y.; Cohoon, M.; de Crecy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R.; et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, J.R.; Chai, B.; Farris, R.J.; Wang, Q.; Kulam-Syed-Mohideen, A.S.; McGarrell, D.M.; Bandela, A.M.; Cardenas, E.; Garrity, G.M.; Tiedje, J.M. The ribosomal database project (RDP-II): introducing myRDP space and quality controlled public data. Nucleic Acids Res. 2007, 35, D169–D172. [Google Scholar] [CrossRef]
- Wuyts, J.; Perriere, G.; Van De Peer, Y. The European ribosomal RNA database. Nucleic Acids Res. 2004, 32, D101–D103. [Google Scholar] [CrossRef]
- Beumer, A.; Robinson, J.B. A broad-host-range, generalized transducing phage (SN-T) acquires 16S rRNA genes from different genera of bacteria. Appl. Environ. Microbiol. 2005, 71, 8301–8304. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Rohwer, F. Method for discovering novel DNA viruses in blood using viral particle selection and shotgun sequencing. Biotechniques 2005, 39, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.A.; Rodriguez-Brito, B.; Wegley, L.; Haynes, M.; Breitbart, M.; Peterson, D.M.; Saar, M.O.; Alexander, S.; Alexander Jr., E.C.; Rohwer, F. Using pyrosequencing to shed light on deep mine microbial ecology. BMC Genomics 2006, 7, 57. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Breitbart, M.; Nulton, J.; Salamon, P.; Lozupone, C.; Jones, R.; Robeson, M.; Edwards, R.A.; Felts, B.; Rayhawk, S.; et al. Metagenomic and small-subunit rRNA analyses reveal the genetic diversity of bacteria, archaea, fungi, and viruses in soil. Appl. Environ. Microbiol. 2007, 73, 7059–7066. [Google Scholar] [CrossRef] [PubMed]
- Wegley, L.; Edwards, R.; Rodriguez-Brito, B.; Liu, H.; Rohwer, F. Metagenomic analysis of the microbial community associated with the coral Porites astreoides. Environ. Microbiol. 2007, 9, 2707–2719. [Google Scholar] [CrossRef]
- Allison, H. Stx-phages: drivers and mediators of the evolution of STEC and STEC-like pathogens. Future Microbiol. 2007, 2, 165–174. [Google Scholar] [CrossRef]
- Mann, N.H.; Clokie, M.R.; Millard, A.; Cook, A.; Wilson, W.H.; Wheatley, P.J.; Letarov, A.; Krisch, H.M. The genome of S-PM2, a "photosynthetic" T4-type bacteriophage that infects marine Synechococcus strains. J. Bacteriol. 2005, 187, 3188–3200. [Google Scholar] [CrossRef] [PubMed]
- Stachowicz, J.J. Mutualism, facilitation, and the structure of ecological communities. Bioscience 2001, 51, 235–246. [Google Scholar] [CrossRef]
- Davis, B.M.; Waldor, M.K. Filamentous phages linked to virulence of Vibrio cholerae. Curr. Opin. Microbiol. 2003, 6, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Foster, S.D.; Dunstan, P.K. The analysis of biodiversity using rank abundance distributions . Biometrics 2009. [Google Scholar]
- Ryan, K.; Ray, C. Sherris Medical Microbiology2004, 4th ed; McGraw Hill: London, UK. [Google Scholar]
- Demirkan, I.; Williams, H.F.; Dhawi, A.; Carter, S.D.; Winstanley, C.; Bruce, K.D.; Hart, C.A. Characterization of a spirochaete isolated from a case of bovine digital dermatitis. J. Appl. Microbiol. 2006, 101, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Del Médico Zajac, M.P.; Ladelfa, M.F.; Kotsias, F.; Muylkens, B.; Thiry, J.; Thiry, E.; Romera, S.A. Biology of bovine herpesvirus 5. Vet. J. 2010, 184, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Muylkens, B.; Thiry, J.; Kirten, P.; Schynts, F.; Thiry, E. Bovine herpesvirus 1 infection and infectious bovine rhinotracheitis. Vet. Res. 2007, 38, 181–209. [Google Scholar] [CrossRef] [PubMed]
- Engels, M.; Ackermann, M. Pathogenesis of ruminant herpesvirus infections. Vet. Microbiol. 1996, 53, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Van Etten, J.L.; Allen, M.J. The Phycodnaviridae: the story of how tiny giants rule the world. Curr. Top. Microbiol. Immunol. 2009, 328, 1–42. [Google Scholar] [PubMed]
- Jancovich, J.K.; Mao, J.; Chinchar, V.G.; Wyatt, C.; Case, S.T.; Kumar, S.; Valente, G.; Subramanian, S.; Davidson, E.W.; Collins, J. P.; et al. Genomic sequence of a ranavirus (family Iridoviridae) associated with salamander mortalities in North America. Virology 2003, 316, 90–103. [Google Scholar] [CrossRef] [PubMed]
- Essbauer, S.; Pfeffer, M.; Meyer, H. Zoonotic poxviruses. Vet. Microbiol. 2010, 140, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Rohrmann, R.D.; Robles, M.; Lopez de Haro, M.; Santos, A. Virial series for fluids of hard hyperspheres in odd dimensions. J. Chem. Phys. 2008, 129, 014510. [Google Scholar] [CrossRef] [PubMed]
- Thoen, C.; LoBue, P.; de Kantor, I. The importance of Mycobacterium bovis as a zoonosis. Vet. Microbiol. 2006, 112, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F. Bacteriophage genomics. Curr. Opin. Microbiol. 2008, 11, 447–453. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F.; Cresawn, S.G.; Hendrix, R.W. Comparative genomics of the mycobacteriophages: insights into bacteriophage evolution. Res. Microbiol. 2008, 159, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Pedulla, M.L.; Ford, M.E.; Houtz, J.M.; Karthikeyan, T.; Wadsworth, C.; Lewis, J.A.; Jacobs-Sera, D.; Falbo, J.; Gross, J.; Pannunzio, N.R.; et al. Origins of highly mosaic mycobacteriophage genomes. Cell 2003, 113, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.L.; James, C.E.; Sergeant, M.J.; Yaxian, Y.; Saunders, J.R.; McCarthy, A.J.; Allison, H.E. Short-tailed stx phages exploit the conserved YaeT protein to disseminate Shiga toxin genes among enterobacteria. J. Bacteriol. 2007, 189, 7223–7233. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: a Laboratory Manual, 2nd ed; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Margulies, M.; Egholm, M.; Altman, W.E.; Attiya, S.; Bader, J.S.; Bemben, L.A.; Berka, J.; Braverman, M.S.; Chen, Y.J.; Chen, Z.; et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 2005, 437, 376–380. [Google Scholar] [PubMed]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Share and Cite
Rooks, D.J.; Smith, D.L.; McDonald, J.E.; Woodward, M.J.; McCarthy, A.J.; Allison, H.E. 454-Pyrosequencing: A Molecular Battiscope for Freshwater Viral Ecology. Genes 2010, 1, 210-226. https://0-doi-org.brum.beds.ac.uk/10.3390/genes1020210
Rooks DJ, Smith DL, McDonald JE, Woodward MJ, McCarthy AJ, Allison HE. 454-Pyrosequencing: A Molecular Battiscope for Freshwater Viral Ecology. Genes. 2010; 1(2):210-226. https://0-doi-org.brum.beds.ac.uk/10.3390/genes1020210
Chicago/Turabian StyleRooks, David J., Darren L. Smith, James E. McDonald, Martin J. Woodward, Alan J. McCarthy, and Heather E. Allison. 2010. "454-Pyrosequencing: A Molecular Battiscope for Freshwater Viral Ecology" Genes 1, no. 2: 210-226. https://0-doi-org.brum.beds.ac.uk/10.3390/genes1020210