An Exonic Switch Regulates Differential Accession of microRNAs to the Cd34 Transcript in Atherosclerosis Progression



Abstract
:1. Background
2. Methods
2.1. Ethics Statement
2.2. Patients and Samples
2.3. Mice
2.4. Reactives
2.5. Histological and Immunohistochemical CD34 Analysis
2.6. qPCR and miRNA Expression Data
2.7. Data Mining and Bioinformatic Analysis
2.8. Statistics
3. Results
3.1. MiR-125: A Complex miRNA Family
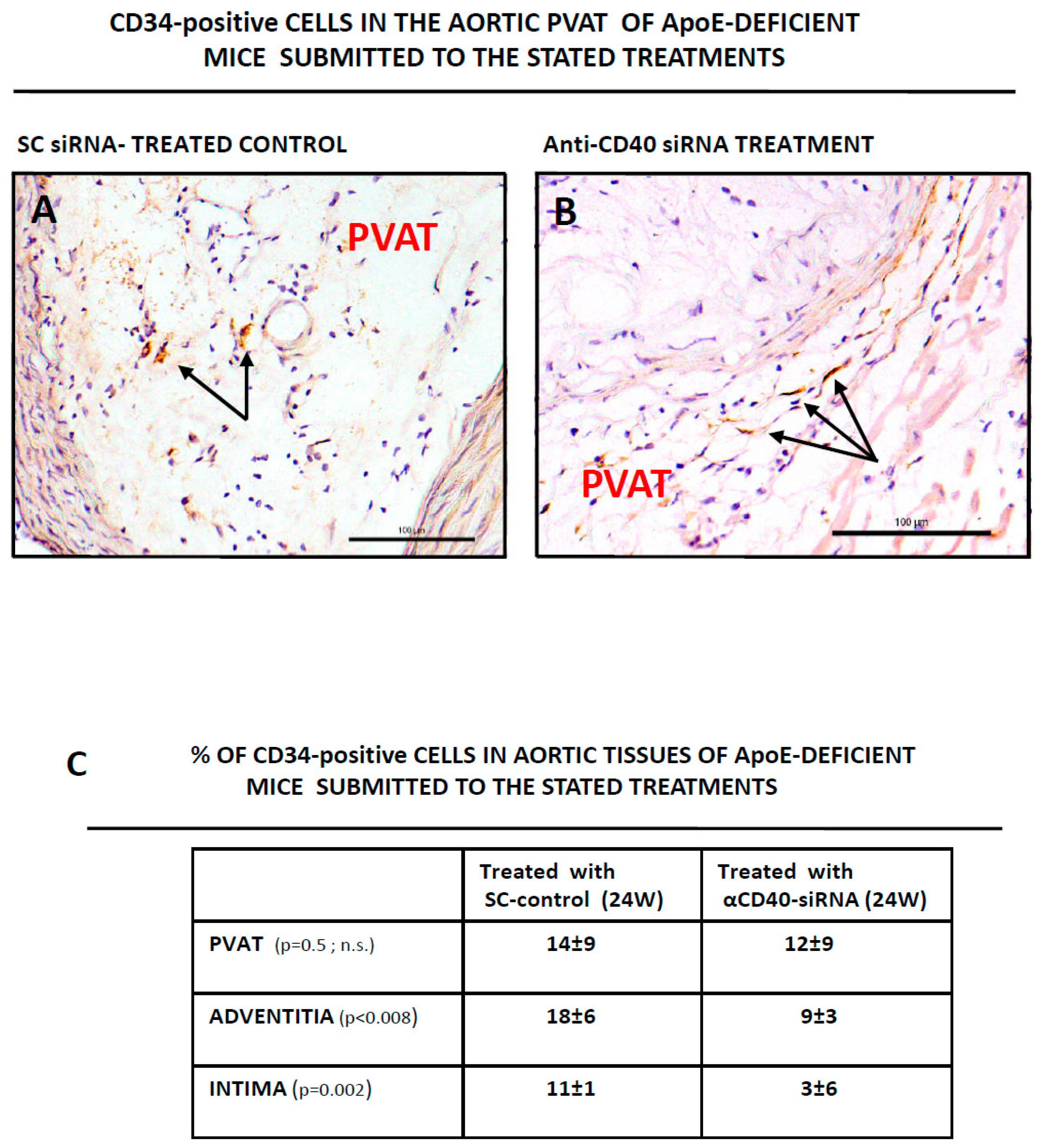
3.2. CD34-Positive CellsAre Clustered in Human Aortic Lesions
3.3. An Exonic Switch Regulates Differential Accession of miR-125, and Other miRNAs, to the CDS or to the 3’UTR of the Cd34 Transcript
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hueso, M.; Cruzado, J.M.; Torras, J.; Navarro, E. ALUminating the Path of Atherosclerosis Progression: Chaos Theory Suggests a Role for Alu Repeats in the Development of Atherosclerotic Vascular Disease. Int. J. Mol. Sci. 2018, 19, 1734. [Google Scholar] [CrossRef] [PubMed]
- Andreou, I.; Tousoulis, D.; Tentolouris, C.; Antoniades, C.; Stefanadis, C. Potential role of endothelial progenitor cells in the pathophysiology of heart failure: Clinical implications and perspectives. Atherosclerosis 2006, 189, 247–254. [Google Scholar] [CrossRef]
- Urbich, C.; Dimmeler, S. Endothelial progenitor cells: Characterization and role in vascular biology. Circ. Res. 2004, 95, 343–353. [Google Scholar] [CrossRef]
- Du, F.; Zhou, J.; Gong, R.; Huang, X.; Pansuria, M.; Virtue, A.; Li, X.; Wang, H.; Yang, X.F. Endothelial progenitor cells in atherosclerosis. Front. Biosci. 2012, 17, 2327–2349. [Google Scholar] [CrossRef]
- Rohde, E.; Malischnik, C.; Thaler, D.; Maierhofer, T.; Linkesch, W.; Lanzer, G.; Guelly, C.; Strunk, D. Blood monocytes mimic endothelial progenitor cells. Stem Cells 2006, 24, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Zalos, G.; Halcox, J.P.; Schenke, W.H.; Waclawiw, M.A.; Quyyumi, A.A.; Finkel, T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N. Engl. J. Med. 2003, 348, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Kuliszewski, M.A.; Li, S.H.; Szmitko, P.E.; Zucco, L.; Wang, C.H.; Badiwala, M.V.; Mickle, D.A.; Weisel, R.D.; Fedak, P.W.; et al. C-reactive protein attenuates endothelial progenitor cell survival, differentiation, and function: Further evidence of a mechanistic link between C-reactive protein and cardiovascular disease. Circulation 2004, 109, 2058–2067. [Google Scholar] [CrossRef] [PubMed]
- Riesinger, L.; Saemisch, M.; Nickmann, M.; Methe, H. CD34(+) circulating cells display signs of immune activation in patients with acute coronary syndrome. Heart Vessel. 2018. [Google Scholar] [CrossRef]
- Leung, K.T.; Chan, K.Y.; Ng, P.C.; Lau, T.K.; Chiu, W.M.; Tsang, K.S.; Li, C.K.; Kong, C.K.; Li, K. The tetraspanin CD9 regulates migration, adhesion, and homing of human cord blood CD34+ hematopoietic stem and progenitor cells. Blood 2011, 117, 1840–1850. [Google Scholar] [CrossRef]
- Suda, J.; Sudo, T.; Ito, M.; Ohno, N.; Yamaguchi, Y.; Suda, T. Two types of murine CD34 mRNA generated by alternative splicing. Blood 1992, 79, 2288–2295. [Google Scholar] [PubMed]
- Lanza, F.; Healy, L.; Sutherland, D.R. Structural and functional features of the CD34 antigen: An update. J. Biol. Regul. Homeost. Agents 2001, 15, 1–13. [Google Scholar]
- Lekka, E.; Hall, J. Noncoding RNAs in disease. FEBS Lett. 2018, 592, 2884–2900. [Google Scholar] [CrossRef]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.O.; Regine, M.A.; Subrata, C.; Long, S. Molecular mechanisms and genetic regulation in atherosclerosis. Int. J. Cardiol. Heart Vasc. 2018, 21, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.C.; Lan, H.Y. MicroRNAs in renal fibrosis. Front. Physiol. 2015, 6, 50. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Ye, Z.M.; Chen, S.; Luo, X.Y.; Chen, S.L.; Mao, L.; Li, Y.; Jin, H.; Yu, C.; Xiang, F.X.; et al. MicroRNA-23a-5p promotes atherosclerotic plaque progression and vulnerability by repressing ATP-binding cassette transporter A1/G1 in macrophages. J. Mol. Cell. Cardiol. 2018, 123, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Rink, C.; Khanna, S. MicroRNA in ischemic stroke etiology and pathology. Physiol. Genom. 2011, 43, 521–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Geng, Q.; Chen, H.; Zhang, J.; Cao, C.; Zhang, F.; Song, J.; Liu, C.; Liang, W. The potential inhibitory effects of miR19b on vulnerable plaque formation via the suppression of STAT3 transcriptional activity. Int. J. Mol. Med. 2018, 41, 859–867. [Google Scholar] [CrossRef]
- Price, N.L.; Rotllan, N.; Canfran-Duque, A.; Zhang, X.; Pati, P.; Arias, N.; Moen, J.; Mayr, M.; Ford, D.A.; Baldan, A.; et al. Genetic Dissection of the Impact of miR-33a and miR-33b during the Progression of Atherosclerosis. Cell Rep. 2017, 21, 1317–1330. [Google Scholar] [CrossRef] [Green Version]
- Chen, B.; Luo, L.; Wei, X.; Gong, D.; Jin, L. Altered Plasma miR-144 as a Novel Biomarker for Coronary Artery Disease. Ann. Clin. Lab. Sci. 2018, 48, 440–445. [Google Scholar]
- Reddy, L.L.; Shah, S.A.; Ponde, C.K.; Rajani, R.M.; Ashavaid, T.F. Circulating miRNA-33: A potential biomarker in patients with Coronary Artery Disease (CAD). Biomarkers 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhao, J.; Sun, D.; Wei, N. MiR-155 inhibits transformation of macrophages into foam cells via regulating CEH expression. Biomed. Pharmacother. 2018, 104, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Di Gregoli, K.; Mohamad Anuar, N.N.; Bianco, R.; White, S.J.; Newby, A.C.; George, S.J.; Johnson, J.L. MicroRNA-181b Controls Atherosclerosis and Aneurysms Through Regulation of TIMP-3 and Elastin. Circ. Res. 2017, 120, 49–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Ouyang, X.P.; Jiang, T.; Zheng, X.L.; He, P.P.; Zhao, G.J. MicroRNA-296: A promising target in the pathogenesis of atherosclerosis? Mol. Med. 2018, 24, 12. [Google Scholar] [CrossRef] [PubMed]
- Hueso, M.; De Ramon, L.; Navarro, E.; Ripoll, E.; Cruzado, J.M.; Grinyo, J.M.; Torras, J. Silencing of CD40 in vivo reduces progression of experimental atherogenesis through an NF-kappaB/miR-125b axis and reveals new potential mediators in the pathogenesis of atherosclerosis. Atherosclerosis 2016, 255, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.M.; Lin, K.Y.; Chen, Y.Q. Diverse functions of miR-125 family in different cell contexts. J. Hematol. Oncol. 2013, 6, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graff, J.W.; Dickson, A.M.; Clay, G.; McCaffrey, A.P.; Wilson, M.E. Identifying functional microRNAs in macrophages with polarized phenotypes. J. Biol. Chem. 2012, 287, 21816–21825. [Google Scholar] [CrossRef]
- Kim, S.W.; Ramasamy, K.; Bouamar, H.; Lin, A.P.; Jiang, D.; Aguiar, R.C. MicroRNAs miR-125a and miR-125b constitutively activate the NF-kappaB pathway by targeting the tumor necrosis factor alpha-induced protein 3 (TNFAIP3, A20). Proc. Natl. Acad. Sci. USA 2012, 109, 7865–7870. [Google Scholar] [CrossRef]
- Chen, T.; Huang, Z.; Wang, L.; Wang, Y.; Wu, F.; Meng, S.; Wang, C. MicroRNA-125a-5p partly regulates the inflammatory response, lipid uptake, and ORP9 expression in oxLDL-stimulated monocyte/macrophages. Cardiovasc. Res. 2009, 83, 131–139. [Google Scholar] [CrossRef] [Green Version]
- Xiu, L.; Xing, Q.; Mao, J.; Sun, H.; Teng, W.; Shan, Z. miRNA-125b-5p Suppresses Hypothyroidism Development by Targeting Signal Transducer and Activator of Transcription 3. Med. Sci. Monit. 2018, 24, 5041–5049. [Google Scholar] [CrossRef]
- Wang, S.; Wu, J.; Ren, J.; Vlantis, A.C.; Li, M.Y.; Liu, S.Y.W.; Ng, E.K.W.; Chan, A.B.W.; Luo, D.C.; Liu, Z.; et al. MicroRNA-125b Interacts with Foxp3 to Induce Autophagy in Thyroid Cancer. Mol. Ther. 2018, 26, 2295–2303. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.B.; Chen, J.; Zhu, J.W.; Yin, X.; You, H.Y.; Lin, Y.R.; Zhu, H.Q. MicroRNA125 inhibits RKO colorectal cancer cell growth by targeting VEGF. Int. J. Mol. Med. 2018, 42, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Natalia, M.A.; Alejandro, G.T.; Virginia, T.J.; Alvarez-Salas, L.M. MARK1 is a Novel Target for miR-125a-5p: Implications for Cell Migration in Cervical Tumor Cells. MicroRNA 2018, 7, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Tong, Z.; Liu, N.; Lin, L.; Guo, X.; Yang, D.; Zhang, Q. miR-125a-5p inhibits cell proliferation and induces apoptosis in colon cancer via targeting BCL2, BCL2L12 and MCL1. Biomed. Pharmacother. 2015, 75, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Hueso, M.; Torras, J.; Carrera, M.; Vidal, A.; Navarro, E.; Grinyo, J. Chronic Kidney Disease is associated with an increase of Intimal Dendritic cells in a comparative autopsy study. J. Inflamm. 2015, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Achuthan, P.; Akanni, W.; Allen, J.; Amode, M.R.; Armean, I.M.; Bennett, R.; Bhai, J.; Billis, K.; Boddu, S.; et al. Ensembl 2019. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaham, L.; Binder, V.; Gefen, N.; Borkhardt, A.; Izraeli, S. MiR-125 in normal and malignant hematopoiesis. Leukemia 2012, 26, 2011–2018. [Google Scholar] [CrossRef] [Green Version]
- Van der Valk, F.M.; Kuijk, C.; Verweij, S.L.; Stiekema, L.C.A.; Kaiser, Y.; Zeerleder, S.; Nahrendorf, M.; Voermans, C.; Stroes, E.S.G. Increased haematopoietic activity in patients with atherosclerosis. Eur. Heart J. 2017, 38, 425–432. [Google Scholar] [CrossRef]
- Sutherland, D.R.; Watt, S.M.; Dowden, G.; Karhi, K.; Baker, M.A.; Greaves, M.F.; Smart, J.E. Structural and partial amino acid sequence analysis of the human hemopoietic progenitor cell antigen CD34. Leukemia 1988, 2, 793–803. [Google Scholar]
- Fina, L.; Molgaard, H.V.; Robertson, D.; Bradley, N.J.; Monaghan, P.; Delia, D.; Sutherland, D.R.; Baker, M.A.; Greaves, M.F. Expression of the CD34 gene in vascular endothelial cells. Blood 1990, 75, 2417–2426. [Google Scholar] [PubMed]
- Schlingemann, R.O.; Rietveld, F.J.; de Waal, R.M.; Bradley, N.J.; Skene, A.I.; Davies, A.J.; Greaves, M.F.; Denekamp, J.; Ruiter, D.J. Leukocyte antigen CD34 is expressed by a subset of cultured endothelial cells and on endothelial abluminal microprocesses in the tumor stroma. Lab. Investig. 1990, 62, 690–696. [Google Scholar] [PubMed]
- Murashov, I.S.; Volkov, A.M.; Kazanskaya, G.M.; Kliver, E.E.; Chernyavsky, A.M.; Nikityuk, D.B.; Lushnikova, E.L. Immunohistochemical Features of Different Types of Unstable Atherosclerotic Plaques of Coronary Arteries. Bull. Exp. Biol. Med. 2018, 166, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Wojtowicz, E.E.; Lechman, E.R.; Hermans, K.G.; Schoof, E.M.; Wienholds, E.; Isserlin, R.; van Veelen, P.A.; Broekhuis, M.J.; Janssen, G.M.; Trotman-Grant, A.; et al. Ectopic miR-125a Expression Induces Long-Term Repopulating Stem Cell Capacity in Mouse and Human Hematopoietic Progenitors. Cell Stem Cell 2016, 19, 383–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, T.; Nakashima, Y.; Yonemitsu, Y.; Sumiyoshi, S.; Chen, Y.X.; Akishima, Y.; Ishii, T.; Iida, M.; Sueishi, K. Angiogenesis and lymphangiogenesis and expression of lymphangiogenic factors in the atherosclerotic intima of human coronary arteries. Hum. Pathol. 2005, 36, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Koutouzis, M.; Nomikos, A.; Nikolidakis, S.; Tzavara, V.; Andrikopoulos, V.; Nikolaou, N.; Barbatis, C.; Kyriakides, Z.S. Statin treated patients have reduced intraplaque angiogenesis in carotid endarterectomy specimens. Atherosclerosis 2007, 192, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Auclair, S.; Milenkovic, D.; Besson, C.; Chauvet, S.; Gueux, E.; Morand, C.; Mazur, A.; Scalbert, A. Catechin reduces atherosclerotic lesion development in apo E-deficient mice: A transcriptomic study. Atherosclerosis 2009, 204, e21–e27. [Google Scholar] [CrossRef]
- Schmidt-Lucke, C.; Rossig, L.; Fichtlscherer, S.; Vasa, M.; Britten, M.; Kamper, U.; Dimmeler, S.; Zeiher, A.M. Reduced number of circulating endothelial progenitor cells predicts future cardiovascular events: Proof of concept for the clinical importance of endogenous vascular repair. Circulation 2005, 111, 2981–2987. [Google Scholar] [CrossRef]
- Boilson, B.A.; Kiernan, T.J.; Harbuzariu, A.; Nelson, R.E.; Lerman, A.; Simari, R.D. Circulating CD34+ cell subsets in patients with coronary endothelial dysfunction. Nat. Clin.Pract. Cardiovasc. Med. 2008, 5, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Fan, J.; Li, H.; Nie, X.; Yin, Z.; Zhao, Y.; Zhang, X.; Yuan, S.; Li, Y.; Chen, C.; Wang, D.W. MiR-665 aggravates heart failure via suppressing CD34-mediated coronary microvessel angiogenesis. Aging 2018, 10, 2459–2479. [Google Scholar] [CrossRef]
- Hou, Q.; Huang, Y.; Luo, Y.; Wang, B.; Liu, Y.; Deng, R.; Zhang, S.; Liu, F.; Chen, D. MiR-351 negatively regulates osteoblast differentiation of MSCs induced by (+)-cholesten-3-one through targeting VDR. Am. J. Transl. Res. 2017, 9, 4963–4973. [Google Scholar] [PubMed]
- Zhang, Y.; Liu, Y.; Zhang, H.; Wang, M.; Zhang, J. Mmu-miR-351 attenuates the survival of cardiac arterial endothelial cells through targeting STAT3 in the atherosclerotic mice. Biochem. Biophys. Res. Commun. 2015, 468, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Greaves, M.F.; Molgaard, H.V. The gene encoding the stem cell antigen, CD34, is conserved in mouse and expressed in haemopoietic progenitor cell lines, brain, and embryonic fibroblasts. Int. Immunol. 1991, 3, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Fackler, M.J.; Civin, C.I.; Sutherland, D.R.; Baker, M.A.; May, W.S. Activated protein kinase C directly phosphorylates the CD34 antigen on hematopoietic cells. J. Biol. Chem. 1990, 265, 11056–11061. [Google Scholar] [PubMed]
- Brummer, A.; Hausser, J. MicroRNA binding sites in the coding region of mRNAs: Extending the repertoire of post-transcriptional gene regulation. BioEssays 2014, 36, 617–626. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| miRNAExpression | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cluster 99b/let7e/125a | Cluster 100/let7a-2/125b-1 | Cluster 99a/let7c-1/125b-2 | |||||||
| 99b | let7e | 125a | 100 | let7a-2 | 125b-1 | 99a | let7c-1 | 125b-2 | |
| C8W (n = 2) | 10.95 ± 0.2 | 8.22 ± 0.2 | 9.43 ± 0.5 | 10.64 ± 0.5 | 13.47 * | 10.47 ± 0.6 | 10.6 ± 0.5 | 10.8 ± 0.6 | 10.47 ± 0.6 |
| SC24W (n = 3) | 4.51 ± 1.3 | 3.35 ± 1.8 | 6.7 ± 0.6 | 5.57 ± 0.8 | 7.20 ± 1.8 | 3.97 ± 0.6 | 6.32 ± 0.3 | 3.03 ± 2.2 | 3.97 ± 0.6 |
| T24W (n = 3) | 6.27 ± 2.8 | 4.99 ± 2.8 | 7.16 ± 1.6 | 6.15 ± 1.8 | 7.5 ± 2.8 | 6.26 ± 2.0 | 6.98 ± 2.1 | 5.74 ± 4.1 | 6.26 ± 2.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hueso, M.; Cruzado, J.M.; Torras, J.; Navarro, E. An Exonic Switch Regulates Differential Accession of microRNAs to the Cd34 Transcript in Atherosclerosis Progression. Genes 2019, 10, 70. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10010070
Hueso M, Cruzado JM, Torras J, Navarro E. An Exonic Switch Regulates Differential Accession of microRNAs to the Cd34 Transcript in Atherosclerosis Progression. Genes. 2019; 10(1):70. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10010070
Chicago/Turabian StyleHueso, Miguel, Josep M. Cruzado, Joan Torras, and Estanis Navarro. 2019. "An Exonic Switch Regulates Differential Accession of microRNAs to the Cd34 Transcript in Atherosclerosis Progression" Genes 10, no. 1: 70. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10010070