Comparative Analysis of the Complete Chloroplast Genome Sequences of Three Closely Related East-Asian Wild Roses (Rosa sect. Synstylae; Rosaceae)



Abstract
:1. Introduction
2. Materials and Methods
2.1. Taxon Sampling and Genome Sequencing
2.2. Chloroplast Genome Assembly and Annotation
2.3. Chloroplast Genome Comparative Analyses
2.4. Analysis of Simple Sequence Repeats
2.5. Phylogenetic Analysis
3. Results
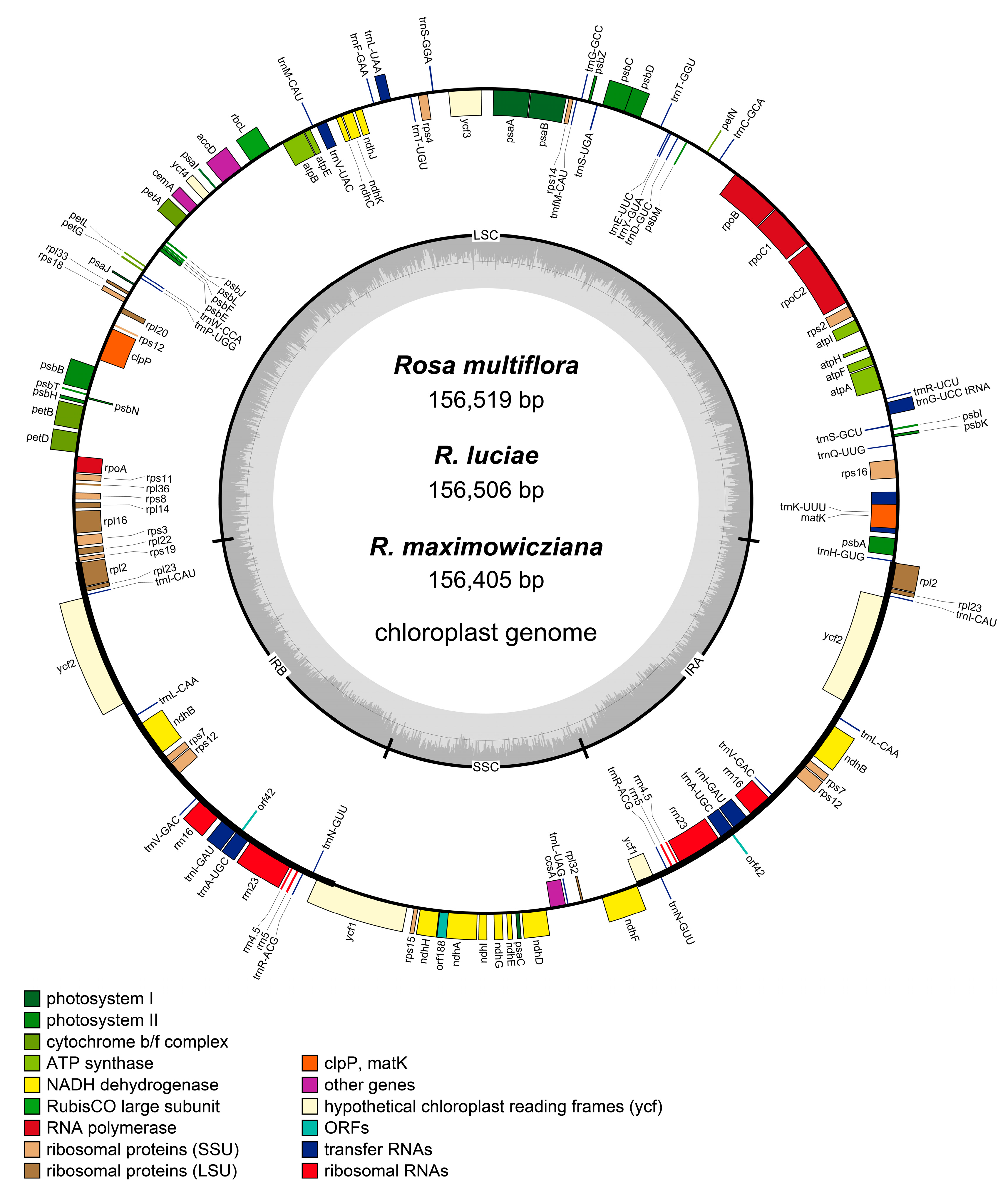
3.1. General Chloroplast Genome Features
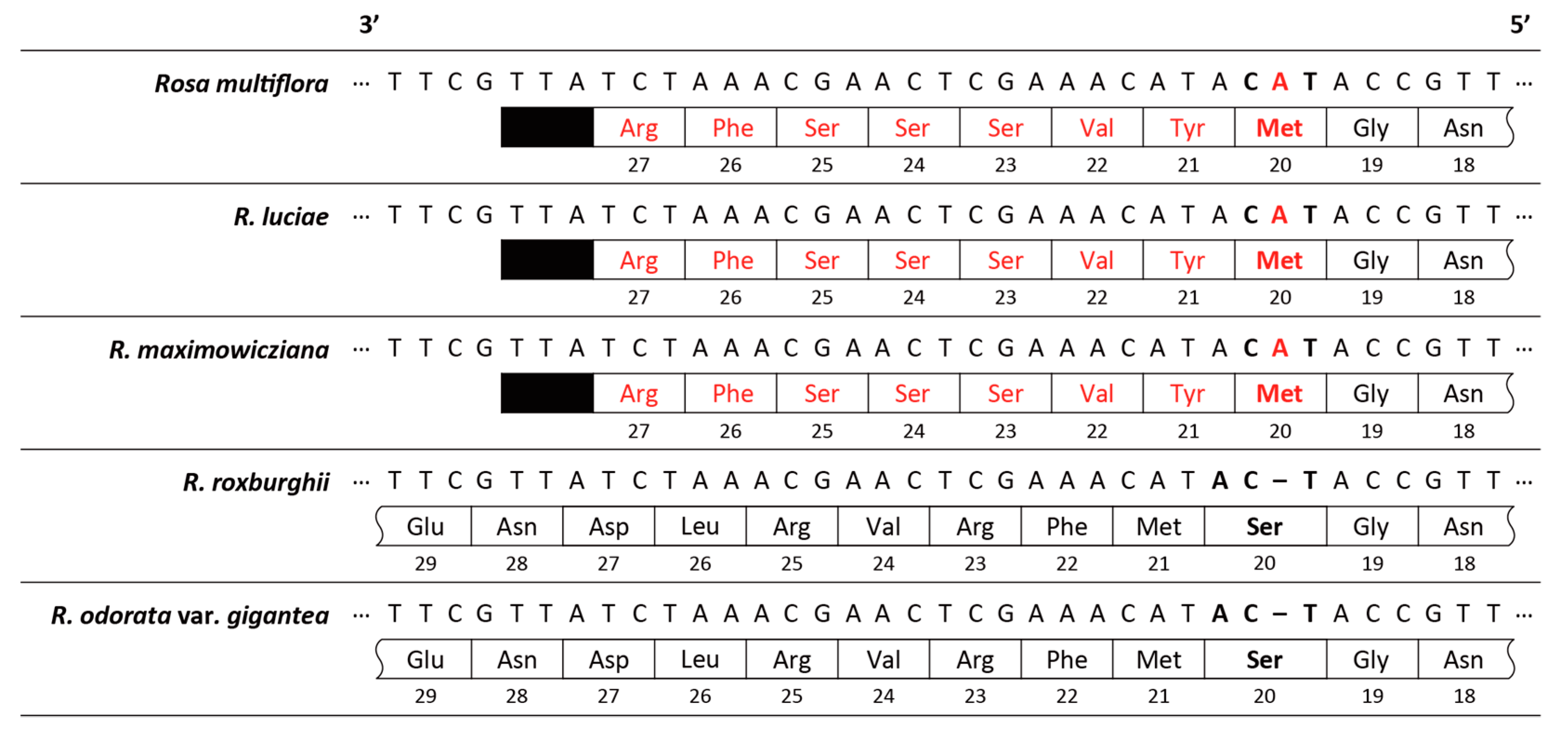
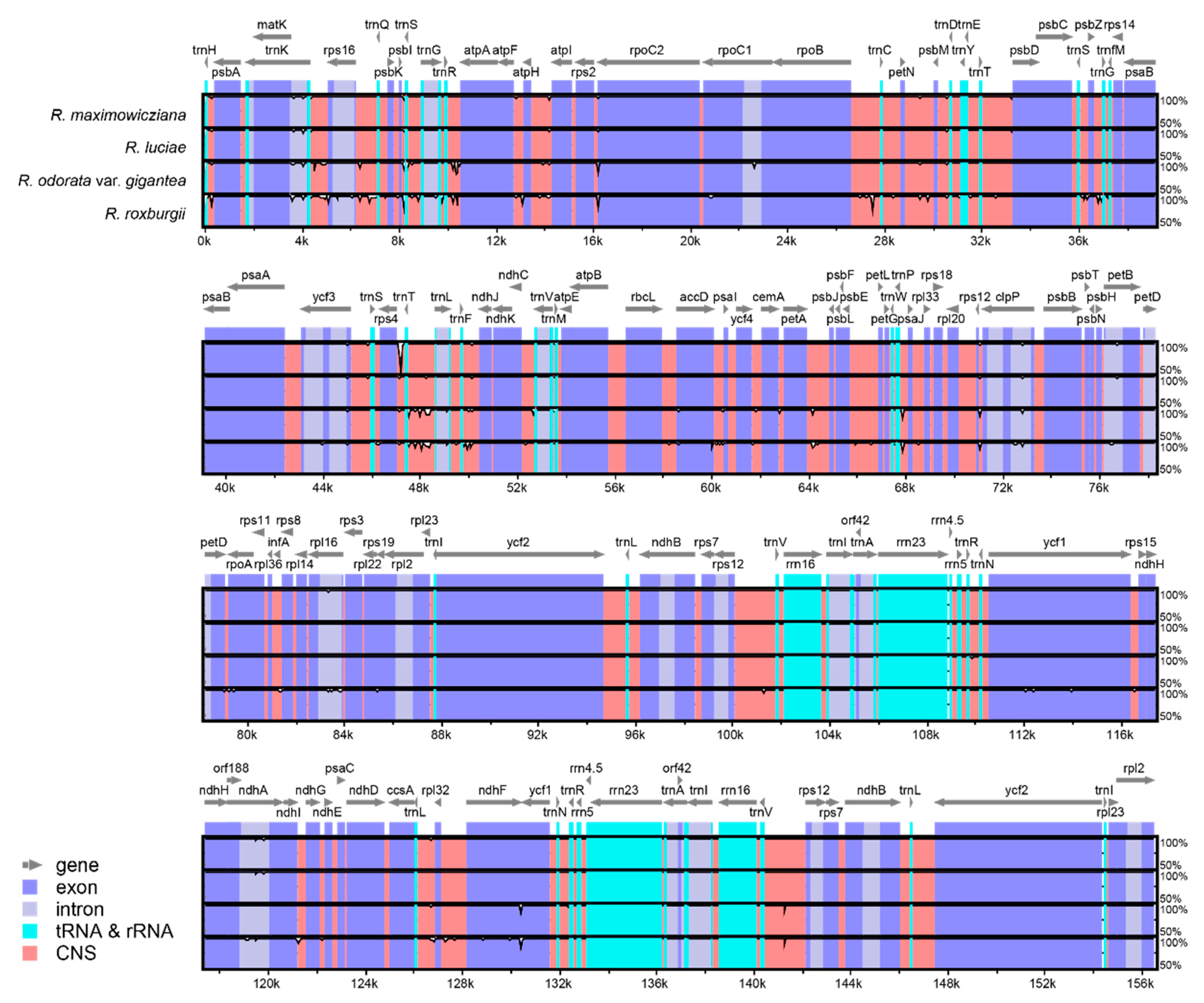
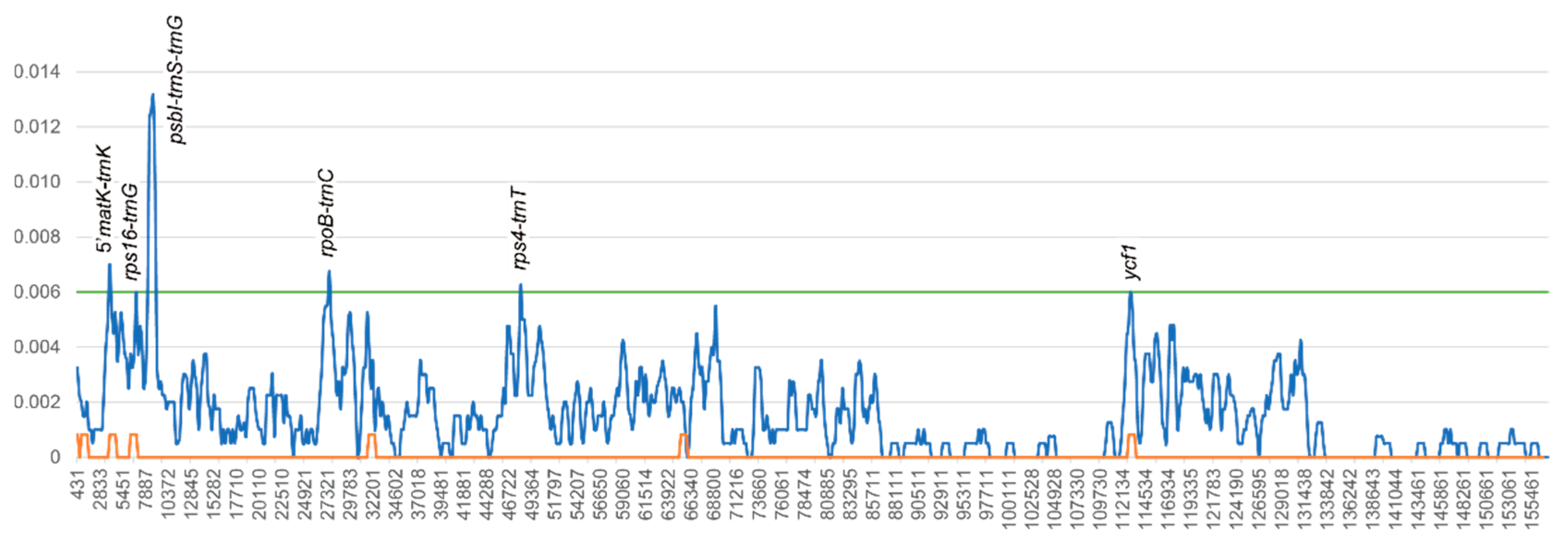
3.2. Interspecific Comparative Analyses
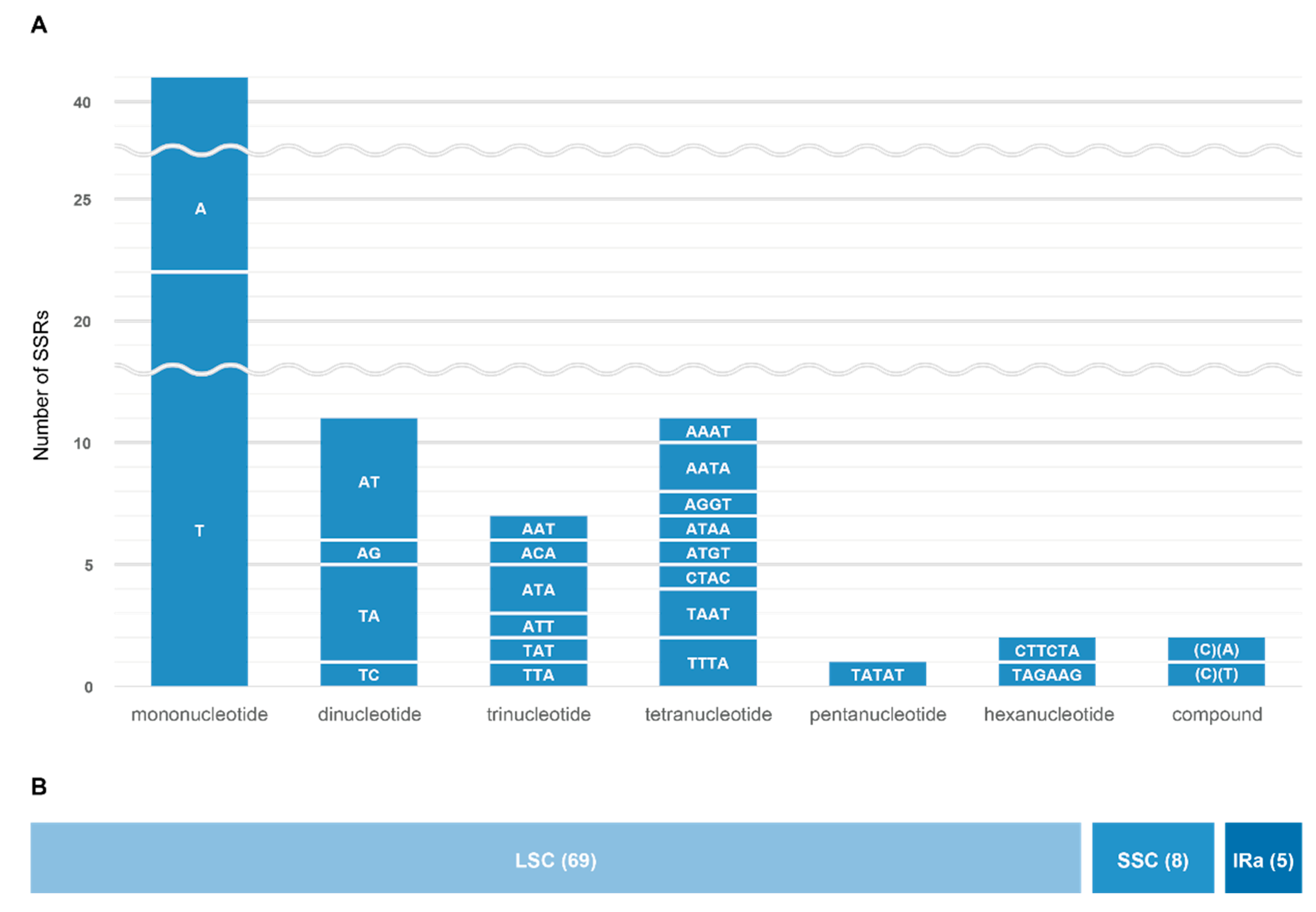
3.3. Simple Sequence Repeats
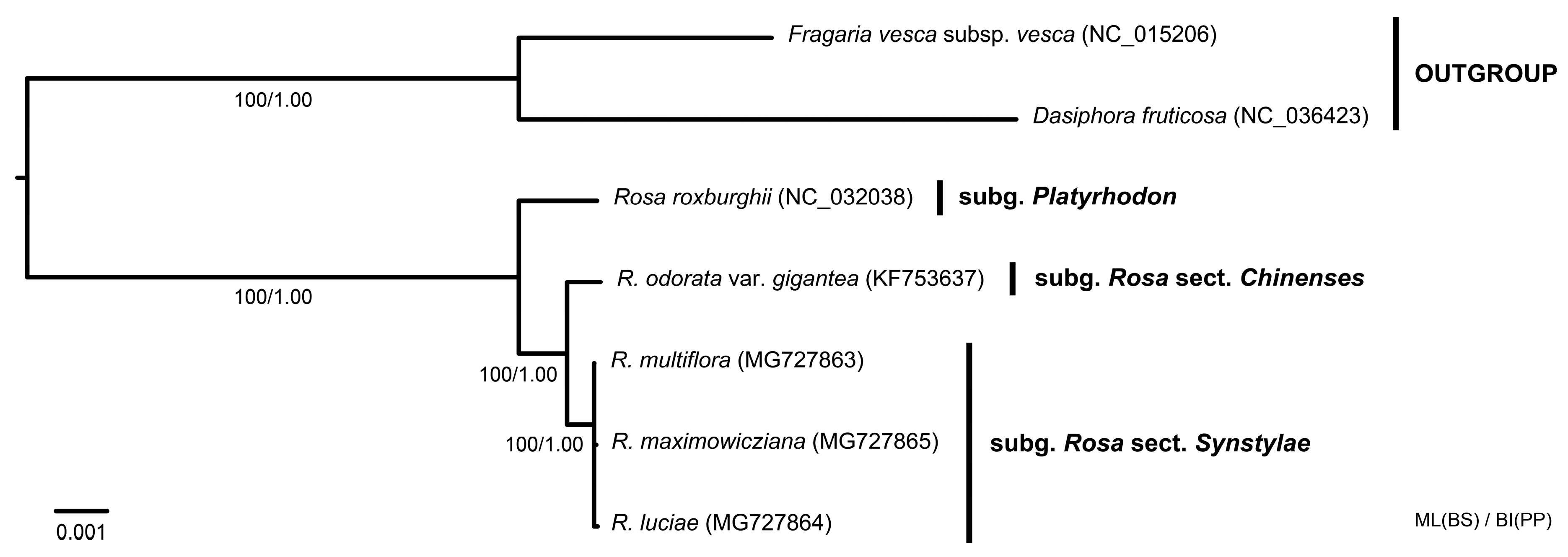
3.4. Phylogenetic Analyses
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Rehder, A. Manual of Cultivated Trees and Shrubs; The Macmillan Company: New York, NY, USA, 1940; pp. 426–451. [Google Scholar]
- Wissemann, V.; Ritz, C.M. The genus Rosa (Rosoideae, Rosaceae) revisited: Molecular analysis of nrITS-1 and atpB-rbcL intergenic spacer (IGS) versus conventional taxonomy. Bot. J. Linn. Soc. 2005, 147, 275–290. [Google Scholar] [CrossRef]
- Christenhusz, M.J.M.; Fay, M.F.; Chase, M.W. Plants of the World: An Illustrated Encyclopedia of Vascular Plants; University of Chicago Press: Chicago, IL, USA, 2017; pp. 262–267. [Google Scholar]
- Rehder, A. Bibliography of Cultivated Trees and Shrubs Hardy in the Cooler Temperate Regions of the Northern Hemisphere; Arnold Arboretum of Harvard University: Jamaica Plain, MA, USA, 1949; pp. 296–317. [Google Scholar]
- Zhu, Z.-M.; Gao, X.-F.; Fougère-Danezan, M. Phylogeny of Rosa sections Chinenses and Synstylae (Rosaceae) based on chloroplast and nuclear markers. Mol. Phylogenetics Evol. 2015, 87, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Ueda, Y.; He, H.; Nishihara, S.; Matsumoto, S. Phylogenetic analysis of Japanese Rosa species using matK sequences. Breed. Sci. 2000, 50, 275–281. [Google Scholar] [CrossRef]
- Fougère-Danezan, M.; Joly, S.; Bruneau, A.; Gao, X.-F.; Zhang, L.-B. Phylogeny and biogeography of wild roses with specific attention to polyploids. Ann. Bot. 2014, 115, 275–291. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Wang, G.; Wang, H.; Xia, T.; Zhang, S.; Wang, Q.; Fang, Y. Phylogenetic relationships in the genus Rosa revisited based on rpl16, trnL-F, and atpB-rbcL sequences. HortScience 2015, 50, 1618–1624. [Google Scholar]
- Debener, T.; Linde, M. Exploring complex ornamental genomes: The rose as a model plant. Crit. Rev. Plant Sci. 2009, 28, 267–280. [Google Scholar] [CrossRef]
- Bruneau, A.; Starr, J.R.; Joly, S. Phylogenetic relationships in the genus Rosa: New evidence from chloroplast DNA sequences and an appraisal of current knowledge. Syst. Bot. 2007, 32, 366–378. [Google Scholar] [CrossRef]
- Matsumoto, S.; Kouchi, M.; Fukui, H.; Ueda, Y. Phylogenetic analyses of the subgenus Eurosa using the its nrDNA sequence. In Proceedings of the XXV International Horticultural Congress, Part 11: Application of Biotechnology and Molecular Biology and Breeding—Gene Expression and Molecular Breeding, Genome Analysis, Brussels, Belgium, 2–7 August 1998; pp. 193–202. [Google Scholar]
- Ohba, H.; Akiyama, S.; Mikanagi, Y. Interspecific hybrids between Japanese species in Rosa section Synstylae (Rosaceae). J. Jap. Bot. 2007, 82, 45–53. [Google Scholar]
- Jeon, J.-H.; Maki, M.; Kim, S.-C. Inferring phylogenetic relationships among recently diverged East Asian species of roses (Rosa section Synstylae; Rosaceae). Unpublished work.
- Ruhlman, T.A.; Jansen, R.K. The plastid genomes of flowering plants. In Chloroplast Biotechnology: Methods and Protocols; Maliga, P., Ed.; Humana Press: Totowa, NJ, USA, 2014; pp. 3–38. [Google Scholar]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef]
- Chang, C.S.; Kim, H.; Chang, G.S. Illustrated Encyclopedia of Fauna & Flora of Korea, Vol. 43, Woody Plants; Ministry of Education: Seoul, Korea, 2011; p. 510.
- Zerbino, D.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Wyman, S.K.; Jansen, R.K.; Boore, J.L. Automatic annotation of organellar genomes with DOGMA. Bioinformatics 2004, 20, 3252–3255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE on-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Shinozaki, K.; Ohme, M.; Tanaka, M.; Wakasugi, T.; Hayashida, N.; Matsubayashi, T.; Zaita, N.; Chunwongse, J.; Obokata, J.; Yamaguchi-Shinozaki, K.; et al. The complete nucleotide sequence of the tobacco chloroplast genome: Its gene organization and expression. EMBO J. 1986, 5, 2043–2049. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Hu, H.; An, J.; Bai, G.; Ren, Q.; Liu, J. Complete chloroplast genome sequence of Rosa roxburghii and its phylogenetic analysis. Mitochondrial DNA Part B 2018, 3, 149–150. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef]
- Yang, J.B.; Li, D.Z.; Li, H.T. Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol. Ecol. Resour. 2014, 14, 1024–1031. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Mayor, C.; Brudno, M.; Schwartz, J.R.; Poliakov, A.; Rubin, E.M.; Frazer, K.A.; Pachter, L.S.; Dubchak, I. VISTA: Visualizing global DNA sequence alignments of arbitrary length. Bioinformatics 2000, 16, 1046–1047. [Google Scholar] [CrossRef]
- Brudno, M.; Do, C.B.; Cooper, G.M.; Kim, M.F.; Davydov, E.; Green, E.D.; Sidow, A.; Batzoglou, S.; Program, N.C.S. LAGAN and multi-LAGAN: Efficient tools for large-scale multiple alignment of genomic DNA. Genome Res. 2003, 13, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Thiel, T.; Michalek, W.; Varshney, R.; Graner, A. Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 2003, 106, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Shulaev, V.; Sargent, D.J.; Crowhurst, R.N.; Mockler, T.C.; Folkerts, O.; Delcher, A.L.; Jaiswal, P.; Mockaitis, K.; Liston, A.; Mane, S.P. The genome of woodland strawberry (Fragaria vesca). Nat. Genet. 2011, 43, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Lu, D.; Han, R.; Wang, L.; Qin, P. The complete chloroplast genome sequence of the shrubby cinquefoil Dasiphora fruticosa (Rosales: Rosaceae). Conserv. Genet. Resour. 2018, 10, 675–678. [Google Scholar] [CrossRef]
- Potter, D.; Eriksson, T.; Evans, R.C.; Oh, S.; Smedmark, J.E.E.; Morgan, D.R.; Kerr, M.; Robertson, K.R.; Arsenault, M.; Dickinson, T.A.; et al. Phylogeny and classification of Rosaceae. Plant Syst. Evol. 2007, 266, 5–43. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [PubMed]
- Millen, R.S.; Olmstead, R.G.; Adams, K.L.; Palmer, J.D.; Lao, N.T.; Heggie, L.; Kavanagh, T.A.; Hibberd, J.M.; Gray, J.C.; Morden, C.W.; et al. Many parallel losses of infA from chloroplast DNA during angiosperm evolution with multiple independent transfers to the nucleus. Plant Cell 2001, 13, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, S.; Bédard, J.; Hirano, M.; Hirabayashi, Y.; Oishi, M.; Imai, M.; Takase, M.; Ide, T.; Nakai, M. Uncovering the protein translocon at the chloroplast inner envelope membrane. Science 2013, 339, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Howarth, D.G.; Gardner, D.E.; Morden, C.W. Phylogeny of Rubus subgenus Idaeobatus (Rosaceae) and its implications toward colonization of the Hawaiian islands. Syst. Bot. 1997, 22, 433–441. [Google Scholar] [CrossRef]
- Dong, W.; Xu, C.; Li, C.; Sun, J.; Zuo, Y.; Shi, S.; Cheng, T.; Guo, J.; Zhou, S. Ycf1, the most promising plastid DNA barcode of land plants. Sci. Rep. 2015, 5, 8348. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Shi, C.; Gao, L.-Z. Plastid genome sequence of a wild woody oil species, Prinsepia utilis, provides insights into evolutionary and mutational patterns of Rosaceae chloroplast genomes. PLoS ONE 2013, 8, e73946. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.J.; Jansen, R.K. Ndhf sequence evolution and the major clades in the sunflower family. Proc. Natl. Acad. Sci. USA 1995, 92, 10379. [Google Scholar] [CrossRef] [PubMed]
- Wendel, J.F.; Doyle, J.J. Phylogenetic incongruence: Window into genome history and molecular evolution. In Molecular Systematics of Plants II; Springer: Berlin, Germany, 1998; pp. 265–296. [Google Scholar]
- Zhang, R.; Hipp, A.L.; Gailing, O. Sharing of chloroplast haplotypes among red oak species suggests interspecific gene flow between neighboring populations. Botany 2015, 93, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Nevill, P.G.; Després, T.; Bayly, M.J.; Bossinger, G.; Ades, P.K. Shared phylogeographic patterns and widespread chloroplast haplotype sharing in Eucalyptus species with different ecological tolerances. Tree Genet. Genom. 2014, 10, 1079–1092. [Google Scholar] [CrossRef]
- Shepherd, L.D.; de Lange, P.J.; Perrie, L.R.; Heenan, P.B. Chloroplast phylogeography of New Zealand Sophora trees (Fabaceae): Extensive hybridization and widespread last glacial maximum survival. J. Biogeogr. 2017, 44, 1640–1651. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Rosa multiflora | Rosa luciae | Rosa maximowicziana |
|---|---|---|---|
| Habitat | Forest edge | Sea shore/Open space | Thicket/Near the water |
| Growth habit | Erect | Prostrate | Decumbent |
| Leaves | Not leathery | Leathery | Not leathery |
| Stipules | Pectinate | Serrate | Serrate |
| Prickles | Sparse | Sparse | Dense |
| Styles | Glabrous | Pubescent | Glabrous |
| Flowering | April–May | June–July | May–June |
| Species | Locality | Voucher Number | GenBank Accession |
|---|---|---|---|
| R. multiflora | Namyangju, Gyeonggi, Korea | JJH258 | MG727863 |
| R. luciae | Seoguipo, Jeju, Korea | SGRW001 | MG727864 |
| R. maximowicziana | Hwaseong, Gyeonggi, Korea | CBRX001 | MG727865 |
| Characteristics | R. multiflora | R. luciae | R. maximowicziana | R. roxburghii | R. odorata var. gigantea |
|---|---|---|---|---|---|
| GenBank accession number | MG727863 | MG727864 | MG727865 | NC_032038 | KF753637 |
| Reference | This study | This study | This study | Wang et al., 2018 [23] | Yang et al., 2014 [25] |
| Total cpDNA size (bp) | 156,519 | 156,506 | 156,405 | 156,749 | 156,634 |
| LSC size (bp) | 85,643 | 85,631 | 85,529 | 85,852 | 85,767 |
| SSC size (bp) | 18,760 | 18,759 | 18,760 | 18,791 | 18,761 |
| IR size (bp) | 26,058 | 26,058 | 26,058 | 26,053 | 26,053 |
| Number of different genes | 114 | 114 | 114 | 115 | 116 (115 *) |
| Number of different protein-coding genes | 80 | 80 | 80 | 81 | 81 |
| Number of different tRNA genes | 30 | 30 | 30 | 30 | 31 (30 *) |
| Number of different rRNA genes | 4 | 4 | 4 | 4 | 4 |
| GC content (%) | 37.2 | 37.2 | 37.2 | 37.2 | 37.2 |
| GC content of LSC (%) | 35.2 | 35.2 | 35.2 | 35.2 | 35.2 |
| GC content of SSC (%) | 31.3 | 31.3 | 31.3 | 31.3 | 31.3 |
| GC content of IR (%) | 42.7 | 42.7 | 42.7 | 42.7 | 42.7 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jeon, J.-H.; Kim, S.-C. Comparative Analysis of the Complete Chloroplast Genome Sequences of Three Closely Related East-Asian Wild Roses (Rosa sect. Synstylae; Rosaceae). Genes 2019, 10, 23. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10010023
Jeon J-H, Kim S-C. Comparative Analysis of the Complete Chloroplast Genome Sequences of Three Closely Related East-Asian Wild Roses (Rosa sect. Synstylae; Rosaceae). Genes. 2019; 10(1):23. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10010023
Chicago/Turabian StyleJeon, Ji-Hyeon, and Seung-Chul Kim. 2019. "Comparative Analysis of the Complete Chloroplast Genome Sequences of Three Closely Related East-Asian Wild Roses (Rosa sect. Synstylae; Rosaceae)" Genes 10, no. 1: 23. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10010023