X-Linked Emery–Dreifuss Muscular Dystrophy: Study Of X-Chromosome Inactivation and Its Relation with Clinical Phenotypes in Female Carriers

 ,
,
Abstract
:1. Introduction
2. Subjects and Methods
2.1. Subjects
2.2. Methods
2.2.1. DNA Analysis
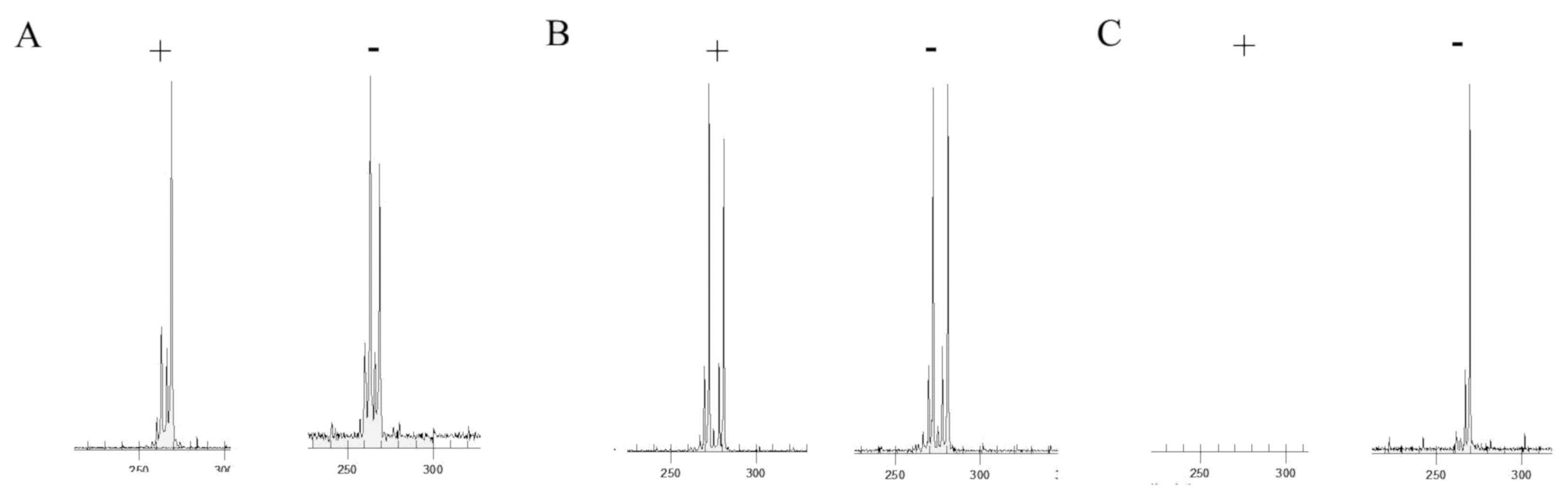
2.2.2. X-Inactivation Assay
- Forward 5’-[D4-PA]TCCAGAATCTGTTCCAGAGCGTGC-3’;
- Reverse 5’-ATGAGGAACAGCAACCTTCACAGC-3’.
2.3. Statistical Analysis
3. Results
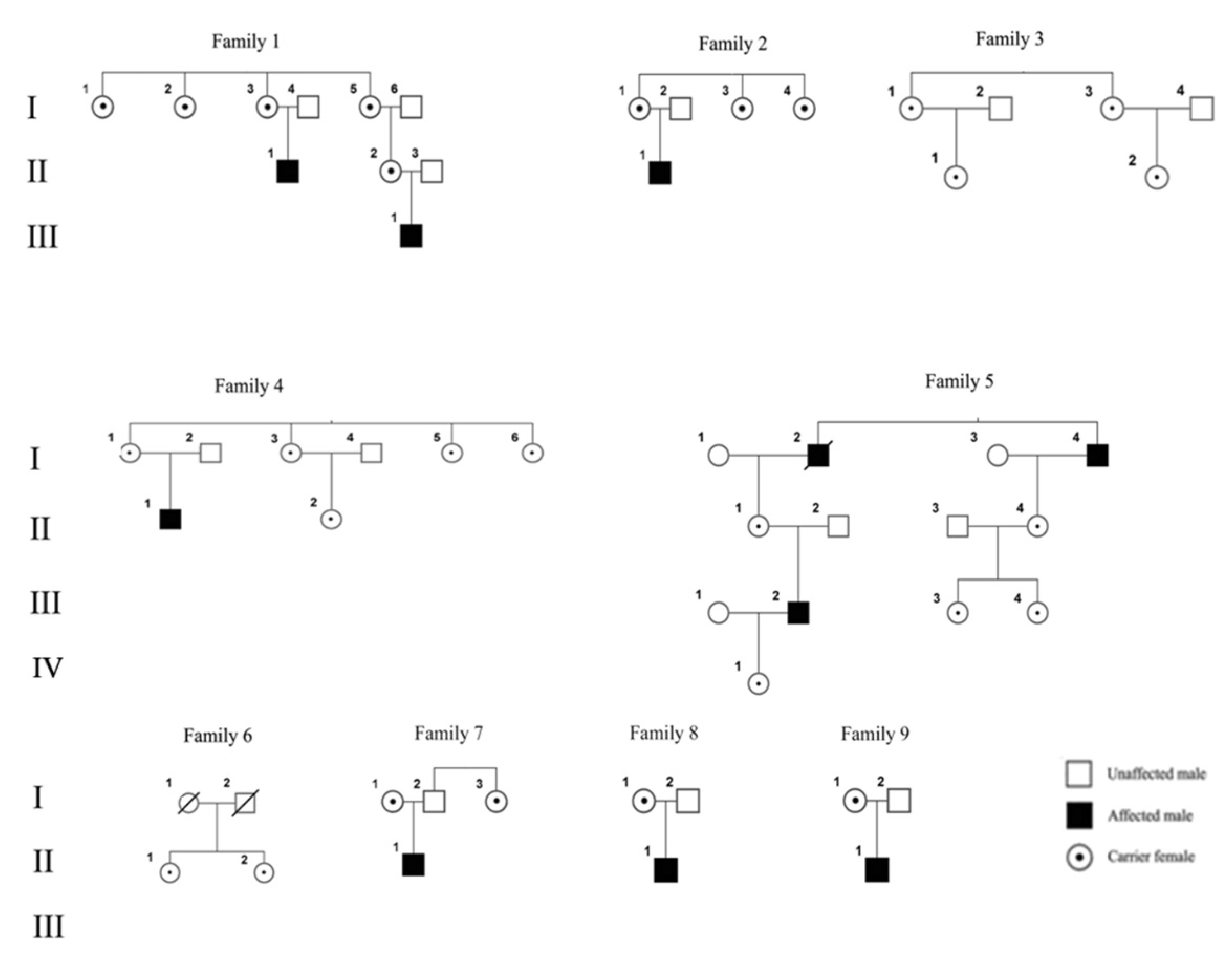
3.1. Subjects
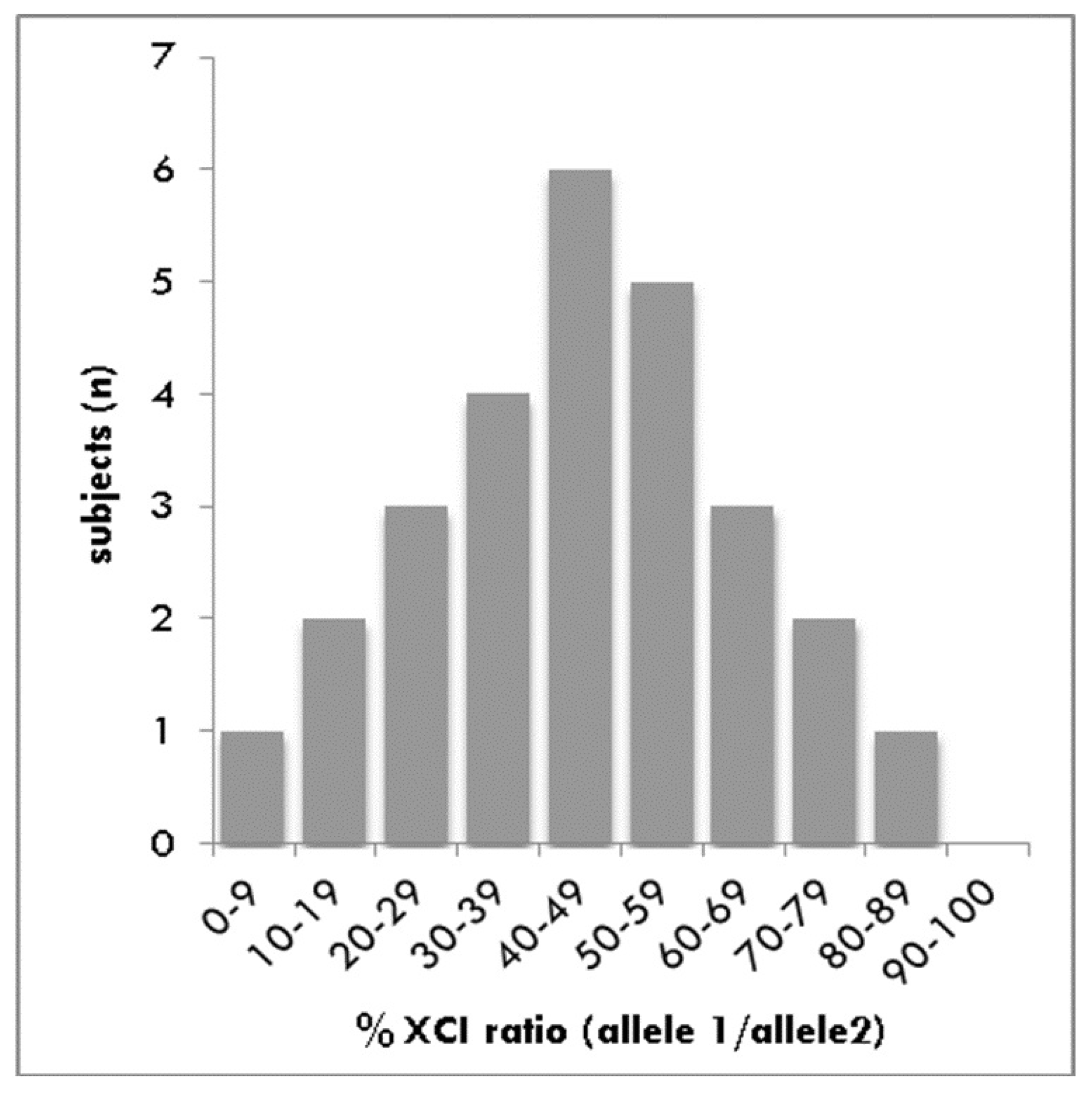
3.2. Analysis of XCI
3.3. Statistical Differences
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bione, S.; Maestrini, E.; Rivella, S.; Mancini, M.; Regis, S.; Romeo, G.; Toniolo, D. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat. Genet. 1994, 8, 323–327. [Google Scholar] [CrossRef] [PubMed]
- Blone, S.; Small, K.; Aksmanovic, V.M.; Morandi, L.; Kress, W.; Yates, J.R.; Warren, S.T.; Toniolo, D.; D’Urso, M.; Ciccodicola, A.; et al. Identification of new mutations in the Emery-Dreifuss muscular dystrophy gene and evidence for genetic heterogeneity of the disease. Hum. Mol. Genet. 1995, 4, 1859–1863. [Google Scholar] [CrossRef] [PubMed]
- Manilal, S.; Manila, S.; Recan, D.; Sewry, C.A.; Hoeltzenbein, M.; Llense, S.; Leturcq, F.; Deburgrave, N.; Barbot, J.-C.; Man, N.T.; et al. Mutations in Emery-Dreifuss muscular dystrophy and their effects on emerin protein expression. Hum. Mol. Genet. 1998, 7, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Manilal, S.; Sewry, C.; Man, N.T.; Muntoni, F.; Morris, G. Diagnosis of X-linked Emery-Dreifuss muscular dystrophy by protein analysis of leucocytes and skin with monoclonal antibodies. Neuromuscul. Disord. 1997, 7, 63–66. [Google Scholar] [CrossRef]
- Mora, M.; Cartegni, L.; Di Blasi, C.; Barresi, R.; Bione, S.; Di Barletta, M.R.; Morandi, L.; Merlini, L.; Nigro, V.; Politano, L.; et al. X-linked emery-dreifuss muscular dystrophy can be diagnosed from skin biopsy or blood sample. Ann. Neurol. 1997, 42, 249–253. [Google Scholar] [CrossRef] [PubMed]
- E Emery, A.; E Dreifuss, F. Unusual type of benign x-linked muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 1966, 29, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E. Emery–Dreifuss muscular dystrophy—A 40 year retrospective. Neuromuscul. Disord. 2000, 10, 228–232. [Google Scholar] [CrossRef]
- Carboni, N.; Mura, M.; Mercuri, E.; Marrosu, G.; Manzi, R.C.; Cocco, E.; Nissardi, V.; Isola, F.; Mateddu, A.; Solla, E.; et al. Cardiac and muscle imaging findings in a family with X-linked Emery–Dreifuss muscular dystrophy. Neuromuscul. Disord. 2012, 22, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Somma, S.; Comi, L.I.; Petretta, V.R.; Giugliano, M.A.M.; Limongelli, F.M.; Divitiis, O.; Nigro, G.; Politano, L.; Papparella, S.; Restucci, B.; et al. Structural Basis of Cardiomyopathy in Duchenne/Becker Carriers. Ann. N. Y. Acad. Sci. 1995, 752, 108–110. [Google Scholar]
- Nigro, G.; Russo, V.; Rago, A.; Papa, A.A.; Carbone, N.; Marchel, M.; Palladino, A.; Hausmanowa-Petrusewicz, I.; Russo, M.G.; Politano, L. Regional and transmural dispersion of repolarisation in patients with Emery-Dreifuss muscular dystrophy. Kardiol. Pol. 2012, 70, 1154–1159. [Google Scholar] [PubMed]
- Russo, V.; Rago, A.; Politano, L.; Papa, A.A.; Di Meo, F.; Russo, M.G.; Golino, P.; Calabrò, R.; Nigro, G. Increased dispersion of ventricular repolarization in emery dreifuss muscular dystrophy patients. Med. Sci. Monit. 2012, 18, CR643–CR647. [Google Scholar] [CrossRef] [PubMed]
- E Buckley, A.; Dean, J.; Mahy, I.R. Cardiac involvement in Emery Dreifuss muscular dystrophy: A case series. Heart 1999, 82, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Bialer, M.G.; McDaniel, N.L.; Kelly, T.E. Progression of cardiac disease in emery-dreifuss muscular dystrophy. Clin. Cardiol. 1991, 14, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Sakata, K.; Shimizu, M.; Ino, H.; Yamaguchi, M.; Terai, H.; Fujino, N.; Hayashi, K.; Kaneda, T.; Inoue, M.; Oda, Y.; et al. High Incidence of Sudden Cardiac Death with Conduction Disturbances and Atrial Cardiomyopathy Caused by a Nonsense Mutation in the STA Gene. Circulation 2005, 111, 3352–3358. [Google Scholar] [CrossRef] [PubMed]
- Peretto, G.; Di Resta, C.; Perversi, J.; Forleo, C.; Maggi, L.; Politano, L.; Barison, A.; Previtali, S.C.; Carboni, N.; Brun, F.; et al. Cardiac and Neuromuscular Features of Patients with LMNA-Related Cardiomyopathy. Ann. Intern. Med. 2019, 171, 458. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Peng, D. Cardiac Involvement in Emery-Dreifuss Muscular Dystrophy and Related Management Strategies. Int. Heart J. 2019, 60, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Faiella, W.; Bessoudo, R. Cardiac manifestations in Emery–Dreifuss muscular dystrophy. Can. Med. Assoc. J. 2018, 190, E1414–E1417. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Di Toro, A.; Giuliani, L.; Favalli, V.; Narula, N.; Grasso, M. Cardiac Phenotypes in Hereditary Muscle Disorders. J. Am. Coll. Cardiol. 2018, 72, 2485–2506. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, M.C.; Siegel, R.J.; Thompson, C.E.; Hopkins, L.C. Sudden death of a carrier of X-linked Emery-Dreifuss muscular dystrophy. Ann. Intern. Med. 1993, 119, 900–905. [Google Scholar] [CrossRef] [PubMed]
- Dickey, R.P.; Ziter, F.A.; Smith, R.A. Emery-Dreifuss muscular dystrophy. J. Pediatr. 1984, 104, 555–559. [Google Scholar] [CrossRef]
- London Medical Research Council (L.M.R.). Aids to the Examination of the Peripheral Nervous System—Medical Research Council Memorandum No. 45; London Her Majesty’s Stationery Office: London, UK, 1976. [Google Scholar]
- Allen, R.C.; Zoghbi, H.Y.; Moseley, A.B.; Rosenblatt, H.M.; Belmont, J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992, 51, 1229–1239. [Google Scholar] [PubMed]
- Viggiano, E.; Picillo, E.; Cirillo, A.; Politano, L. Comparison of X-chromosome inactivation in Duchenne muscle/myocardium-manifesting carriers, non-manifesting carriers and related daughters. Clin. Genet. 2013, 84, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Ørstavik, K.H. X chromosome inactivation in clinical practice. Hum. Genet. 2009, 126, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Ørstavik, K.H. Skewed X inactivation in healthy individuals and in different diseases. Acta Paediatr. 2006, 95, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Brunl, P.; Polltano, L.; Plluso, G.; Cappa, V.; Covone, A.E.; Nigro, V.; Ciccodicola, A.; Romeo, G.; D’Urso, M. SSCP detection of novel mutations in patients with Emery-Dreifuss muscular dystrophy: Definition of a small C-terminal region required for emerin function. Hum. Mol. Genet. 1995, 4, 2003–2004. [Google Scholar]
- Nigro, G.; Russo, V.; Ventriglia, V.M.; Della Coppa, N.; Palladino, A.; Nigro, V.; Calabrò, R.; Nigro, G.; Politano, L. Early onset of cardiomyopathy and primary prevention of sudden death in X-linked Emery-Dreifuss muscular dystrophy. Neuromuscul. Disord. 2010, 20, 174–177, Epub 2010 Feb 10. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Hylek, E.M.; A Phillips, K.; Chang, Y.; Henault, L.E.; Selby, J.V.; Singer, D.E. Prevalence of diagnosed atrial fibrillation in adults: National implications for rhythm management and stroke prevention: The AnTicoagulation and Risk Factors in Atrial Fibrillation (ATRIA) Study. JAMA 2001, 285, 2370–2375. [Google Scholar] [CrossRef] [PubMed]
- Upshaw, C.B. Comparison of the prevalence of first-degree atrioventricular block in African-American and in Caucasian patients: An electrocardiographic study III. J. Natl. Med. Assoc. 2004, 96, 756–760. [Google Scholar] [PubMed]
- Lindberg, T.; Bohman, D.M.; Elmstahl, S.; Jogréus, C.; Berglund, J.S. Prevalence of unknown and untreated arrhythmias in an older outpatient population screened by wireless long-term recording ECG. Clin. Interv. Aging 2016, 11, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Heeringa, J.; Van Der Kuip, D.A.; Hofman, A.; Kors, J.A.; Van Herpen, G.; Stricker, B.H.; Stijnen, T.; Lip, G.Y.; Witteman, J.C. Prevalence, incidence and lifetime risk of atrial fibrillation: The Rotterdam study. Eur. Heart J. 2006, 27, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Busque, L.; Mio, R.; Mattioli, J.; Brais, E.; Blais, N.; LaLonde, Y.; Maragh, M.; Gilliland, D. Nonrandom X-inactivation patterns in normal females: Lyonization ratios vary with age. Blood 1996, 88, 59–65. [Google Scholar] [CrossRef] [PubMed]
- El Kassar, N.; Hetet, G.; Brière, J.; Grandchamp, B. X-chromosome inactivation in healthy females: Incidence of excessive lyonization with age and comparison of assays involving DNA methylation and transcript polymorphisms. Clin. Chem. 1998, 44, 61–67. [Google Scholar] [PubMed]
- Espinós, C.; I Lorenzo, J.; Casaña, P.; Martínez, F.; Aznar, J.A. Haemophilia B in a female caused by skewed inactivation of the normal X-chromosome. Haematologica 2000, 85, 1092–1095. [Google Scholar] [PubMed]
- Devriendt, K.; Matthijs, G.; Legius, E.; Schollen, E.; Blockmans, D.; Van Geet, C.; Degreef, H.; Cassiman, J.J.; Fryns, J.P. Skewed X-chromosome inactivation in female carriers of dyskeratosis congenita. Am. J. Hum. Genet. 1997, 60, 581–587. [Google Scholar] [PubMed]
- Viggiano, E.; Picillo, E.; Politano, L. DMD Phenotype in Girls with a de novo Balanced X;3 Autosome Translocation: A Case Report. J. Genet. Syndr. Gene Ther. 2013, 4, 2. [Google Scholar]
- Juan-Mateu, J.; Rodríguez, M.J.; Nascimento, A.; Jimenez-Mallebrera, C.; González-Quereda, L.; Rivas, E.; Paradas, C.; Madruga, M.; Sanchez-Ayaso, P.; Jou, C.; et al. Prognostic value of X-chromosome inactivation in symptomatic female carriers of dystrophinopathy. Orphanet J. Rare Dis. 2012, 7, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yutzey, K.E.; Kirby, M.L. Wherefore heart thou? Embryonic origins of cardiogenic mesoderm. Dev. Dyn. 2002, 223, 307–320. [Google Scholar] [CrossRef] [PubMed]
- Azofeifa, J.; Cremer, M.; Waldherr, R. X-chromosome methylation ratios as indicators of chromosomal activity: Evidence of intraindividual divergencies among tissues of different embryonal origin. Hum. Genet 1996, 97, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Fialkow, P.J. Primordial cell pool size and lineage relationships of five human cell types. Ann. Hum. Genet. 1973, 37, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Kirby, M.L.; Stewart, D.E. Neural crest origin of cardiac ganglion cells in the chick embryo: Identification and extirpation. Dev. Boil. 1983, 97, 433–443. [Google Scholar] [CrossRef]
- Virágh, S.; E Challice, C. The development of the conduction system in the mouse embryo heart. II. Histogenesis of the atrioventricular node and bundle. Dev. Boil. 1977, 56, 397–411. [Google Scholar] [CrossRef]
- Jongbloed, M.R.M.; Mahtab, E.A.F.; Blom, N.A.; Schalij, M.J.; Groot, A.C.G.-D. Development of the Cardiac Conduction System and the Possible Relation to Predilection Sites of Arrhythmogenesis. Sci. World J. 2008, 8, 239–269. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Nagara, H.; Mawatari, S.; Kondo, A.; Sato, H. Emery-Dreifuss muscular dystrophy. An autopsy case. J. Neurol. Sci. 1987, 79, 23–31. [Google Scholar] [CrossRef]
- Waters, D.D.; Nutter, D.O.; Hopkins, L.C.; Dorney, E.R. Cardiac Features of an Unusual X-Linked Humeroperoneal Neuromuscular Disease. N. Engl. J. Med. 1975, 293, 1017–1022. [Google Scholar] [CrossRef] [PubMed]
- Voit, T.; Krogmann, O.; Lenard, H.; Neuen-Jacob, E.; Wechsler, W.; Goebel, H.; Rahlf, G.; Lindinger, A.; Nienaber, C. Emery-Dreifuss Muscular Dystrophy: Disease Spectrum and Differential Diagnosis. Neuropediatrics 1988, 19, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Cartegni, L.; Di Barletta, M.R.; Barresi, R.; Squarzoni, S.; Sabatelli, P.; Maraldi, N.; Mora, M.; Di Blasi, C.; Cornelio, F.; Merlini, L.; et al. Heart-specific localization of emerin: New insights into Emery-Dreifuss muscular dystrophy. Hum. Mol. Genet. 1997, 6, 2257–2264. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Family Number | ID Subject | Age at Last Control (Years) | XCw Inactivation (%) | Signs/Symptoms | EMD Mutation |
|---|---|---|---|---|---|
| N. 1 | |||||
| I-1 | 5327 | 58 | 54.0 | 1st degree AV block | c. 130 C>T |
| I-2 | 5328 | 51 | 49 | no | |
| I-4 | 5325 | 49 | 30 | no | |
| I-3 | 5326 | 47 | 70 | no | |
| II-2 | 5322 | 28 | 28 | no | |
| N. 2 | |||||
| I-1 | 4811 | 32 | 35.4 | no | c. 564–565 del CT* |
| I-2 | 4810 | 24 | 40.7 | no | |
| I-3 | 4809 | 21 | 49.7 | no | |
| N. 3 | |||||
| I-1 | 9265 | 50 | 64.8 | no | c. 1A>G |
| I-3 | 9266 | 46 | 41.9 | no | |
| II-1 | 9264 | 31 | 65.5 | no | |
| II-2 | 9267 | 25 | 50.9 | no | |
| N. 4 | |||||
| I-1 | 9577 | 52 | 91.8 | 1st degree AV block | c. IVS4+2G>Cc. 399+2G>C |
| II-2 | 9261 | 35 | 44.8 | no | |
| I-5 | 9262 | 22 | 34.3 | no | |
| I-6 | 9263 | 20 | 0 | no | |
| N. 5 | |||||
| IV-1 | 9259 | 29 | n.i. | no | c. 153delC |
| III-3 | 9258 | 53 | 89.2 | no | |
| III-4 | 9257 | 49 | 50. | no | |
| N. 6 | |||||
| II-1 | 9269 | 37 | 40.7 | no | c. 451dup |
| II-2 | 9270 | 25 | 68.9 | no | |
| N. 7 | |||||
| I-1 | 6418 | 42 | 90.8 | no | c. 106 A>T ** |
| I-2 | 5008 | 40 | 70 | no | |
| N. 8 | |||||
| I-1 | 2907 | 50 | 35.3 | 1st degree AV block | c. 740 C<T*** |
| N. 9 | |||||
| I-1 | 202 | 62 | 20 | Atrial fibrillation | c. 740 C<T*** |
| Isolated Cases | ID Subject | Age at Last Control (Years) | XCw Inactivation (%) | Cardiac Findings | EMDMutation |
| 1 | 8581 | 50 | n.i. | no | c. 106 A>T* |
| 2 | 9268 | 40 | 48.9 | no | c.192_192delinsTC |
| 3 | 9256 | 44 | 78.8 | no | c.IVS3 −27del18 |
| 4 | 9637 | 51 | 74.6 | 2nd degree AV block | c.IVS2+1G>A |
| 5 | 9578 | 51 | 18.7 | no | c.256 C>T |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Viggiano, E.; Madej-Pilarczyk, A.; Carboni, N.; Picillo, E.; Ergoli, M.; del Gaudio, S.; Marchel, M.; Nigro, G.; Palladino, A.; Politano, L. X-Linked Emery–Dreifuss Muscular Dystrophy: Study Of X-Chromosome Inactivation and Its Relation with Clinical Phenotypes in Female Carriers. Genes 2019, 10, 919. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10110919
Viggiano E, Madej-Pilarczyk A, Carboni N, Picillo E, Ergoli M, del Gaudio S, Marchel M, Nigro G, Palladino A, Politano L. X-Linked Emery–Dreifuss Muscular Dystrophy: Study Of X-Chromosome Inactivation and Its Relation with Clinical Phenotypes in Female Carriers. Genes. 2019; 10(11):919. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10110919
Chicago/Turabian StyleViggiano, Emanuela, Agnieszka Madej-Pilarczyk, Nicola Carboni, Esther Picillo, Manuela Ergoli, Stefania del Gaudio, Michal Marchel, Gerardo Nigro, Alberto Palladino, and Luisa Politano. 2019. "X-Linked Emery–Dreifuss Muscular Dystrophy: Study Of X-Chromosome Inactivation and Its Relation with Clinical Phenotypes in Female Carriers" Genes 10, no. 11: 919. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10110919