Production and Application of Stable Isotope-Labeled Internal Standards for RNA Modification Analysis

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Labeling of Escherichia coli
2.3. Labeling of Saccharomyces cerevisiae
2.4. Isolation of Total RNA for Stable Isotope-Labeled Internal Standards (SILIS) Production
2.5. Sample Preparation and RNA Isolation
2.5.1. Dictyostelium Discoideum
2.5.2. Caenorhabditis Elegans
2.5.3. Cell Culture
2.5.4. Mouse Brain and Liver
2.6. Purification of Ribosomal RNA and Transfer RNA by Size Exclusion Chromatography
2.7. Messenger RNA Purification
2.8. Enzymatic Digestion of RNA
2.9. High-Resolution Mass Spectrometry
2.10. Dilution of RNA Digests for 10× SILIS Generation
2.11. Liquid Chromatography–Mass Spectrometry (LC–MS/MS) Analysis
3. Results
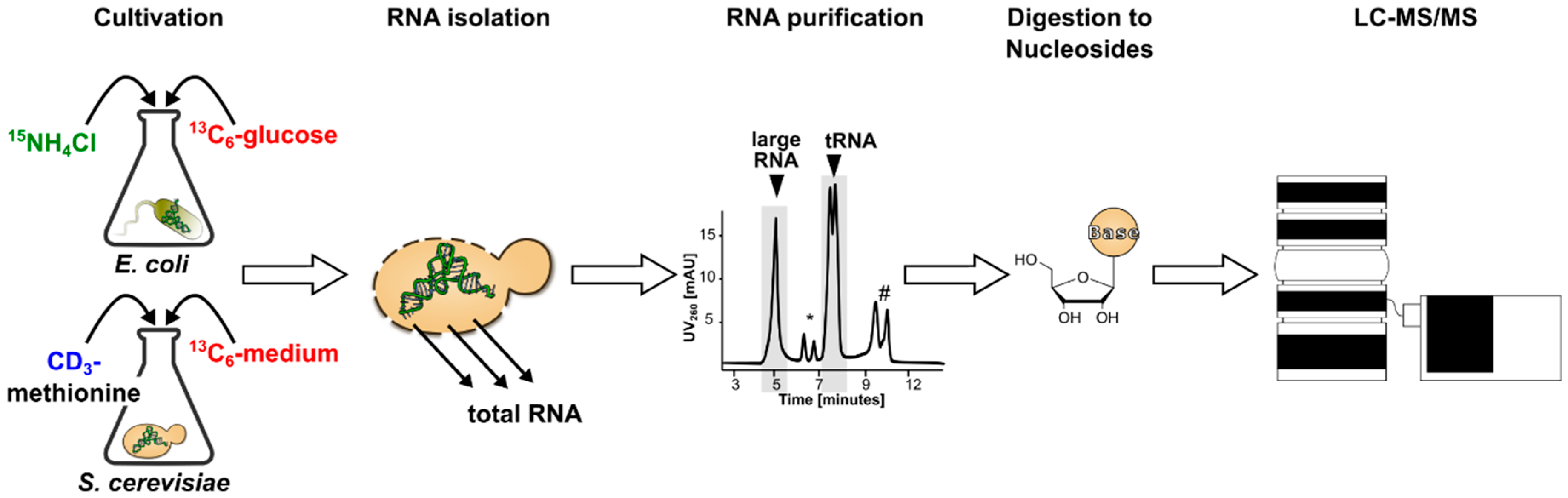
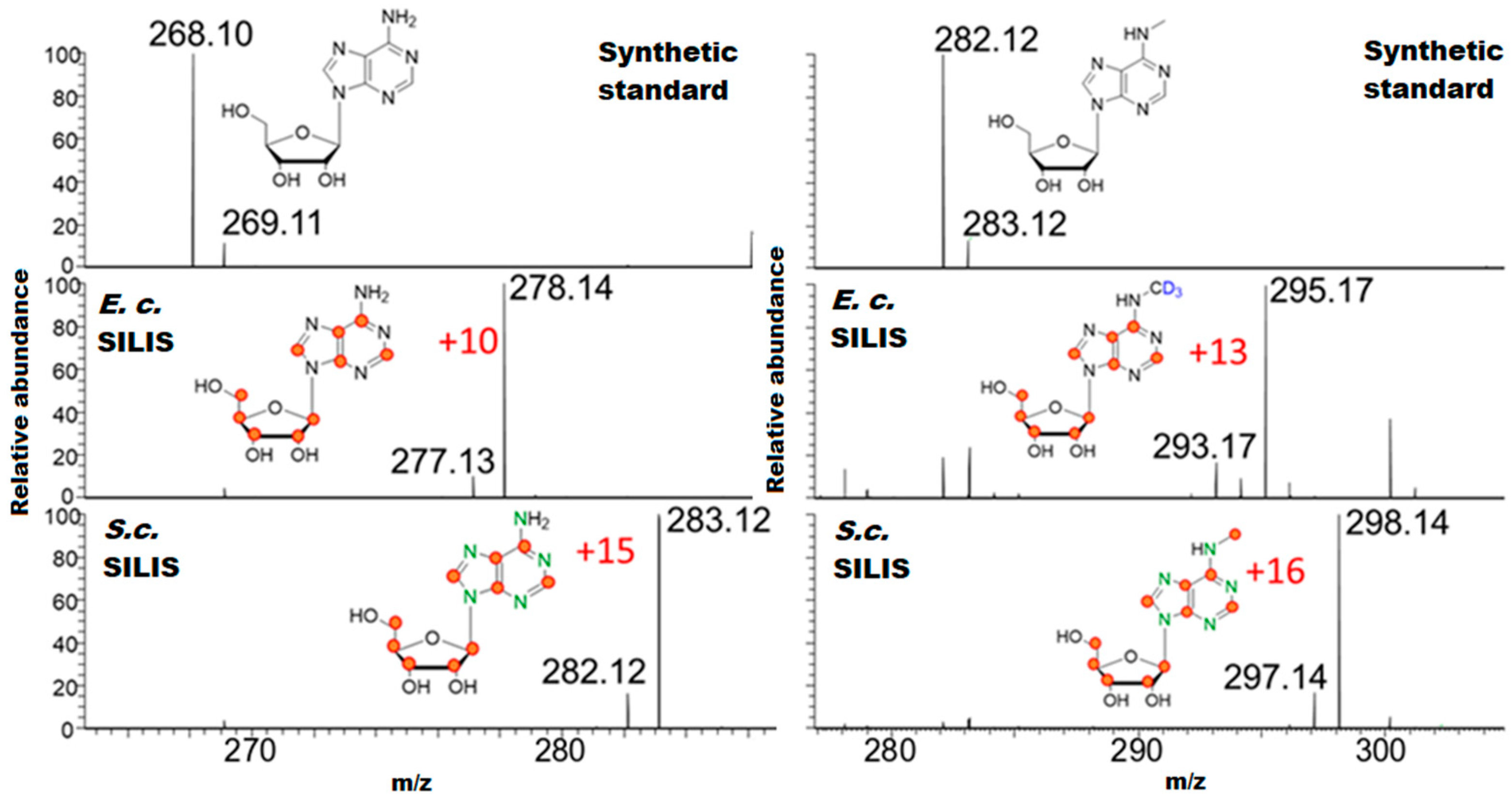
3.1. Generation and Characterization of SILIS
Guideline for SILIS Generation
3.2. Absolute Quantification with SILIS
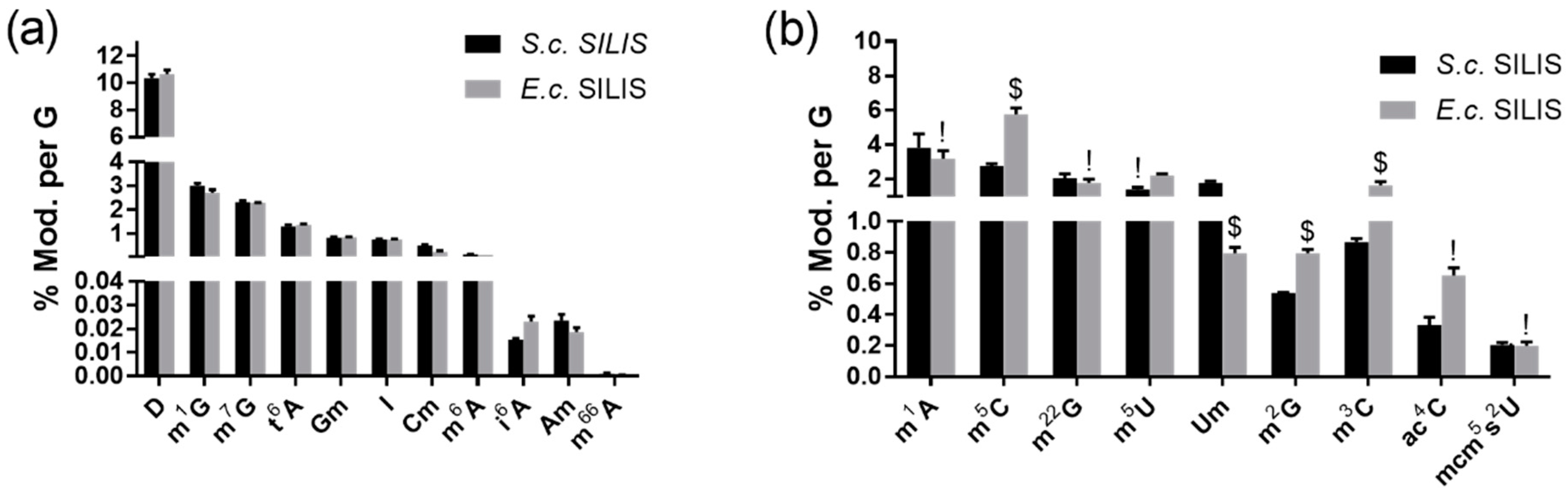
3.2.1. The Origin of the SILIS Has No Impact on the Quantification Result
3.2.2. Modified Nucleosides in Pseudomonas Aeruginosa Total tRNA
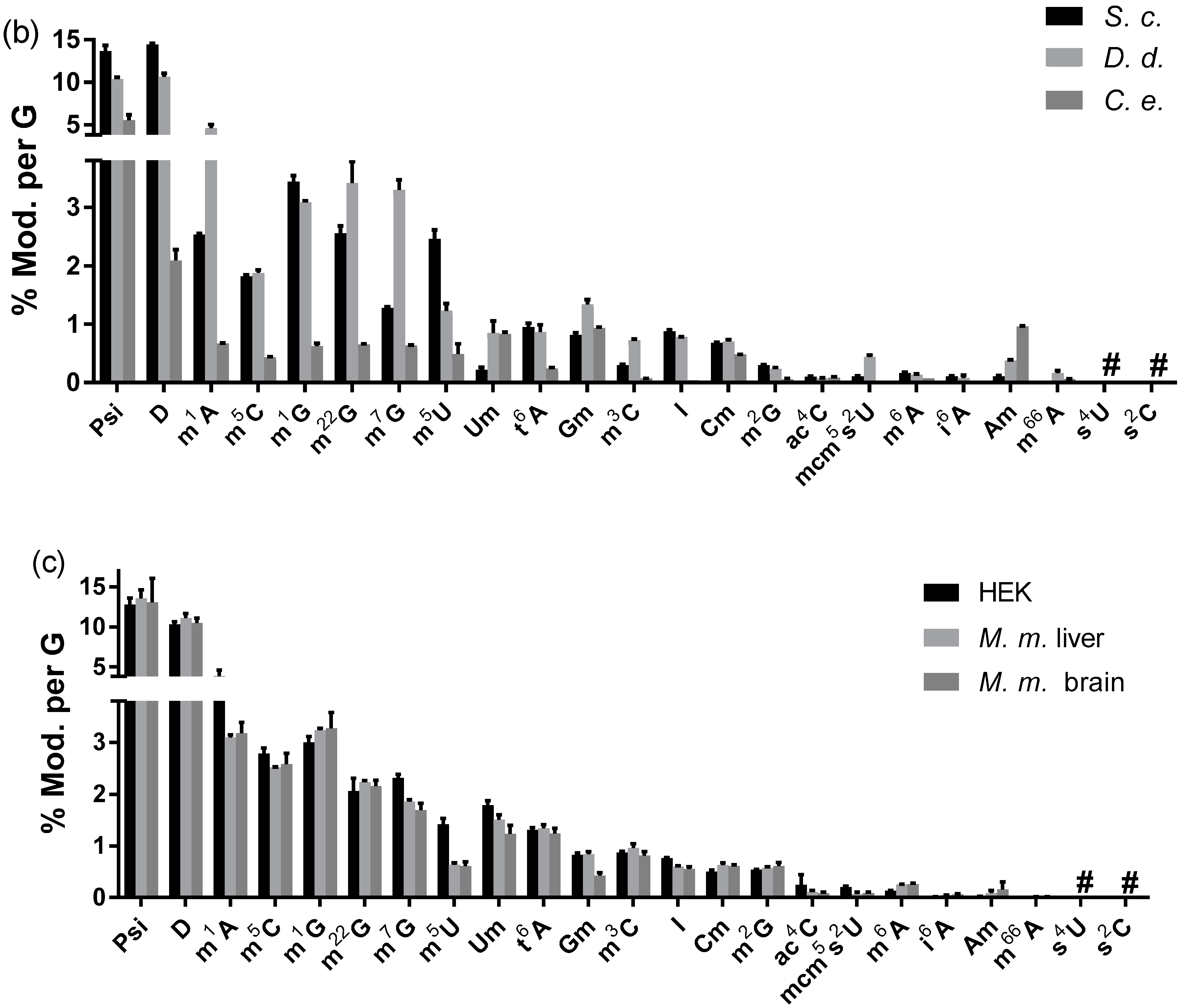
3.2.3. Modified Nucleosides in Total tRNA from Various Eukaryotes
3.2.4. The Abundance of Modified Nucleosides in Messenger RNA Is Different among Tissues
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hartmann, R.K.; Gossringer, M.; Spath, B.; Fischer, S.; Marchfelder, A. The making of tRNAs and more—RNase P and tRNase Z. Prog. Mol. Biol. Transl. Sci. 2009, 85, 319–368. [Google Scholar] [PubMed]
- Soll, D. Enzymatic modification of transfer RNA. Science 1971, 173, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Blanco, S.; Dietmann, S.; Flores, J.V.; Hussain, S.; Kutter, C.; Humphreys, P.; Lukk, M.; Lombard, P.; Treps, L.; Popis, M.; et al. Aberrant methylation of tRNAs links cellular stress to neuro-developmental disorders. EMBO J. 2014, 33, 2020–2039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pereira, M.; Francisco, S.; Varanda, A.S.; Santos, M.; Santos, M.A.S.; Soares, A.R. Impact of tRNA modifications and tRNA-modifying enzymes on proteostasis and human disease. Int. J. Mol. Sci. 2018, 19, 3738. [Google Scholar] [CrossRef] [PubMed]
- van Delft, P.; Akay, A.; Huber, S.M.; Bueschl, C.; Rudolph, K.L.M.; Di Domenico, T.; Schuhmacher, R.; Miska, E.A.; Balasubramanian, S. The profile and dynamics of RNA modifications in animals. ChemBioChem 2017, 18, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Carlile, T.M.; Rojas-Duran, M.F.; Gilbert, W.V. Transcriptome-wide identification of pseudouridine modifications using Pseudo-seq. Curr Protoc Mol Biol 2015, 112, 4.25.1–4.25.24. [Google Scholar]
- Li, X.; Xiong, X.; Yi, C. Epitranscriptome sequencing technologies: Decoding RNA modifications. Nat. Methods 2016, 14, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Helm, M.; Motorin, Y. Detecting RNA modifications in the epitranscriptome: Predict and validate. Nat. Rev. Genet. 2017, 18, 275–291. [Google Scholar] [CrossRef] [PubMed]
- Lobue, P.A.; Yu, N.; Jora, M.; Abernathy, S.; Limbach, P.A. Improved Application of RNAModMapper—An RNA modification mapping software tool—for analysis of liquid chromatography tandem mass spectrometry (LC-MS/MS) data. Methods 2018. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Pomerantz, S.C.; McCloskey, J.A. Detection of ribose-methylated nucleotides in enzymatic hydrolysates of RNA by thermospray liquid chromatography-mass spectrometry. J. Chromatogr. 1991, 562, 225–235. [Google Scholar] [CrossRef]
- Stokvis, E.; Rosing, H.; Beijnen, J.H. Stable isotopically labeled internal standards in quantitative bioanalysis using liquid chromatography/mass spectrometry: Necessity or not? Rapid Commun. Mass Spectrom. 2005, 19, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S. Isotope effect in negative ion chemical ionization mass spectrometry of deuterium labelled lormetazepam. Biomed. Environ. Mass Spectrom. 1987, 14, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Brandmayr, C.; Wagner, M.; Bruckl, T.; Globisch, D.; Pearson, D.; Kneuttinger, A.C.; Reiter, V.; Hienzsch, A.; Koch, S.; Thoma, I.; et al. Isotope-based analysis of modified tRNA nucleosides correlates modification density with translational efficiency. Angew. Chem. Int. Ed. Engl. 2012, 51, 11162–11165. [Google Scholar] [CrossRef] [PubMed]
- Kellner, S.; Ochel, A.; Thuring, K.; Spenkuch, F.; Neumann, J.; Sharma, S.; Entian, K.D.; Schneider, D.; Helm, M. Absolute and relative quantification of RNA modifications via biosynthetic isotopomers. Nucleic Acids Res. 2014, 42, e142. [Google Scholar] [CrossRef]
- Li, P.; Li, Z.; Beck, W.D.; Callahan, P.M.; Terry, A.V., Jr.; Bar-Peled, M.; Bartlett, M.G. Bio-generation of stable isotope-labeled internal standards for absolute and relative quantitation of phase II drug metabolites in plasma samples using LC-MS/MS. Anal. Bioanal. Chem. 2015, 407, 4053–4063. [Google Scholar] [CrossRef]
- Argus, J.P.; Yu, A.K.; Wang, E.S.; Williams, K.J.; Bensinger, S.J. An optimized method for measuring fatty acids and cholesterol in stable isotope-labeled cells. J. Lipid Res. 2017, 58, 460–468. [Google Scholar] [CrossRef]
- Wang, P.; Guo, L.; Jaini, R.; Klempien, A.; McCoy, R.M.; Morgan, J.A.; Dudareva, N.; Chapple, C. A 13C isotope labeling method for the measurement of lignin metabolic flux in Arabidopsis stems. Plant Methods 2018, 14, 51. [Google Scholar] [CrossRef]
- Sarin, L.P.; Kienast, S.D.; Leufken, J.; Ross, R.L.; Dziergowska, A.; Debiec, K.; Sochacka, E.; Limbach, P.A.; Fufezan, C.; Drexler, H.C.A.; et al. Nano LC-MS using capillary columns enables accurate quantification of modified ribonucleosides at low femtomol levels. RNA 2018, 24, 1403–1417. [Google Scholar] [CrossRef]
- Kellner, S.; DeMott, M.S.; Cheng, C.P.; Russell, B.S.; Cao, B.; You, D.; Dedon, P.C. Oxidation of phosphorothioate DNA modifications leads to lethal genomic instability. Nat. Chem. Biol. 2017, 13, 888–894. [Google Scholar] [CrossRef] [Green Version]
- Thiaville, J.J.; Kellner, S.M.; Yuan, Y.; Hutinet, G.; Thiaville, P.C.; Jumpathong, W.; Mohapatra, S.; Brochier-Armanet, C.; Letarov, A.V.; Hillebrand, R.; et al. Novel genomic island modifies DNA with 7-deazaguanine derivatives. Proc. Natl. Acad. Sci. USA 2016, 113, E1452–E1459. [Google Scholar] [CrossRef] [Green Version]
- Heiss, M.; Reichle, V.F.; Kellner, S. Observing the fate of tRNA and its modifications by nucleic acid isotope labeling mass spectrometry: NAIL-MS. RNA Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Reichle, V.F.; Kaiser, S.; Heiss, M.; Hagelskamp, F.; Borland, K.; Kellner, S. Surpassing limits of static RNA modification analysis with dynamic NAIL-MS. Methods 2018. [Google Scholar] [CrossRef] [PubMed]
- Chow, F.W.-N.; Koutsovoulos, G.; Ovando-Vázquez, C.; Neophytou, K.; Laetsch, D.R.; Bermúdez-Barrientos, J.R.; Kumar, S.; Robertson, E.; Claycomb, J.M.; Blaxter, M.; et al. Secretion of an Argonaute protein by a parasitic nematode and the evolution of its siRNA guides. bioRxiv 2019. [Google Scholar] [CrossRef]
- Chionh, Y.H.; Ho, C.H.; Pruksakorn, D.; Ramesh Babu, I.; Ng, C.S.; Hia, F.; McBee, M.E.; Su, D.; Pang, Y.L.; Gu, C.; et al. A multidimensional platform for the purification of non-coding RNA species. Nucleic Acids Res. 2013, 41, e168. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.M.; Chionh, Y.H.; Hia, F.; Gu, C.; Kellner, S.; McBee, M.E.; Ng, C.S.; Pang, Y.L.; Prestwich, E.G.; Lim, K.S.; et al. A Platform for discovery and quantification of modified ribonucleosides in RNA: Application to stress-induced reprogramming of tRNA modifications. Methods Enzymol. 2015, 560, 29–71. [Google Scholar] [PubMed]
- Reichle, V.F.; Weber, V.; Kellner, S. NAIL-MS in E. coli determines the source and fate of methylation in tRNA. Chembiochem 2018. [Google Scholar] [CrossRef] [PubMed]
- Klepper, F.; Jahn, E.M.; Hickmann, V.; Carell, T. Synthesis of the transfer-RNA nucleoside queuosine by using a chiral allyl azide intermediate. Angew. Chem. Int. Ed. Engl. 2007, 46, 2325–2327. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Clark, W.; Luo, G.; Wang, X.; Fu, Y.; Wei, J.; Wang, X.; Hao, Z.; Dai, Q.; Zheng, G.; et al. ALKBH1-Mediated tRNA demethylation regulates translation. Cell 2016, 167, 1897. [Google Scholar] [CrossRef]
- Jaroensuk, J.; Atichartpongkul, S.; Chionh, Y.H.; Wong, Y.H.; Liew, C.W.; McBee, M.E.; Thongdee, N.; Prestwich, E.G.; DeMott, M.S.; Mongkolsuk, S.; et al. Methylation at position 32 of tRNA catalyzed by TrmJ alters oxidative stress response in Pseudomonas aeruginosa. Nucleic Acids Res. 2016, 44, 10834–10848. [Google Scholar] [CrossRef]
- Kaiser, S.; Rimbach, K.; Eigenbrod, T.; Dalpke, A.H.; Helm, M. A modified dinucleotide motif specifies tRNA recognition by TLR7. RNA 2014, 20, 1351–1355. [Google Scholar] [CrossRef] [Green Version]
- Boccaletto, P.; Machnicka, M.A.; Purta, E.; Piatkowski, P.; Baginski, B.; Wirecki, T.K.; de Crecy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A database of RNA modification pathways. 2017 update. Nucleic Acids Res. 2018, 46, D303–D307. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Liu, J.; He, C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015, 29, 1343–1355. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Li, H.B.; Yin, Z.; Flavell, R.A. Recent advances in dynamic m6A RNA modification. Open Biol 2016, 6, 160003. [Google Scholar] [CrossRef] [PubMed]
- Patil, D.P.; Pickering, B.F.; Jaffrey, S.R. Reading m6A in the transcriptome: m6A-binding proteins. Trends Cell Biol. 2018, 28, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Legrand, C.; Tuorto, F.; Hartmann, M.; Liebers, R.; Jacob, D.; Helm, M.; Lyko, F. Statistically robust methylation calling for whole-transcriptome bisulfite sequencing reveals distinct methylation patterns for mouse RNAs. Genome Res. 2017, 27, 1589–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matorin, Y.; Helm, M. Methods for RNA modification mapping using deep sequencing: Established and new emerging technologies. Genes, accepted for publication.
- Xu, L.; Liu, X.; Sheng, N.; Oo, K.S.; Liang, J.; Chionh, Y.H.; Xu, J.; Ye, F.; Gao, Y.G.; Dedon, P.C.; et al. Three distinct 3-methylcytidine (m3C) methyltransferases modify tRNA and mRNA in mice and humans. J. Biol. Chem. 2017, 292, 14695–14703. [Google Scholar] [CrossRef] [PubMed]
- Marchand, V.; Ayadi, L.; Ernst, F.G.M.; Hertler, J.; Bourguignon-Igel, V.; Galvanin, A.; Kotter, A.; Helm, M.; Lafontaine, D.L.J.; Motorin, Y. AlkAniline-Seq: Profiling of m7G and m3C RNA modifications at single nucleotide resolution. Angew. Chem. Int. Ed. Engl. 2018. [Google Scholar] [CrossRef] [PubMed]
- Favre, A.; Michelson, A.M.; Yaniv, M. Photochemistry of 4-thiouridine in Escherichia coli transfer RNA1Val. J. Mol. Biol. 1971, 58, 367–379. [Google Scholar] [CrossRef]
- Ramabhadran, T.V.; Fossum, T.; Jagger, J. Escherichia coli mutant lacking 4-thiouridine in its transfer ribonucleic acid. J. Bacteriol. 1976, 128, 671–672. [Google Scholar]
- Xu, F.; Vostal, J.G. Inactivation of bacteria via photosensitization of vitamin K3 by UV-A light. FEMS Microbiol. Lett. 2014, 358, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Guy, M.P.; Shaw, M.; Weiner, C.L.; Hobson, L.; Stark, Z.; Rose, K.; Kalscheuer, V.M.; Gecz, J.; Phizicky, E.M. Defects in tRNA anticodon loop 2′-O-methylation are implicated in nonsyndromic X-linked intellectual disability due to mutations in FTSJ1. Hum. Mutat. 2015, 36, 1176–1187. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.J.; Esberg, A.; Huang, B.; Bjork, G.R.; Bystrom, A.S. Eukaryotic wobble uridine modifications promote a functionally redundant decoding system. Mol. Cell. Biol. 2008, 28, 3301–3312. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification | Yeast tRNA | Yeast Large RNA | E. coli tRNA | E. coli Large RNA |
|---|---|---|---|---|
| 1-methyladenosine (m1A) | ✓ | ✓ | 0 | 0 |
| 1-methylguanosine (m1G) | ✓ | ✓ | ✓ | ✓ |
| 2-methyladenosine (m2A) | 0 | 0 | ✓ | 0 |
| 2-thiocytidine (s2C) | 0 | 0 | ✓ | 0 |
| 2′-O-methyladenosine (Am) | ✓ | ✓ | ✓ | ✓ |
| 2′-O-methylcytidine (Cm) | ✓ | ✓ | ✓ | ✓ |
| 2′-O-methylguanosine (Gm) | ✓ | ✓ | ✓ | ✓ |
| 2′-O-methyluridine (Um) | ✓ | ✓ | ✓l | 0 |
| 3-methylcytidine (m3C) | ✓ | ✓ | 0 | 0 |
| 3-methyluridine (m3U) | 0 | ✓ | 0 | ✓ l |
| 4-thiouridine (s4U) | 0 | 0 | ✓ | 0 |
| 5-carbamoylmethyl-2-thiouridine (ncm5s2U) | ✓ | 0 | 0 | 0 |
| 5-methoxycarbonylmethyluridine (mcm5U) | ✓ | 0 | 0 | 0 |
| 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U) | ✓ | 0 | 0 | 0 |
| 5-methylcytidine (m5C) | ✓ | ✓ | 0 | ✓ |
| 5-methyluridine (m5U) | ✓ | 0 | ✓ | 0 |
| 7-methylguanosine (m7G) | ✓ | ✓ | ✓ | ✓ |
| N2-methylguanosine (m2G) | ✓ | ✓ | 0 | ✓ |
| N2,N2-dimethylguanosine (m22G) | ✓ | ✓ | 0 | 0 |
| N4-acetylcytidine (ac4C) | ✓ | ✓ | 0 | 0 |
| 2-methylthioadenosine (ms2A) | ✓ | 0 | 0 | 0 |
| N6-isopentenyladenosine (i6A) | ✓ | 0 | ✓ | 0 |
| N6-methyladenosine (m6A) | ✓ | ✓ | ✓ | ✓ |
| N6,N6-dimethyladenosine (m66A) | ✓ | ✓ | ✓ | ✓ |
| N6-threonylcarbamoyladenosine (t6A) | ✓ | 0 | ✓ | 0 |
| Dihydrouridine (D) | ✓ | ✓ | ✓ | 0 |
| Inosine (I) | ✓ | ✓ | ✓ | ✓ |
| Pseudouridine (Psi or Ψ) | ✓ | ✓ | ✓ 2 | ✓ 2 |
| Queuosine (Q) | ✓ | 0 | 0 | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borland, K.; Diesend, J.; Ito-Kureha, T.; Heissmeyer, V.; Hammann, C.; Buck, A.H.; Michalakis, S.; Kellner, S. Production and Application of Stable Isotope-Labeled Internal Standards for RNA Modification Analysis. Genes 2019, 10, 26. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10010026
Borland K, Diesend J, Ito-Kureha T, Heissmeyer V, Hammann C, Buck AH, Michalakis S, Kellner S. Production and Application of Stable Isotope-Labeled Internal Standards for RNA Modification Analysis. Genes. 2019; 10(1):26. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10010026
Chicago/Turabian StyleBorland, Kayla, Jan Diesend, Taku Ito-Kureha, Vigo Heissmeyer, Christian Hammann, Amy H. Buck, Stylianos Michalakis, and Stefanie Kellner. 2019. "Production and Application of Stable Isotope-Labeled Internal Standards for RNA Modification Analysis" Genes 10, no. 1: 26. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10010026