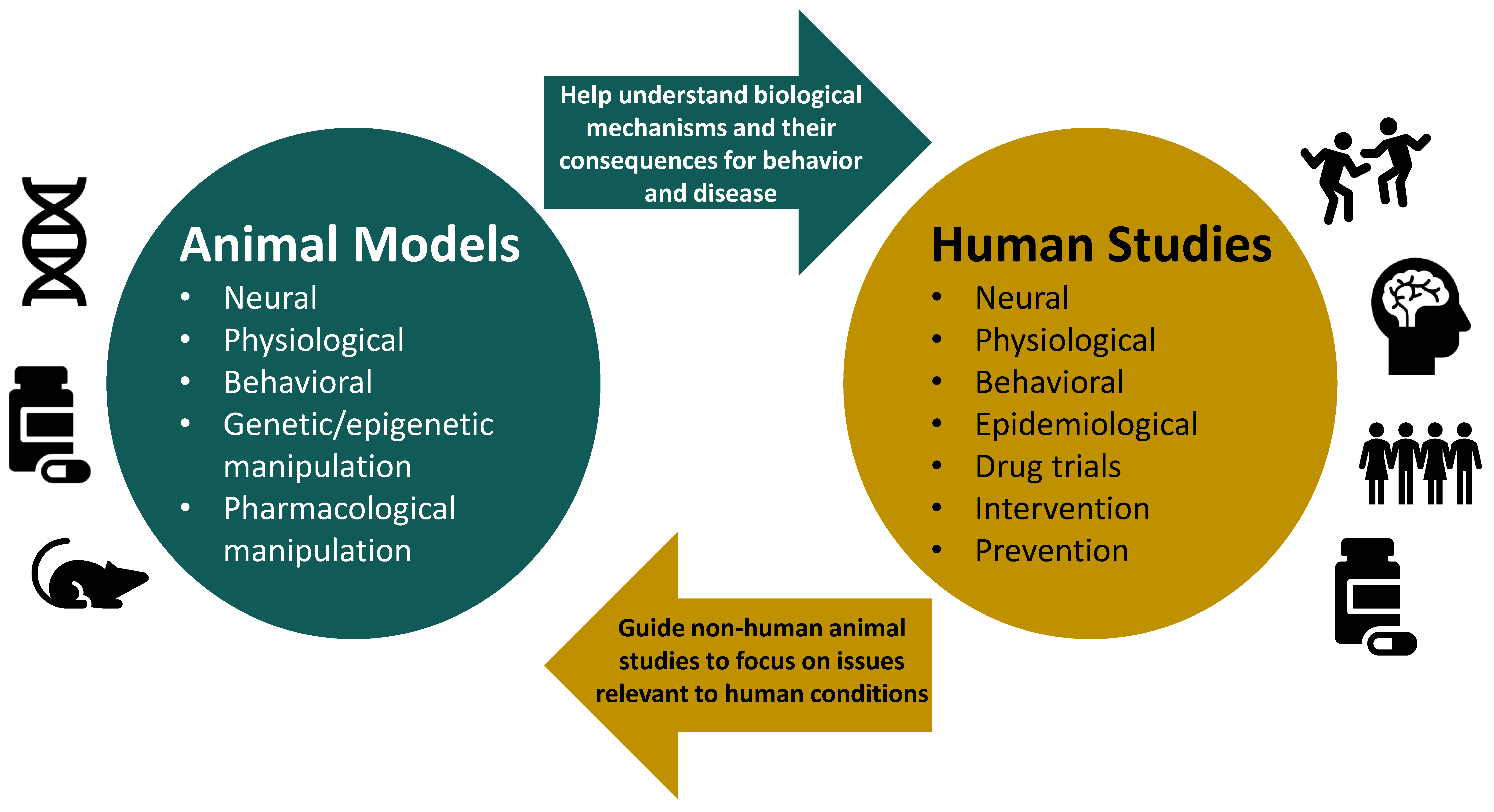
Animal Models and Their Contribution to Our Understanding of the Relationship Between Environments, Epigenetic Modifications, and Behavior

{kind=link}
{kind=link}
Abstract
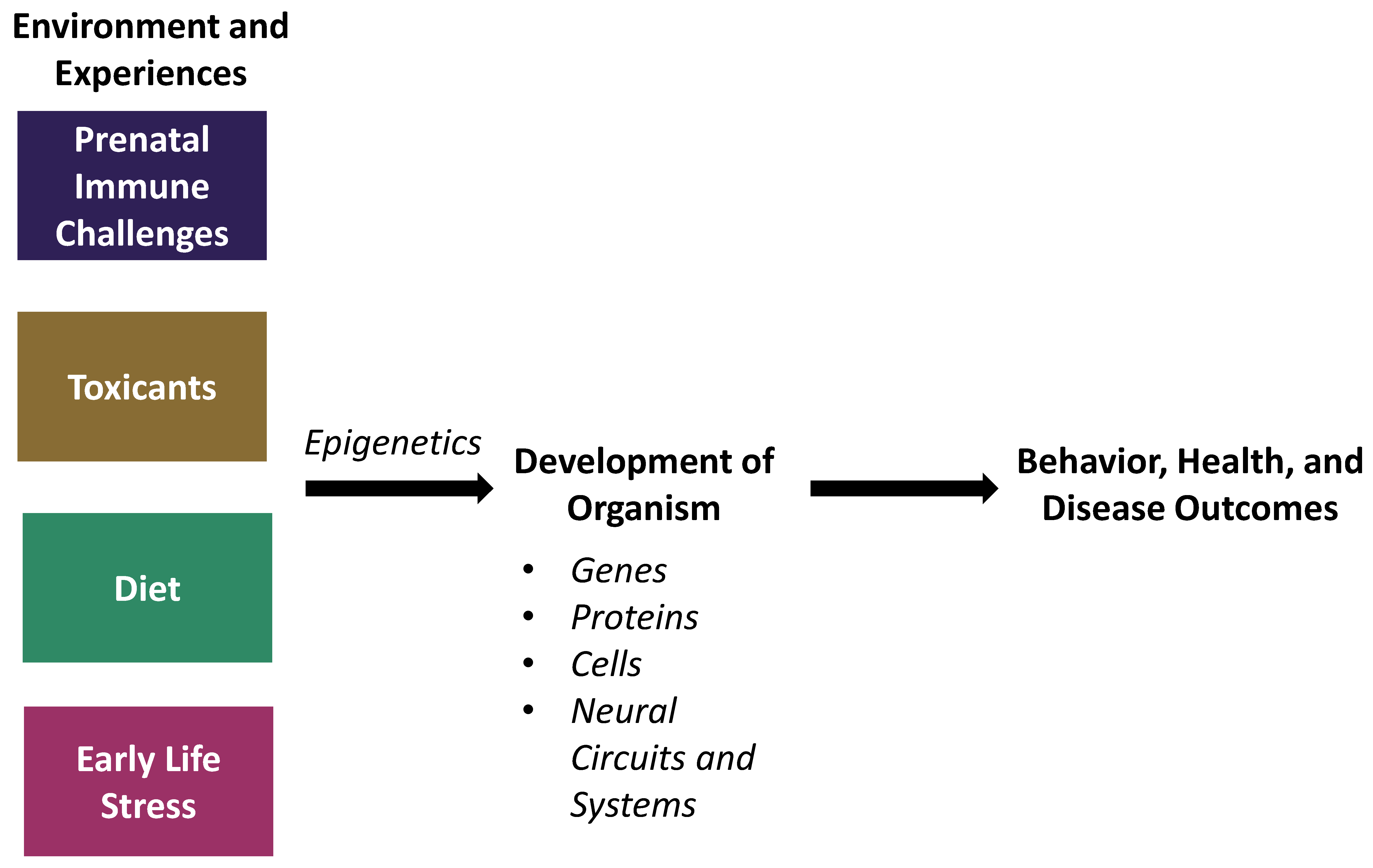
:1. Introduction
2. Prenatal Immune Challenges
3. Environmental Toxicants
4. Diet
5. Early-Life Stress
6. Concluding Remarks and Recommendations for Future Research
Funding
Conflicts of Interest
References
- Ericsson, A.C.; Crim, M.J.; Franklin, C.L. A brief history of animal modeling. Mo. Med. 2013, 201–205. [Google Scholar]
- Bennett, A.J. Gene environment interplay: Nonhuman primate models in the study of resilience and vulnerability. Dev. Psychobiol. 2007, 50, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, K.C.K.; Robinson, P.N.; MacRae, C.A. Animal-based studies will be essential for precision medicine. Sci. Transl. Med. 2016. [Google Scholar] [CrossRef]
- Bennett, A.J.; Lesch, K.P.; Heils, A.; Long, J.C.; Lorenz, J.G.; Shoaf, S.E.; Champoux, M.; Suomi, S.J.; Linnoila, M.V.; Higley, J.D. Early experience and serotonin transporter gene variation interact to influence primate CNS function. Mol. Psychiatry 2002, 7, 118–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caspi, A.; Sugden, K.; Moffitt, T.; Taylor, A.; Craig, I.; Harrington, H.; McClay, J.; Mill, J.; Martin, J.; Braithwaite, A.; et al. Influence of life stress on depression: Moderation by a polymorphism in the 5-HTT gene. Science 2003, 301, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Doherty, T.S.; Roth, T.L. Insight from animal models of environmentally driven epigenetic changes in the developing and adult brain. Dev. Psychopathol. 2016, 28, 1229–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pogribny, I.P.; Karpf, A.R.; James, S.R.; Melnyk, S.; Han, T.; Tryndyak, V.P. Epigenetic alterations in the brains of Fisher 344 rats induced by long-term administration of folate/methyl-deficient diet. Brain Res. 2008, 1237, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Dolinoy, D.C.; Huang, D.; Jirtle, R.L. Maternal nutrient supplementation counteracts Bisphenol A-induced DNA hypomethylation in early development. Proc. Natl. Acad. Sci. USA 2007, 104, 13056–13061. [Google Scholar] [CrossRef]
- Tollenaar, M.; O’Donnell, K.; Garg, E.; Nguyen, T.; Meaney, M.; Beijers, R.; Zijlmans, M.; de Weerth, C. F48. Epigenetic markers of the intergenerational transmission of stress. Biol. Psychiatry 2018, 83, S256. [Google Scholar] [CrossRef]
- Parker, K.J.; Buckmaster, C.L.; Sundlass, K.; Schatzberg, A.F.; Lyons, D.M. Maternal mediation, stress inoculation, and the development of neuroendocrine stress resistance in primates. Proc. Natl. Acad. Sci. USA 2006, 103, 3000–3005. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.S.; Patterson, P.H. Maternal infection and schizophrenia: Implications for prevention. Schizophr. Bull. 2011, 37, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Susser, E.S. In utero infection and adult schizophrenia. Ment. Retard. Dev. Disabil. Res. Rev. 2002, 8, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Khandaker, G.M.; Zimbron, J.; Lewis, G.; Jones, P.B. Prenatal maternal infection, neurodevelopment and adult schizophrenia: A systematic review of population-based studies. Psychol. Med. 2013, 43, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Kundakovic, M. Fearing the mother’s virus: The lasting consequences of prenatal immune activation on the epigenome and brain function. Biol. Psychiatry 2017, 81, e25. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.; Yee, B.K.; Feldon, J. The neurodevelopmental impact of prenatal infections at different times of pregnancy: The earlier the worse? Neuroscientist 2007, 13, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Weber-Stadlbauer, U.; Richetto, J.; Labouesse, M.A.; Bohacek, J.; Mansuy, I.M.; Meyer, U. Transgenerational transmission and modification of pathological traits induced by prenatal immune activation. Mol. Psychiatry 2017, 22, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Zerbo, O.; Qian, Y.; Yoshida, C.; Grether, J.; Van de Water, J.; Croen, L. Maternal infection during pregnancy and autism spectrum disorders. J. Autism Dev. Disord. 2015, 45, 4015–4025. [Google Scholar] [CrossRef]
- Smith, S.E.P.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef]
- Wu, W.-L.; Hsiao, E.Y.; Yan, Z.; Mazmanian, S.K.; Patterson, P.H. The placental interleukin-6 signaling controls fetal brain development and behavior. Brain Behav. Immun. 2017, 62, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.N.C.; Gartlon, J.E.; Minassian, A.; Perry, W.; Geyer, M.A. Animal and Translational Models for CNS Drug Discovery; Academic Press: San Diego, CA, USA, 2008; Chapter 8; pp. 199–261. [Google Scholar]
- Lubow, R. Latent inhibition: Effect of frequency of nonreinforced preexposure of the CS. J. Comp. Physiol. Psychol. 1966, 60, 454–457. [Google Scholar] [CrossRef]
- Swerdlow, N.R.; Braff, D.L.; Hartston, H.; Perry, W.; Geyer, M.A. Latent inhibition in schizophrenia. Schizophr. Res. 1996, 20, 91–103. [Google Scholar] [CrossRef]
- Carroll, L.S.; Owen, M.J. Genetic overlap between autism, schizophrenia and bipolar disorder. Genome Med. 2009, 1, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crespi, B.; Stead, P.; Elliot, M. Comparative genomics of autism and schizophrenia. Proc. Natl. Acad. Sci. USA 2010, 107, 1736–1741. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, S.E.; Gillis, J.; Kramer, M.; Lihm, J.; Yoon, S.; Berstein, Y.; Mistry, M.; Pavlidis, P.; Solomon, R.; Ghiban, E.; et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry 2014, 19, 652–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibbs, R.A.; Weinstock, G.M.; Metzker, M.L.; Muzny, D.M.; Sodergren, E.J.; Scherer, S.; Scott, G.; Steffen, D.; Worley, K.C.; Burch, P.E.; et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 2004, 428, 493–521. [Google Scholar] [CrossRef] [PubMed]
- Dahlgren, J.; Samuelsson, A.-M.; Jansson, T.; Holmäng, A. Interleukin-6 in the maternal circulation reaches the rat fetus in mid-gestation. Pediatr. Res. 2006, 60, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Hodge, D.R.; Cho, E.C.; Copeland, T.D.; Guszczynski, T.; Yang, E.; Seth, A.K.; Farrar, W.L. IL-6 enhances the nuclear translocation of DNA cytosine-5-methyltransferase 1 (DNMT1) via phosphorylation of the nuclear localization sequence by the AKT kinase. Cancer Genom. Proteom. 2007, 4, 387–398. [Google Scholar]
- McCullough, L.E.; Miller, E.E.; Calderwood, L.E.; Shivappa, N.; Steck, S.E.; Forman, M.R.; Mendez, M.A.; Maguire, R.; Fuemmeler, B.F.; Kollins, S.H.; et al. Maternal inflammatory diet and adverse pregnancy outcomes: Circulating cytokines and genomic imprinting as potential regulators? Epigenetics 2017, 12, 688–697. [Google Scholar] [CrossRef]
- Richetto, J.; Massart, R.; Weber-Stadlbauer, U.; Szyf, M.; Riva, M.A.; Meyer, U. Genome-wide DNA methylation changes in a mouse model of infection-mediated neurodevelopmental disorders. Biol. Psychiatry 2017, 81, 265–276. [Google Scholar] [CrossRef]
- Zaretsky, M.V.; Alexander, J.M.; Byrd, W.; Bawdon, R.E. Transfer of inflammatory cytokines across the placenta. Obstet. Gynecol. 2004, 103, 546. [Google Scholar] [CrossRef]
- Ziats, M.N.; Rennert, O.M. Expression profiling of autism candidate genes during human brain development implicates central immune signaling pathways. PLoS ONE 2011, 6, e24691. [Google Scholar] [CrossRef] [PubMed]
- Labouesse, M.A.; Dong, E.; Grayson, D.R.; Guidotti, A.; Meyer, U. Maternal immune activation induces GAD1 and GAD2 promoter remodeling in the offspring prefrontal cortex. Epigenetics 2015, 10, 1143–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skortsova, K.; Taberlay, P.; Clark, S.; Stirzaker, C. Role of 5-Hydroxymethylation and TET enzymes in remodelling the epigenome. Exp. Med. 2016, 34, 1–9. [Google Scholar]
- Kinde, B.; Gabel, H.W.; Gilbert, C.S.; Griffith, E.C.; Greenberg, M.E. Reading the unique DNA methylation landscape of the brain: Non-CpG methylation, hydroxymethylation, and MeCP2. Proc. Natl. Acad. Sci. USA 2015, 112, 6800–6806. [Google Scholar] [CrossRef] [PubMed]
- Szyf, M.; Bick, J. DNA Methylation: A mechanism for embedding early life experiences in the genome. Child Dev. 2013, 84, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Vaissière, T.; Sawan, C.; Herceg, Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. 2008, 659, 40–48. [Google Scholar] [CrossRef]
- Del Blanco, B.; Barco, A. Impact of environmental conditions and chemicals on the neuronal epigenome. Curr. Opin. Chem. Biol. 2018, 45, 157–165. [Google Scholar] [CrossRef]
- Nilsson, E.E.; Skinner, M.K. Environmentally induced epigenetic transgenerational inheritance of disease susceptibility. Transl. Res. 2015, 165, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Jiao, Z.; Zheng, S.; Li, M.; Zhang, J.; Feng, Y.; Yin, J.; Shao, B. Long-term effects of Bisphenol AF (BPAF) on hormonal balance and genes of hypothalamus-pituitary-gonad axis and liver of zebrafish (Danio rerio), and the impact on offspring. Chemosphere 2015, 128, 252–257. [Google Scholar] [CrossRef]
- Tran, N.Q.V.; Miyake, K. Neurodevelopmental disorders and environmental toxicants: Epigenetics as an underlying mechanism. Int. J. Genomics 2017. [Google Scholar] [CrossRef]
- Kundakovic, M.; Jaric, I. The epigenetic link between prenatal adverse environments and neurodevelopmental disorders. Genes 2017, 8, 104. [Google Scholar] [CrossRef] [PubMed]
- Kundakovic, M.; Gudsnuk, K.; Herbstman, J.B.; Tang, D.; Perera, F.P.; Champagne, F.A. DNA methylation of BDNF as a biomarker of early-life adversity. Proc. Natl. Acad. Sci. USA 2015, 112, 6807–6813. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, D.S.; Gray, L.E.; Wilson, V.S. Modeling the interaction of binary and ternary mixtures of estradiol with Bisphenol A and Bisphenol AF in an in vitro estrogen-mediated transcriptional activation assay (T47D-KBluc). Toxicol. Sci. 2010, 116, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Burns, K.A.; Arao, Y.; Luh, C.J.; Korach, K.S. Differential estrogenic actions of endocrine-disrupting chemicals Bisphenol A, Bisphenol AF, and zearalenone through estrogen receptor α and β in vitro. Environ. Health Perspect. 2012, 120, 1029. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Perera, L.; Coons, L.A.; Burns, K.A.; Tyler Ramsey, J.; Pelch, K.E.; Houtman, R.; van Beuningen, R.; Teng, C.T.; Korach, K.S. Differential in vitro biological action, coregulator interactions, and molecular dynamic analysis of Bisphenol A (BPA), BPAF, and BPS ligand-ERα complexes. Environ. Health Perspect. 2018, 126, 017012. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, A.; Liu, X.; Okada, H.; Shimohigashi, M.; Shimohigashi, Y. Bisphenol AF is a full agonist for the estrogen receptor ERα but a highly specific antagonist for ERβ. Environ. Health Perspect. 2010, 118, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Calafat, A.M.; Ye, X.; Wong, L.Y.; Reidy, J.A.; Needham, L.L. Exposure of the U.S. population to Bisphenol A and 4-tertiary-octylphenol: 2003-2004. Environ. Health Perspect. 2008, 116, 39–44. [Google Scholar] [CrossRef]
- Kundakovic, M.; Champagne, F.A. Epigenetic perspective on the developmental effects of Bisphenol A. Brain Behav. Immun. 2011, 25, 1084–1093. [Google Scholar] [CrossRef]
- Mouneimne, Y.; Nasrallah, M.; Khoueiry-Zgheib, N.; Nasreddine, L.; Nakhoul, N.; Ismail, H.; Abiad, M.; Koleilat, L.; Tamim, H. Bisphenol A urinary level, its correlates, and association with cardiometabolic risks in Lebanese urban adults. Environ. Monit. Assess. 2017, 189, 517. [Google Scholar] [CrossRef]
- Lee, S.; Kim, Y.; Shin, T.-Y.; Kim, S.-H. Neurotoxic effects of Bisphenol AF on calcium-induced ROS and MAPKs. Neurotox. Res. 2013, 23, 249–259. [Google Scholar] [CrossRef]
- Cao, J.; Mickens, J.A.; McCaffrey, K.A.; Leyrer, S.M.; Patisaul, H.B. Neonatal Bisphenol A exposure alters sexually dimorphic gene expression in the postnatal rat hypothalamus. NeuroToxicology 2012, 33, 23–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eid, A.; Zawia, N. Consequences of lead exposure, and it’s emerging role as an epigenetic modifier in the aging brain. NeuroToxicology 2016, 56, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Nicolopoulou-Stamati, P.; Maipas, S.; Kotampasi, C.; Stamatis, P.; Hens, L. Chemical pesticides and human health: The urgent need for a new concept in agriculture. Front. Public Health 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-H.; Kabir, E.; Jahan, S.A. Exposure to pesticides and the associated human health effects. Sci. Total Environ. 2017, 575, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Anway, M.D.; Leathers, C.; Skinner, M.K. Endocrine disruptor vinclozolin induced epigenetic transgenerational adult-onset disease. Endocrinology 2006, 147, 5515–5523. [Google Scholar] [CrossRef] [PubMed]
- Anway, M.D.; Memon, M.A.; Uzumcu, M.; Skinner, M.K. Transgenerational effect of the endocrine disruptor vinclozolin on male spermatogenesis. J. Androl. 2006, 27, 868–879. [Google Scholar] [CrossRef] [PubMed]
- Manikkam, M.; Tracey, R.; Guerrero-Bosagna, C.; Skinner, M.K. Pesticide and insect repellent mixture (permethrin and DEET) induces epigenetic transgenerational inheritance of disease and sperm epimutations. Reprod. Toxicol. 2012, 34, 708–719. [Google Scholar] [CrossRef] [Green Version]
- Anderson, O.S.; Sant, K.E.; Dolinoy, D.C. Nutrition and epigenetics: an interplay of dietary methyl donors, one-carbon metabolism and DNA methylation. J. Nutr. Biochem. 2012, 23, 853–859. [Google Scholar] [CrossRef] [Green Version]
- Kovacheva, V.P.; Mellott, T.J.; Davison, J.M.; Wagner, N.; Lopez-Coviella, I.; Schnitzler, A.C.; Blusztajn, J.K. Gestational choline deficiency causes global and Igf2 gene DNA hypermethylation by up-regulation of Dnmt1 expression. J. Biol. Chem. 2007, 282, 31777–31788. [Google Scholar] [CrossRef]
- Guarasci, F.; D’Aquila, P.; Mandalà, M.; Garasto, S.; Lattanzio, F.; Corsonello, A.; Passarino, G.; Bellizzi, D. Aging and nutrition induce tissue-specific changes on global DNA methylation status in rats. Mech. Ageing Dev. 2018, 174, 47–54. [Google Scholar] [CrossRef]
- Vucetic, Z.; Carlin, J.L.; Totoki, K.; Reyes, T.M. Epigenetic dysregulation of the dopamine system in diet-induced obesity. J. Neurochem. 2012, 120. [Google Scholar] [CrossRef] [PubMed]
- Chidambaram, B. Folate in pregnancy. J. Pediatr. Neurosci. 2012, 7, 81. [Google Scholar] [CrossRef] [PubMed]
- Paternain, L.; Martisova, E.; Campión, J.; Martínez, J.A.; Ramírez, M.J.; Milagro, F.I. Methyl donor supplementation in rats reverses the deleterious effect of maternal separation on depression-like behaviour. Behav. Brain Res. 2016, 299, 51. [Google Scholar] [CrossRef]
- Wolff, G.L.; Kodell, R.L.; Moore, S.R.; Cooney, C.A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998, 12, 949–957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoek, H.W.; Brown, A.S.; Susser, E. The Dutch famine and schizophrenia spectrum disorders. Soc. Psychiatry Psychiatr. Epidemiol. 1998, 33, 373–379. [Google Scholar] [CrossRef]
- Roseboom, T.; de Rooij, S.; Painter, R. The Dutch famine and its long-term consequences for adult health. Early Hum. Dev. 2006, 82, 485–491. [Google Scholar] [CrossRef]
- Lambrot, R.; Xu, C.; Saint-phar, S.; Chountalos, G.; Cohen, T.; Paquet, M.; Suderman, M.; Hallett, M.; Kimmins, S. Low paternal dietary folate alters the mouse sperm epigenome and is associated with negative pregnancy outcomes. Nat. Comm. 2013, 4, 2889. [Google Scholar] [CrossRef]
- McCoy, C.R.; Jackson, N.L.; Brewer, R.L.; Moughnyeh, M.M.; Smith, J.D.L.; Clinton, S.M. A paternal methyl donor depleted diet leads to increased anxiety- and depression-like behavior in adult rat offspring. Biosci. Rep. 2018, 38, BSR20180730. [Google Scholar] [CrossRef]
- Sivanathan, S.; Thavartnam, K.; Arif, S.; Elegino, T.; McGowan, P.O. Chronic high fat feeding increases anxiety-like behaviour and reduces transcript abundance of glucocorticoid signalling genes in the hippocampus of female rats. Behav. Brain Res. 2015, 286, 265–270. [Google Scholar] [CrossRef]
- Shen, W.; Wang, C.; Xia, L.; Fan, C.; Dong, H.; Deckelbaum, R.J.; Qi, K. Epigenetic modification of the leptin promoter in diet-induced obese mice and the effects of N-3 polyunsaturated fatty acids. Sci. Rep. 2014, 4, 5282. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Reinberg, D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001, 15, 2343–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyward, F.D.; Gilliam, D.; Coleman, M.A.; Gavin, C.F.; Wang, J.; Kaas, G.; Trieu, R.; Lewis, J.; Moulden, J.; Sweatt, J.D. Obesity weighs down memory through a mechanism involving the neuroepigenetic dysregulation of Sirt1. J. Neurosci. 2016, 36, 1324–1335. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Molet, J.; Ivy, A.; Baram, T.Z. New insights into early-life stress and behavioral outcomes. Curr. Opin. Behav. Sci. 2017, 14, 133–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunnar, M.; Quevedo, K. The neurobiology of stress and development. Ann. Rev. Psychol. 2006, 58, 145–173. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.; Binder, E.B. Current research trends in early life stress and depression: Review of human studies on sensitive periods, gene-environment interactions, and epigenetics. Exp. Neurol. 2012, 233, 102–111. [Google Scholar] [CrossRef] [PubMed]
- Cicchetti, D.; Hetzel, S.; Rogosch, F.A.; Handley, E.D.; Toth, S.L. An investigation of child maltreatment and epigenetic mechanisms of mental and physical health risk. Dev. Psychopathol. 2016, 28, 1305–1317. [Google Scholar] [CrossRef] [Green Version]
- Klengel, T.; Mehta, D.; Anacker, C.; Rex-Haffner, M.; Pruessner, J.C.; Pariante, C.M.; Pace, T.W.W.; Mercer, K.B.; Mayberg, H.S.; Bradley, B.; et al. Allele-specific FKBP5 DNA demethylation mediates gene-childhood trauma interactions. Nat. Neurosci. 2013, 16, 33–41. [Google Scholar] [CrossRef]
- Nelson, C.A. Hazards to early development: The biological embedding of early life adversity. Neuron 2017, 96, 262–266. [Google Scholar] [CrossRef]
- Naumova, O.Y.; Lee, M.; Koposov, R.; Szyf, M.; Dozier, M.; Grigorenko, E.L. Differential patterns of whole-genome DNA methylation in institutionalized children and children raised by their biological parents. Dev. Psychopathol. 2012, 24, 143–155. [Google Scholar] [CrossRef]
- Romens, S.E.; McDonald, J.; Svaren, J.; Pollak, S.D. Associations between early life stress and gene methylation in children. Child Dev. 2015, 86, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Provencal, N.; Massart, R.; Nemoda, Z.A.; Suomi, S. Epigenetics and Neuroendocrinology: Clinical Focus on Psychiatry; Springer International Publishing: New York, NY, USA, 2016; pp. 165–190. [Google Scholar]
- McGowan, P.O.; Sasaki, A.; D’Alessio, A.C.; Dymov, S.; Labonté, B.; Szyf, M.; Turecki, G.; Meaney, M.J. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 2009, 12, 342–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farrell, C.; Doolin, K.; O’ Leary, N.; Jairaj, C.; Roddy, D.; Tozzi, L.; Morris, D.; Harkin, A.; Frodl, T.; Nemoda, Z.; et al. DNA methylation differences at the glucocorticoid receptor gene in depression are related to functional alterations in hypothalamic–pituitary–adrenal axis activity and to early life emotional abuse. Psychiatry Res. 2018, 265, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.M.; Doherty, T.S.; Roth, T.L. Pharmacological manipulation of DNA methylation in adult female rats normalizes behavioral consequences of early-life maltreatment. Front. Behav. Neurosci. 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Bockmühl, Y.; Patchev, A.V.; Madejska, A.; Hoffmann, A.; Sousa, J.C.; Sousa, N.; Holsboer, F.; Almeida, O.F.X.; Spengler, D. Methylation at the CpG island shore region upregulates Nr3c1 promoter activity after early-life stress. Epigenetics 2015, 10, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivy, A.S.; Brunson, K.L.; Sandman, C.; Baram, T.Z. Dysfunctional nurturing behavior in rat dams with limited access to nesting material: A clinically relevant model for early-life stress. Neuroscience 2008, 154, 1132–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molet, J.; Maras, P.M.; Avishai-Eliner, S.; Baram, T.Z. Naturalistic rodent models of chronic early-life stress. Dev. Psychobiol. 2014, 56, 1675–1688. [Google Scholar] [CrossRef] [Green Version]
- Moloney, R.D.; Stilling, R.M.; Dinan, T.G.; Cryan, J.F. Early-life stress-induced visceral hypersensitivity and anxiety behavior is reversed by histone deacetylase inhibition. Neurogastroenterol. Motil. 2015, 27, 1831–1836. [Google Scholar] [CrossRef]
- Roth, T.L.; Lubin, F.D.; Funk, A.J.; Sweatt, J.D. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol. Psychiatry 2009, 65, 760–769. [Google Scholar] [CrossRef]
- Murgatroyd, C.; Patchev, A.V.; Wu, Y.; Micale, V.; Bockmühl, Y.; Fischer, D.; Holsboer, F.; Wotjak, C.T.; Almeida, O.F.X.; Spengler, D. Dynamic DNA methylation programs persistent adverse effects of early-life stress. Nat. Neurosci. 2009, 12, 1559–1566. [Google Scholar] [CrossRef]
- Lehmann, J.; Feldon, J. Long-term biobehavioral effects of maternal separation in the rat: consistent or confusing? Rev. Neurosci. 2000, 11, 383–408. [Google Scholar] [CrossRef] [PubMed]
- Millstein, R.A.; Holmes, A. Effects of repeated maternal separation on anxiety- and depression-related phenotypes in different mouse strains. Neurosci. Biobehav. Rev. 2007, 31, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.-D.; Bath, K.G.; Joels, M.; Korosi, A.; Larauche, M.; Lucassen, P.J.; Morris, M.J.; Raineki, C.; Roth, T.L.; Sullivan, R.M.; et al. Chronic early life stress induced by limited bedding and nesting (LBN) material in rodents: critical considerations of methodology, outcomes and translational potential. Stress 2017, 20, 421–448. [Google Scholar] [CrossRef] [PubMed]
- Doherty, T.S.; Blaze, J.; Keller, S.M.; Roth, T.L. Phenotypic outcomes in adolescence and adulthood in the scarcity-adversity model of low nesting resources outside the home cage. Dev. Psychobiol. 2017, 59, 703–714. [Google Scholar] [CrossRef]
- Gilles, E.E.; Schultz, L.; Baram, T.Z. Abnormal corticosterone regulation in an immature rat model of continuous chronic stress. Pediatr. Neurol. 1996, 15, 114–119. [Google Scholar] [CrossRef]
- Rice, C.J.; Sandman, C.A.; Lenjavi, M.R.; Baram, T.Z. A novel mouse model for acute and long-lasting consequences of early life stress. Endocrinology 2008, 149, 4892–4900. [Google Scholar] [CrossRef] [PubMed]
- Weaver, I.C.G.; Cervoni, N.; Champagne, F.A.; D’Alessio, A.C.; Sharma, S.; Seckl, J.R.; Dymov, S.; Szyf, M.; Meaney, M.J. Epigenetic programming by maternal behavior. Nat. Neurosci. 2004, 7, 847–854. [Google Scholar] [CrossRef]
- Blaze, J.; Asok, A.; Borrelli, K.; Tulbert, C.; Bollinger, J.; Ronca, A.E.; Roth, T.L. Intrauterine exposure to maternal stress alters Bdnf IV DNA methylation and telomere length in the brain of adult rat offspring. Int. J. Dev. Neurosci. 2017, 62, 56–62. [Google Scholar] [CrossRef] [Green Version]
- Class, Q.A.; Abel, K.M.; Khashan, A.S.; Rickert, M.E.; Dalman, C.; Larsson, H.; Hultman, C.M.; Långström, N.; Lichtenstein, P.; D’Onofrio, B.M. Offspring Psychopathology Following Preconception, Prenatal and Postnatal Maternal Bereavement Stress; Cambridge University Press: England, 2014; Volume 44, pp. 71–84. [Google Scholar]
- Cao-Lei, L.; Massart, R.; Suderman, M.J.; Machnes, Z.; Elgbeili, G.; Laplante, D.P.; Szyf, M.; King, S. DNA methylation signatures triggered by prenatal maternal stress exposure to a natural disaster: Project ice storm. PLoS ONE 2014, 9, e107653. [Google Scholar] [CrossRef]
- Peña, C.J.; Monk, C.; Champagne, F.A. Epigenetic effects of prenatal stress on 11β-hydroxysteroid dehydrogenase-2 in the placenta and fetal brain. PLoS ONE 2012, 7, e39791. [Google Scholar] [CrossRef]
- Mueller, B.R.; Bale, T.L. Sex-specific programming of offspring emotionality after stress early in pregnancy. J. Neurosci. 2008, 28, 9055–9065. [Google Scholar] [CrossRef]
- Chan, J.C.; Nugent, B.M.; Bale, T.L. Parental advisory: Maternal and paternal stress can impact offspring neurodevelopment. Biol. Psychiatry 2018, 83, 886–894. [Google Scholar] [CrossRef] [PubMed]
- Franklin, T.B.; Russig, H.; Weiss, I.C.; Gräff, J.; Linder, N.; Michalon, A.; Vizi, S.; Mansuy, I.M. Epigenetic transmission of the impact of early stress across generations. Biol. Psychiatry 2010, 68, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Scorza, P.; Duarte, C.S.; Hipwell, A.E.; Posner, J.; Ortin, A.; Canino, G.; Monk, C. Research Review: Intergenerational transmission of disadvantage: epigenetics and parents’ childhoods as the first exposure. J. Child Psychol. Psychiatry 2018. [Google Scholar] [CrossRef]
- Ward, I.; Zucchi, F.; C Robbins, J.; A Falkenberg, E.; Olson, D.; Benzies, K.; Metz, G. Transgenerational programming of maternal behaviour by prenatal stress. BMC Pregnancy Childbirth 2013, 13. [Google Scholar] [CrossRef]
- Gapp, K.; Bohacek, J.; Grossmann, J.; Brunner, A.M.; Manuella, F.; Nanni, P.; Mansuy, I.M. Potential of environmental enrichment to prevent transgenerational effects of paternal trauma. Neuropsychopharmacology 2016, 41, 2749–2758. [Google Scholar] [CrossRef]
- Skinner, M.K.; Manikkam, M.; Guerrero-Bosagna, C. Epigenetic transgenerational actions of environmental factors in disease etiology. Trends Endocrinol. Metab. 2010, 21, 214–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elwenspoek, M.M.C.; Hengesch, X.; Leenen, F.A.D.; Schritz, A.; Sias, K.; Schaan, V.K.; Mériaux, S.B.; Schmitz, S.; Bonnemberger, F.; Schächinger, H.; et al. Proinflammatory T cell status associated with early life adversity. J. Immunol. 2017, 199, 4046–4055. [Google Scholar] [CrossRef] [PubMed]
- Tyrrell, J.; Melzer, D.; Henley, W.; Galloway, T.S.; Osborne, N.J. Associations between socioeconomic status and environmental toxicant concentrations in adults in the USA: NHANES 2001-2010. Environ. Int. 2013, 59, 328–335. [Google Scholar] [CrossRef]
- Darmon, N.; Drewnowski, A. Does social class predict diet quality? Am. J. Clin. Nutr. 2008, 87, 1107–1117. [Google Scholar] [CrossRef]
- Baum, A.; Garofalo, J.P.; Yali, A.M. Socioeconomic status and chronic stress. Does stress account for SES effects on health? Ann. N. Y. Acad. Sci. 1999, 896, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Troller-Renfree, S.; Zeanah, C.H.; Nelson, C.A.; Fox, N.A. Neural and cognitive factors influencing the emergence of psychopathology: Insights from the Bucharest early intervention project. Child Dev. Perspect. 2018, 12, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Non, A.L.; Hollister, B.M.; Humphreys, K.L.; Childebayeva, A.; Esteves, K.; Zeanah, C.H.; Fox, N.A.; Nelson, C.A.; Drury, S.S. DNA methylation at stress-related genes is associated with exposure to early life institutionalization. Am. J. Phys. Anthropol. 2016, 161, 84. [Google Scholar] [CrossRef] [PubMed]
- Elwenspoek, M.M.C.; Kuehn, A.; Muller, C.P.; Turner, J.D. The effects of early life adversity on the immune system. Psychoneuroendocrinology 2017, 82, 140–154. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phillips, N.L.H.; Roth, T.L. Animal Models and Their Contribution to Our Understanding of the Relationship Between Environments, Epigenetic Modifications, and Behavior. Genes 2019, 10, 47. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10010047
Phillips NLH, Roth TL. Animal Models and Their Contribution to Our Understanding of the Relationship Between Environments, Epigenetic Modifications, and Behavior. Genes. 2019; 10(1):47. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10010047
Chicago/Turabian StylePhillips, Natalia Ledo Husby, and Tania L. Roth. 2019. "Animal Models and Their Contribution to Our Understanding of the Relationship Between Environments, Epigenetic Modifications, and Behavior" Genes 10, no. 1: 47. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10010047