Comparative Study of Gut Microbiota in Wild and Captive Giant Pandas (Ailuropoda melanoleuca)

Abstract
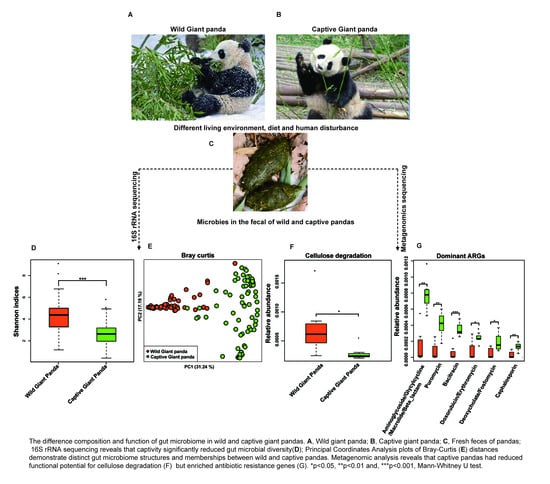
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Methods
2.1. Sample Collection, DNA Extraction, and Sequencing
2.2. S Amplicon Data Analysis
2.3. Metagenome Analysis
2.3.1. Quality Control
2.3.2. De Novo Assembly and Construction of a Non-Redundant Gut Microbiome Gene Set
2.3.3. Read Normalization
2.3.4. Gene Abundance Profiling
2.3.5. Gene Functional Annotation
2.3.6. Analysis of Resistance Genes and Virulence Factors
2.3.7. α Diversity, β Diversity, and Statistical Comparisons of the Abundances of CAZy Families, Select Gene Groups, and Viruses
2.3.8. Phylogenetic Analysis of Metagenome Dataset
Enzyme Activity Assay
Ethics Approval and Consent to Participate
3. Results
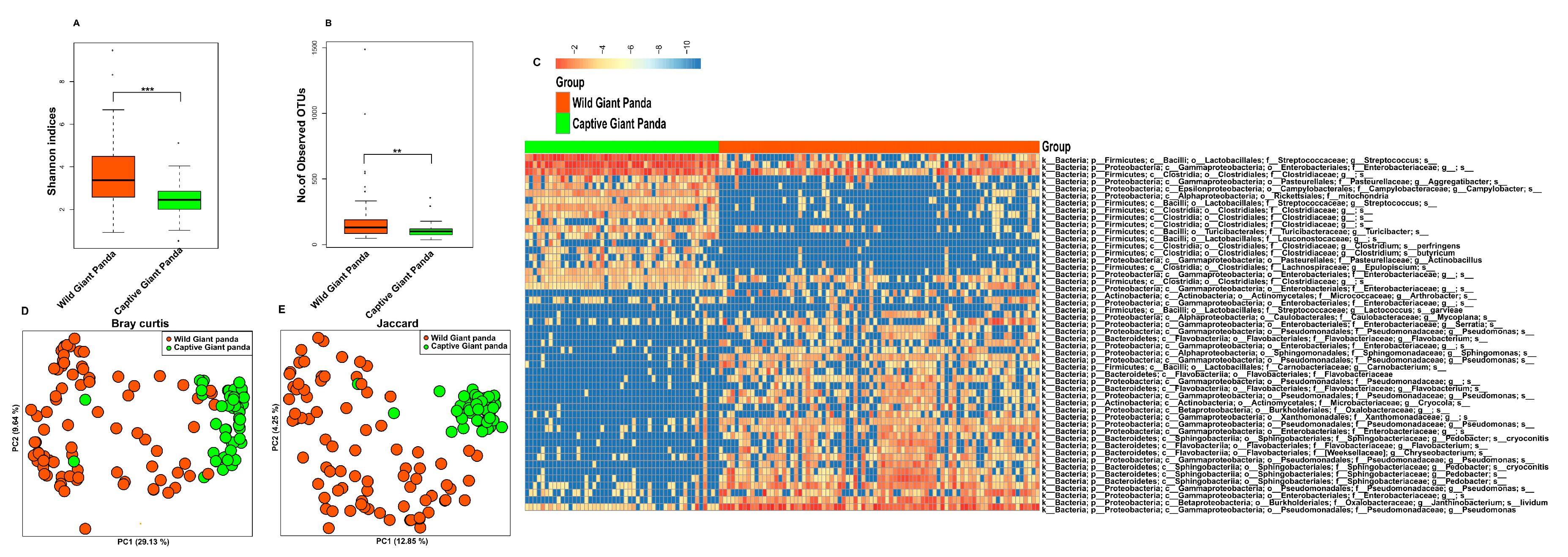
3.1. Captivity Reduced Microbial Diversity and Altered the Gut Microbiome Structure of Giant Pandas
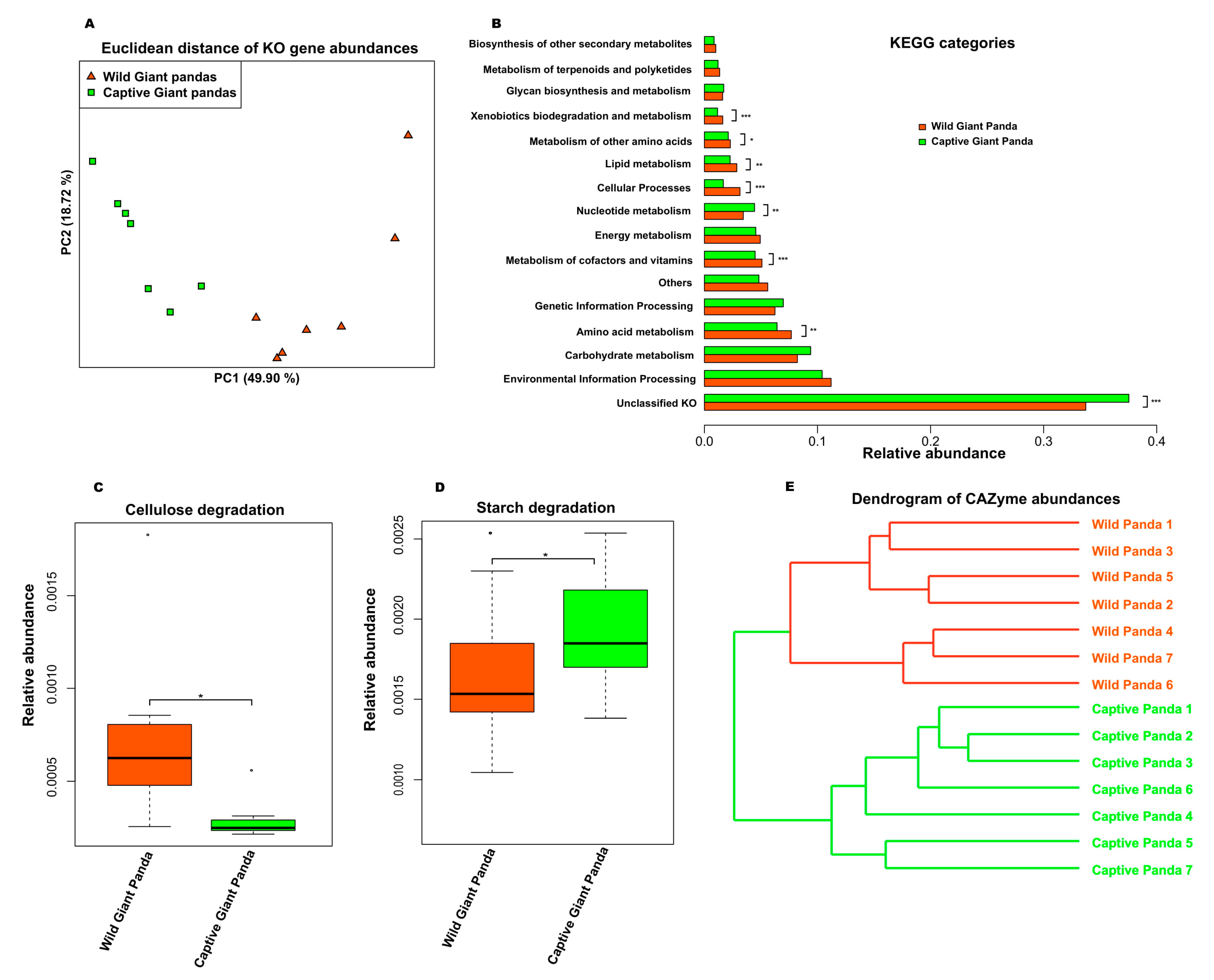
3.2. Captivity Reduced Functional Potential for Cellulose Degradation but Increased Metabolic Pathways for Starch Metabolism
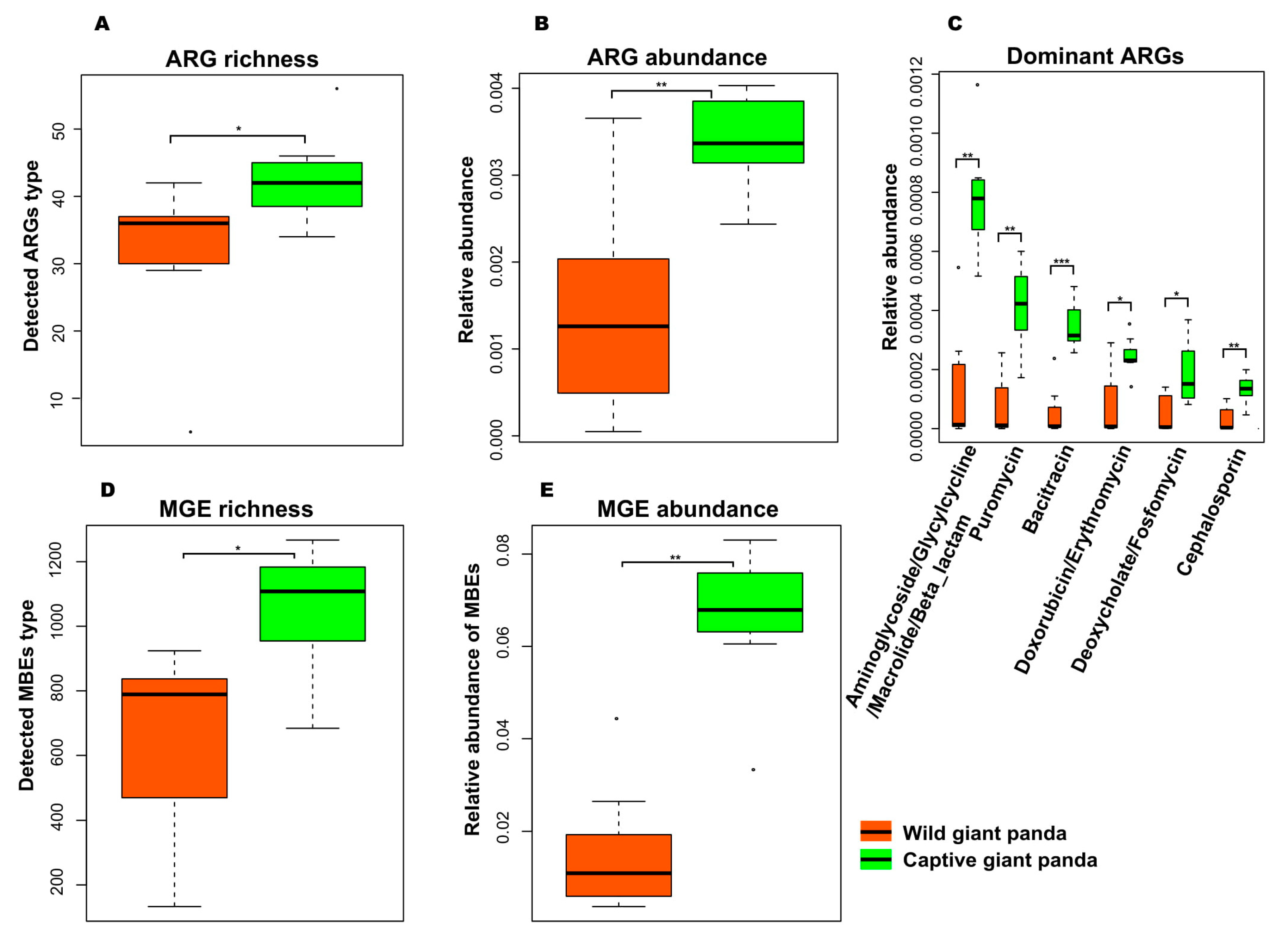
3.3. Captivity Enriched Antibiotic Resistance Genes in the Microbiomes of Captive Pandas
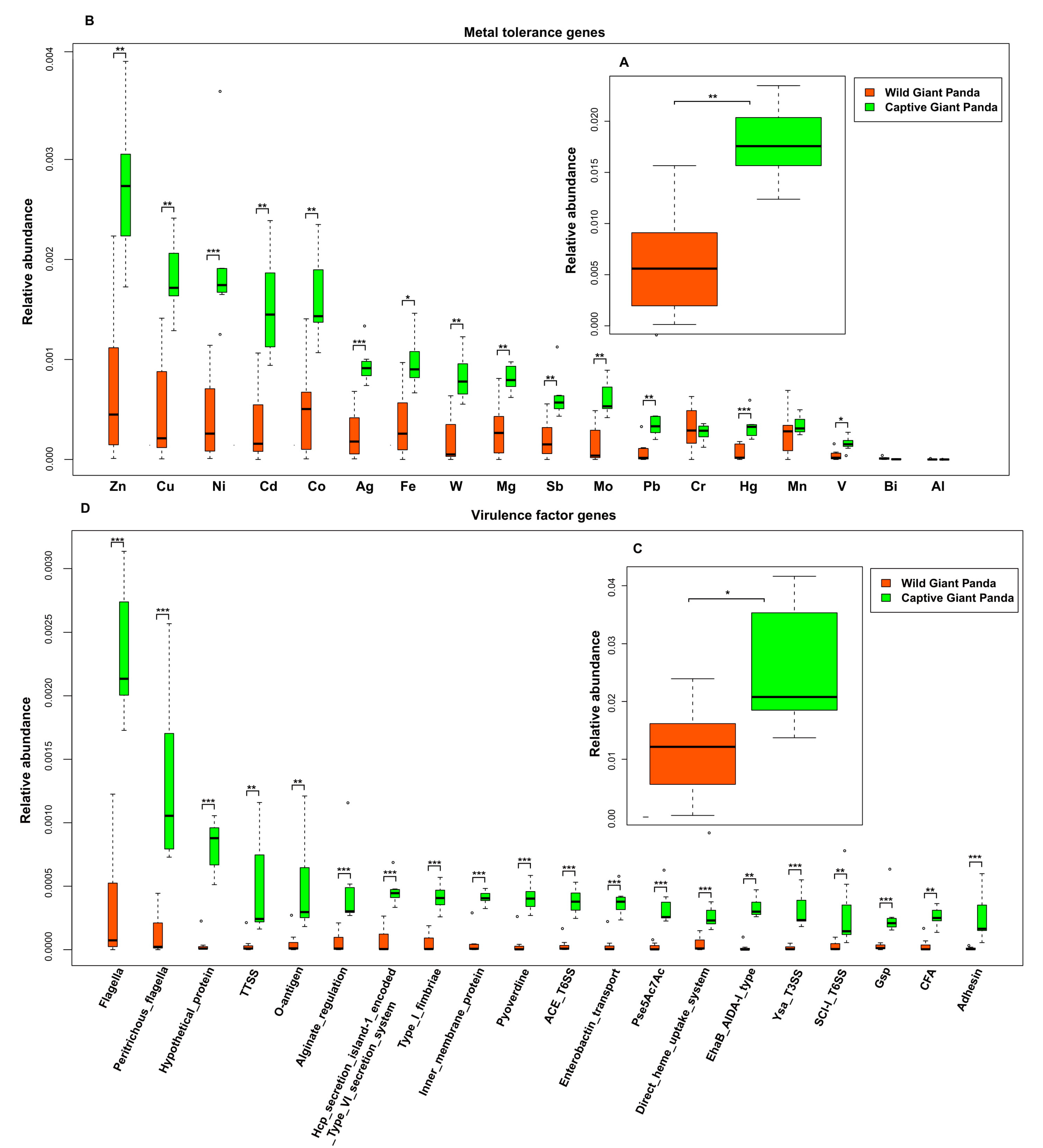
3.4. Captivity Enriched Heavy Metal Tolerance Genes in the Microbiomes of Captive Pandas
3.5. Captivity Enriched Virulence Factors in the Microbiomes of Captive Pandas
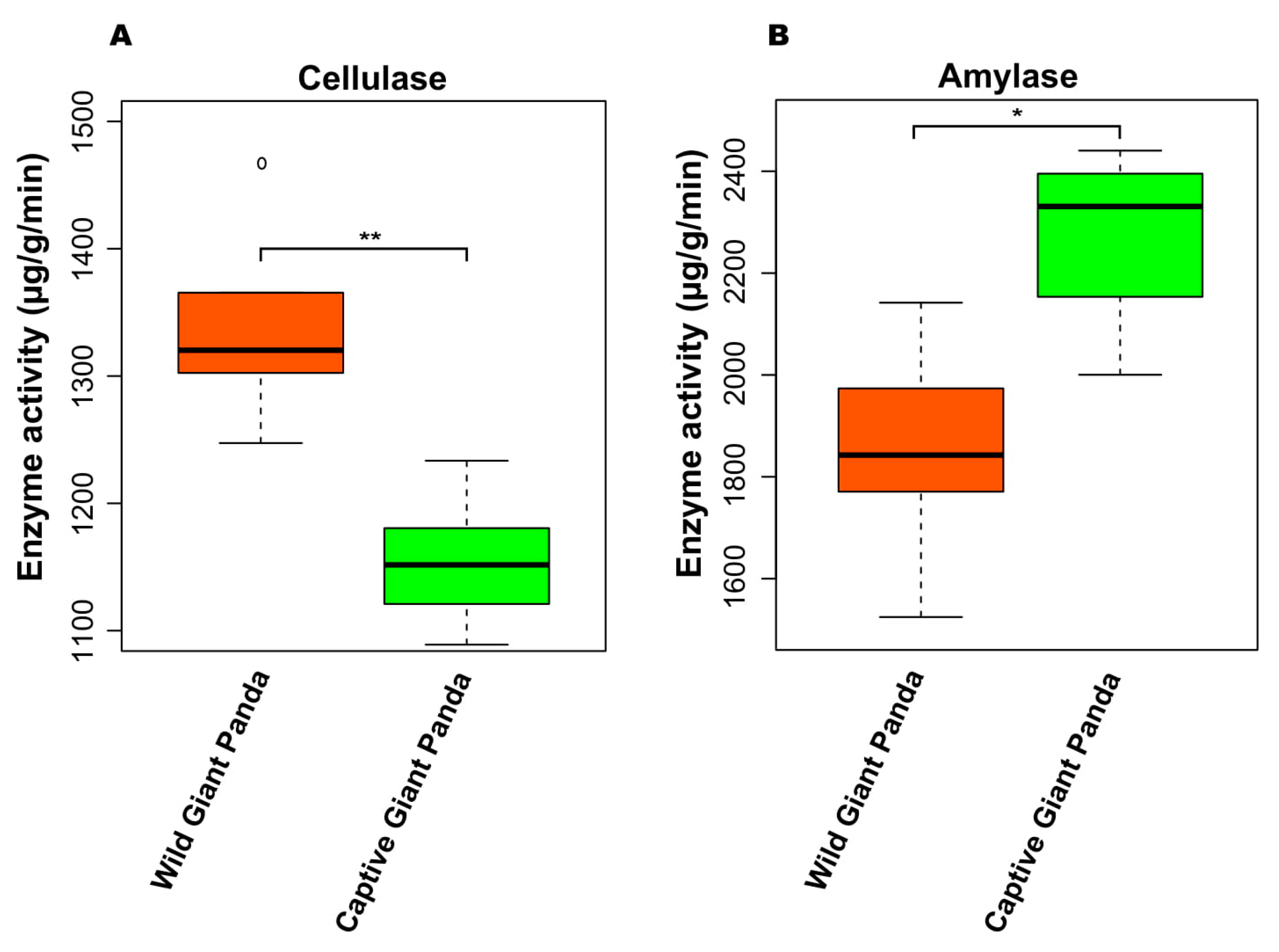
3.6. Enzyme Activity Between Giant Pandas
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| FNNR | Fengtongzhai National Nature Reserve |
| WNNR | Wolong National Nature Reserve |
| CCRC | China Conservation and Research Center for the Giant Panda |
| OUT | operational taxonomic unit |
| BWA | Burrows-Wheeler Aligner |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| ARG | antibiotic resistance genes |
| MGE | mobile genetic elements |
| MRG | metal resistance genes |
| VFDB | virulence factor database |
| TTSS | type III secretion system |
References
- Williams, C.L.; Dill-Mcfarland, K.A.; Vandewege, M.W.; Sparks, D.L.; Willard, S.T.; Kouba, A.J.; Suen, G.; Brown, A.E.J.F.I.M. Dietary Shifts May Trigger Dysbiosis and Mucous Stools in Giant Pandas (Ailuropoda melanoleuca). Front. Microbiol. 2016, 7, 661. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Mainka, S.A. Review of Mortality of the Giant Panda (Ailuropoda melanoleuca). J. Zoo Wildl. Med. 1993, 24, 425–429. [Google Scholar]
- Janssen, D.L.; Morris, P.; Sutherland-Smith, M.; Greenberg, M.; Li, D.; Mauroo, N.; Lucy, S. Medical management of captive adult and geriatric giant pandas. In Giant Pandas: Biology, Veterinary Medicine and Management; Zhang, A., Wildt, D.E., Janssen, D.L., Zhang, H., Ellis, S., Eds.; Cambridge University Press: Cambridge, UK, 2006; pp. 353–376. [Google Scholar] [CrossRef]
- Li, R.Q.; Fan, W.; Tian, G.; Zhu, H.M.; He, L.; Cai, J.; Huang, Q.F.; Cai, Q.L.; Li, B.; Bai, Y.Q.; et al. The sequence and de novo assembly of the giant panda genome. Nature 2010, 463, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wu, Q.; Dai, J.; Zhang, S.; Wei, F. Evidence of cellulose metabolism by the giant panda gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108, 17714–17719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, K.R.; Yeoman, C.J.; Kent, A.; Righini, N.; Carbonero, F.; Estrada, A.; Gaskins, H.R.; Stumpf, R.M.; Yildirim, S.; Torralba, M.; et al. Habitat degradation impacts black howler monkey (Alouatta pigra) gastrointestinal microbiomes. ISME J. 2013, 7, 1344–1353. [Google Scholar] [CrossRef]
- Bahrndorff, S.; Alemu, T.; Alemneh, T.; Lund Nielsen, J. The Microbiome of Animals: Implications for Conservation Biology. Int. J. Genom. 2016, 2016, 5304028. [Google Scholar] [CrossRef]
- Kong, F.; Zhao, J.; Han, S.; Zeng, B.; Yang, J.; Si, X.; Yang, B.; Yang, M.; Xu, H.; Li, Y. Characterization of the gut microbiota in the red panda (Ailurus fulgens). PLoS ONE 2014, 9, e87885. [Google Scholar] [CrossRef]
- Clayton, J.B.; Vangay, P.; Huang, H.; Ward, T.; Hillmann, B.M.; Al-Ghalith, G.A.; Travis, D.A.; Long, H.T.; Tuan, B.V.; Minh, V.V. Captivity humanizes the primate microbiome. Proc. Natl. Acad. Sci. USA 2016, 113, 10376. [Google Scholar] [CrossRef]
- Borbóngarcía, A.; Reyes, A.; Vivesflórez, M.; Caballero, S. Captivity Shapes the Gut Microbiota of Andean Bears: Insights into Health Surveillance. Front. Microbiol. 2017, 8, 1316. [Google Scholar] [CrossRef]
- Guan, Y.; Yang, H.; Han, S.; Feng, L.; Wang, T.; Ge, J.J.A.E. Correction to: Comparison of the gut microbiota composition between wild and captive sika deer (Cervus nippon hortulorum) from feces by high-throughput sequencing. AMB Express 2018, 8, 15. [Google Scholar] [CrossRef]
- Metcalf, J.L.; Song, S.J.; Morton, J.T.; Weiss, S.; Seguinorlando, A.; Joly, F.; Feh, C.; Taberlet, P.; Coissac, E.; Amir, A. Evaluating the impact of domestication and captivity on the horse gut microbiome. Sci. Rep. 2017, 7, 15497. [Google Scholar] [CrossRef]
- Roura, A.; Doyle, S.R.; Nande, M.; Strugnell, J.M. You Are What You Eat: A Genomic Analysis of the Gut Microbiome of Captive and Wild Octopus vulgaris Paralarvae and Their Zooplankton Prey. Front. Physiol. 2017, 8, 362. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, C.; Courcelle, M.; Redivo, B.; Derome, N. Structural and compositional mismatch between captive and wild Atlantic salmon (Salmo salar) parrs gut microbiota highlights the relevance of integrating molecular ecology for management and conservation methods. Evol. Appl. 2018, 11, 1671–1685. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Gao, H.; Wu, G.; Qin, W.; Song, P.; Wang, L.; Chen, J.; Cai, Z.; Zhang, T. Comparison of gut microbiota diversity between wild and captive bharals (Pseudois nayaur). BMC Vet. Res. 2019, 15, 243. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Chi, X.; Qin, W.; Wang, L.; Zhang, T.J.J.o.A.M. Comparison of the Gut Microbiota Composition between the Wild and Captive Tibetan Wild Ass ( Equus kiang ). J. Appl. Microbiol. 2019, 126, 1869–1878. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K.M.; Nguyen, B.N.; Neumann, L.M.; Miller, M.; Buss, P.; Daniels, S.; Ahn, M.J.; Crandall, K.A.; Pukazhenthi, B. Gut microbiome differences between wild and captive black rhinoceros—Implications for rhino health. Sci. Rep. 2019, 9, 7570. [Google Scholar] [CrossRef]
- Schmidt, E.; Mykytczuk, N.; Schulte-Hostedde, A. Effects of the captive and wild environment on diversity of the gut microbiome of deer mice (Peromyscus maniculatus). ISME J. 2019, 13, 1293. [Google Scholar] [CrossRef]
- Kohl, K.D.; Brun, A.; Magallanes, M.; Brinkerhoff, J.; Laspiur, A.; Acosta, J.C.; Caviedes-Vidal, E.; Bordenstein, S.R. Gut microbial ecology of lizards: Insights into diversity in the wild, effects of captivity, variation across gut regions and transmission. Mol. Ecol. 2017, 26, 1175–1189. [Google Scholar] [CrossRef]
- Wei, F.; Wu, Q.; Hu, Y.; Huang, G.; Nie, Y.; Yan, L. Conservation metagenomics: A new branch of conservation biology. Sci. China Life Sci. 2019, 62, 168–178. [Google Scholar] [CrossRef]
- Xue, Z.; Zhang, W.; Wang, L.; Hou, R.; Zhang, M.; Fei, L.; Zhang, X.; Huang, H.; Bridgewater, L.C.; Jiang, Y.; et al. The bamboo-eating giant panda harbors a carnivore-like gut microbiota, with excessive seasonal variations. MBio 2015, 6, e00022-15. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, W.; Hou, R.; Zhang, L.; Schmitz-Esser, S.; Sun, H.; Xie, J.; Zhang, Y.; Wang, C.; Li, L.; et al. Age-associated microbiome shows the giant panda lives on hemicelluloses, not on cellulose. ISME J. 2018, 12, 1319–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, R.; Xu, L.; Hu, T.; Chen, H.; Zhu, L. The “wildness” of the giant panda gut microbiome and its relevance to effective translocation. Glob. Ecol. Conserv. 2019, 18, e00644. [Google Scholar] [CrossRef]
- Zhang, Z.; He, G.; Wang, X.; Zhong, S.; Zhang, A.; Li, G. The study on the giant panda^ s intestinal flora. Acta Theriol. Sin. 1995, 15, 170–175. [Google Scholar]
- Qiao, M.; Connor, T.; Shi, X.; Huang, J.; Huang, Y.; Zhang, H.; Ran, J. Population genetics reveals high connectivity of giant panda populations across human disturbance features in key nature reserve. Ecol. Evol. 2019, 9, 1809–1819. [Google Scholar] [CrossRef]
- Alnasir, J.; Shanahan, H.P. Investigation into the annotation of protocol sequencing steps in the sequence read archive. Gigascience 2015, 4, 23. [Google Scholar] [CrossRef]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a Dual-Index Sequencing Strategy and Curation Pipeline for Analyzing Amplicon Sequence Data on the MiSeq Illumina Sequencing Platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194. [Google Scholar] [CrossRef]
- Breiman, L. Random forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt Removes Adapter Sequences From High-Throughput Sequencing Reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, Z.; Lu, Y.; Li, L.; Zhao, Q.; Feng, Q.; Gao, Z.; Lu, H.; Hu, T.; Yao, N.; Liu, K.; et al. The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat. Genet. 2013, 45, 456–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Marc, L.; Bjoern, U.J.B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Noguchi, H.; Park, J.; Takagi, T. MetaGene: Prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006, 34, 5623–5630. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Liu, B.; Pop, M. ARDB-Antibiotic Resistance Genes Database. Nucleic Acids Res. 2009, 37, D443–D447. [Google Scholar] [CrossRef] [PubMed]
- Leplae, R.; Lima-Mendez, G.; Toussaint, A. ACLAME: A CLAssification of Mobile genetic Elements, update 2010. Nucleic Acids Res. 2010, 38, D57–D61. [Google Scholar] [CrossRef] [PubMed]
- Pal, C.; Bengtsson-Palme, J.; Rensing, C.; Kristiansson, E.; Larsson, D.G. BacMet: Antibacterial biocide and metal resistance genes database. Nucleic Acids Res. 2014, 42, D737–D743. [Google Scholar] [CrossRef]
- Chen, L.H.; Zheng, D.D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis-10 years on. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef]
- Rampelli, S.; Schnorr, S.L.; Consolandi, C.; Turroni, S.; Severgnini, M.; Peano, C.; Brigidi, P.; Crittenden, A.N.; Henry, A.G.; Candela, M. Metagenome Sequencing of the Hadza Hunter-Gatherer Gut Microbiota. Curr. Biol. 2015, 25, 1682–1693. [Google Scholar] [CrossRef] [Green Version]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. PAST: Paleontological Statistics Software Package for Education and Data Analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Pope, P.B.; Denman, S.E.; Jones, M.; Tringe, S.G.; Barry, K.; Malfatti, S.A.; McHardy, A.C.; Cheng, J.F.; Hugenholtz, P.; McSweeney, C.S.; et al. Adaptation to herbivory by the Tammar wallaby includes bacterial and glycoside hydrolase profiles different from other herbivores. Proc. Natl. Acad. Sci. USA 2010, 107, 14793–14798. [Google Scholar] [CrossRef] [Green Version]
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for Glycogenomics. Nucleic Acids Res. 2009, 37, D233. [Google Scholar] [CrossRef]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9. [Google Scholar] [CrossRef]
- Abou-Taleb, K.A.; Mashhoor, W.A.; Nasr, S.A.; Sharaf, M.S.; Abdel-Azeem, H.H. Nutritional and Environmental Factors Affecting Cellulase Production by Two Strains of Cellulolytic Bacilli. Aust. J. Basic Appl. Sci. 2009, 3, 2429–2436. [Google Scholar]
- Jia, H.; Chen, X.R.; Lu, G.X.; Kong, Y.L.; Yang, C.D.J.A.P.S. Isolation of cellulose-degrading bacteria and determination of their degradation activity. Int. J. Microbiol. 2016, 2012, 578925. [Google Scholar]
- Black, H. Determination of Sodium Carboxymethylcellulose in Detergent Mixtures by Anthrone Method. Anal. Chem. 1951, 23, 1792–1795. [Google Scholar] [CrossRef]
- Miller, A.L.G. Use of Dinitrosalicylic Acid Reagent for Determination of Reducing Sugar. Anal. Biochem. 1959, 31, 426–428. [Google Scholar] [CrossRef]
- van Hoek, A.H.A.M.; Mevius, D.; Guerra, B.; Mullany, P.; Roberts, A.P.; Aarts, H.J.M. Acquired antibiotic resistance genes: An overview. Front. Microbiol. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Li, L.G.; Xia, Y.; Zhang, T. Co-occurrence of antibiotic and metal resistance genes revealed in complete genome collection. ISME J. 2017, 11, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Dierenfeld, E.S.; Qiu, X.; Mainka, S.A.; Liu, W.X. Giant panda diets fed in five Chinese facilities: An assessment. Zoo Biol. 1995, 14, 211–222. [Google Scholar] [CrossRef]
- Schaller, G.B.; Hu, J.C.; Pan, W.S.; Zhu, J. The Giant Panda of Wolong; University of Chicago Press: Chicago, IL, USA, 1985. [Google Scholar]
- Frédéric, D.; Metcalf, J.L.; Laura, W.P.; Se Jin, S.; Antonio, G.; Rob, K. Convergence of gut microbiomes in myrmecophagous mammals. Mol. Ecol. 2014, 23, 1301–1317. [Google Scholar]
- Muegge, B.D.; Justin, K.; Dan, K.; Clemente, J.C.; Antonio, G.; Luigi, F.; Bernard, H.; Rob, K.; Gordon, J.I. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 2011, 332, 970. [Google Scholar] [CrossRef]
- Ley, R.E.; Hamady, M.; Lozupone, C.; Turnbaugh, P.J.; Ramey, R.R.; Bircher, J.S.; Schlegel, M.L.; Tucker, T.A.; Schrenzel, M.D.; Knight, R. Evolution of mammals and their gut microbes. Science 2008, 320, 1647–1651. [Google Scholar] [CrossRef]
- Wei, F.W.; Feng, Z.J.; Wang, Z.; Li, M. Feeding strategy and resource partitioning between giant and red pandas. Mammalia 1999, 63, 417–430. [Google Scholar] [CrossRef]
- Guo, W.; Mishra, S.; Zhao, J.; Tang, J.; Zeng, B.; Kong, F.; Ning, R.; Li, M.; Zhang, H.; Zeng, Y. Metagenomic Study Suggests That the Gut Microbiota of the Giant Panda (Ailuropoda melanoleuca) May Not Be Specialized for Fiber Fermentation. Front. Microbiol. 2018, 9, 229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome Med. 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Mulder, I.E.; Schmidt, B.; Stokes, C.R.; Lewis, M.; Bailey, M.; Aminov, R.I.; Prosser, J.I.; Gill, B.P.; Pluske, J.R.; Mayer, C.D.; et al. Environmentally-acquired bacteria influence microbial diversity and natural innate immune responses at gut surfaces. BMC Biol. 2009, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xia, P.; Wang, H.; Yu, H.; Giesy, J.P.; Zhang, Y.; Mora, M.A.; Zhang, X. Effects of captivity and artificial breeding on microbiota in feces of the red-crowned crane (Grus japonensis). Sci. Rep. 2016, 6, 33350. [Google Scholar] [CrossRef] [Green Version]
- Sonnenburg, E.D.; Smits, S.A.; Tikhonov, M.; Higginbottom, S.K.; Wingreen, N.S.; Sonnenburg, J.L. Diet-induced extinctions in the gut microbiota compound over generations. Nature 2016, 529, 212–215. [Google Scholar] [CrossRef] [Green Version]
- Pal, C.; Bengtsson-Palme, J.; Kristiansson, E.; Larsson, D.G. The structure and diversity of human, animal and environmental resistomes. Microbiome 2016, 4, 54. [Google Scholar] [CrossRef]
- Chen, Y.P.; Zheng, Y.J.; Liu, Q.; Song, Y.; An, Z.S.; Ma, Q.Y.; Ellison, A.M. Atmospheric deposition exposes Qinling pandas to toxic pollutants. Ecol. Appl. 2017, 27, 343–348. [Google Scholar] [CrossRef]
- Chen, Y.P.; Maltby, L.; Liu, Q.; Song, Y.; Zheng, Y.J.; Ellison, A.M.; Ma, Q.Y.; Wu, X.M. Captive pandas are at risk from environmental toxins. Front. Ecol. Environ. 2016, 14, 363–367. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.P.; Li, B.; Jiang, X.T.; Wang, Y.L.; Xia, Y.; Li, A.D.; Zhang, T. Catalogue of antibiotic resistome and host-tracking in drinking water deciphered by a large scale survey. Microbiome 2017, 5. [Google Scholar] [CrossRef]
- Hu, Y.; Yang, X.; Li, J.; Lv, N.; Liu, F.; Wu, J.; Lin, I.Y.; Wu, N.; Weimer, B.C.; Gao, G.F. The transfer network of bacterial mobile resistome connecting animal and human microbiome. Appl. Environ. Microbiol. 2016, 82, 6672. [Google Scholar] [CrossRef]
- de Been, M.; Lanza, V.F.; de Toro, M.; Scharringa, J.; Dohmen, W.; Du, Y.; Hu, J.; Lei, Y.; Li, N.; Tooming-Klunderud, A.; et al. Dissemination of cephalosporin resistance genes between Escherichia coli strains from farm animals and humans by specific plasmid lineages. PLoS Genet. 2014, 10, e1004776. [Google Scholar] [CrossRef] [PubMed]
- Mckenzie, V.J.; Song, S.J.; Delsuc, F.; Prest, T.L.; Oliverio, A.M.; Korpita, T.M.; Alexiev, A.; Amato, K.R.; Metcalf, J.L.; Kowalewski, M. The Effects of Captivity on the Mammalian Gut Microbiome. Integr. Comp. Biol. 2017, 57, 690–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, W.; Mishra, S.; Wang, C.; Zhang, H.; Ning, R.; Kong, F.; Zeng, B.; Zhao, J.; Li, Y. Comparative Study of Gut Microbiota in Wild and Captive Giant Pandas (Ailuropoda melanoleuca). Genes 2019, 10, 827. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10100827
Guo W, Mishra S, Wang C, Zhang H, Ning R, Kong F, Zeng B, Zhao J, Li Y. Comparative Study of Gut Microbiota in Wild and Captive Giant Pandas (Ailuropoda melanoleuca). Genes. 2019; 10(10):827. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10100827
Chicago/Turabian StyleGuo, Wei, Sudhanshu Mishra, Chengdong Wang, Hemin Zhang, Ruihong Ning, Fanli Kong, Bo Zeng, Jiangchao Zhao, and Ying Li. 2019. "Comparative Study of Gut Microbiota in Wild and Captive Giant Pandas (Ailuropoda melanoleuca)" Genes 10, no. 10: 827. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10100827