The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye



1
Institute of Ophthalmology, UCL, London EC1V 9EL, UK
2
Moorfields Eye Hospital NHS Foundation Trust, London EC1V 2PD, UK
3
Great Ormond Street Hospital for Children NHS Foundation Trust, London WC1N 3JH, UK
4
Department of Genetics & Genomics, Instituto de Investigación Sanitaria-Fundación Jiménez Díaz University Hospital—Universidad Autónoma de Madrid (IIS-FJD, UAM), 28040 Madrid, Spain
5
Centre for Biomedical Network Research on Rare Diseases (CIBERER), 28029 Madrid, Spain
*
Author to whom correspondence should be addressed.
Genes 2019, 10(12), 1050; https://0-doi-org.brum.beds.ac.uk/10.3390/genes10121050
Submission received: 15 November 2019
/
Revised: 9 December 2019
/
Accepted: 12 December 2019
/
Published: 17 December 2019
(This article belongs to the Special Issue Recent Advances in Inherited Eye Disease)
Abstract
:The transcription factor PAX6 is essential in ocular development in vertebrates, being considered the master regulator of the eye. During eye development, it is essential for the correct patterning and formation of the multi-layered optic cup and it is involved in the developing lens and corneal epithelium. In adulthood, it is mostly expressed in cornea, iris, and lens. PAX6 is a dosage-sensitive gene and it is highly regulated by several elements located upstream, downstream, and within the gene. There are more than 500 different mutations described to affect PAX6 and its regulatory regions, the majority of which lead to PAX6 haploinsufficiency, causing several ocular and systemic abnormalities. Aniridia is an autosomal dominant disorder that is marked by the complete or partial absence of the iris, foveal hypoplasia, and nystagmus, and is caused by heterozygous PAX6 mutations. Other ocular abnormalities have also been associated with PAX6 changes, and genotype-phenotype correlations are emerging. This review will cover recent advancements in PAX6 regulation, particularly the role of several enhancers that are known to regulate PAX6 during eye development and disease. We will also present an updated overview of the mutation spectrum, where an increasing number of mutations in the non-coding regions have been reported. Novel genotype-phenotype correlations will also be discussed.
1. Introduction
The human paired box 6, as coded by the PAX6 gene, is a member of the PAX family of transcription factors, which are evolutionarily highly conserved among metazoans and are characterized by the presence of a conserved DNA-binding domain, the paired domain [1]. Positional cloning identified it in the early 1990s [2]. At the same time, murine Pax6 was also identified by the screening of mouse embryonic expression libraries [3] and associated as the causal gene of an heterozygous Sey mouse strain (Sey+/−) [4], which presented with microphthalmia, iris hypoplasia, cataracts, and corneal opacifications, resembling human developmental eye disorder aniridia [5]. Further PAX6 homologous genes were later identified in zebrafish (pax6a and pax6b) [6], quail [7], and Drosophila (ey, toy, eyg, and toe) [8].
PAX6 is considered to be the master regulator of the eye, since the overexpression of the gene resulted in ectopic eye formation in both Drosophila and Xenopus [9,10]. During early eye development, PAX6 is expressed on the surface and neural ectoderm. By week five in human gestation, it is expressed throughout the optic vesicle, which then invaginates to form the bi-layered optic cup, where PAX6 is found in both neural and pigmented retinal layers [11]. It is also highly expressed in the anterior segment structures that are derived from the surface ectoderm, including the lens vesicle and corneal epithelium [11,12]. Postnatally, PAX6 is restricted to retinal ganglion, amacrine and horizontal cells, lens, cornea, conjunctiva, iris, and ciliary body [12,13]. Outside the eye, it is expressed in the pancreas, nasal epithelia, and several distinct regions of the central nervous system (CNS), like the forebrain, hindbrain, and spinal cord [12].
Defects in PAX6 gene can affect eye development and result in a broad range of clinical phenotypes, with the most common being aniridia, a pan ocular disorder that is primarily characterised by the absence or hypoplasia of the iris, nystagmus, and foveal hypoplasia, accompanied by cataracts, glaucoma and corneal keratopathy [14]. Other ocular phenotypes include microphthalmia, optic nerve anomalies, or anterior segment dysgenesis [15]. Systemic features can include neurodevelopmental abnormalities, like autism and attention deficit hyperactivity (ADHD) disorders, language impairment [16,17,18], and in some cases the absence or malformations of the pineal and pituitary gland [19,20,21,22]. Defects in PAX6 have also been associated with obesity and diabetes mellitus due to its role in pancreas development [23,24].
The majority of PAX6 mutations are heterozygous and they result in loss of one allele causing PAX6 haploinsufficiency. This review will highlight recent advances in PAX6 function and regulation and will particularly focus on the spectrum of mutations in the eye and resultant genotype-phenotype correlations.
2. PAX6
PAX6 (OMIM 607108) locus was mapped to the chromosome region 11p13, being 22 Kb in size, but with regulatory regions spanning ~450 Kb of genomic DNA [25,26]. The gene contains 14 exons, with the first three being non-coding (Figure 1A) [27].
PAX6 encodes a protein whose canonical form is approximately 46 KDa and is formed by 422 amino acids. It has two DNA-binding domains, the paired domain (PD) and the homeodomain (HD), -connected by a linker region (Figure 1B). At the C-terminal, immediately downstream of the HD, there is also a proline-serine-threonine-rich transactivation domain (PSTD), which is required for initiating transcription and it modulates DNA binding by the HD [28,29]. The PD is, in turn, comprised of a N-terminal subdomain (NTS or PAI) and a C-terminal subdomain (CTS or RED) (Figure 1B) [30]. Both subdomains bind the respective consensus DNA sequences and the two major PAX6 isoforms, canonical PAX6 and PAX6(5a), modulate their activity [31,32].
PAX6(5a) is formed by the alternative splicing of exon 5a between exons 5 and 6, which results in a slightly larger isoform with 436 amino acids (48 KDa) (Figure 1B). The extra 14 amino acids encoded by exon 5a are inserted in the NTS of the paired domain, blocking its DNA-binding activity, and unmasking DNA-binding activity of the CTS [31,33].
A third isoform has been reported in quail and mice—Pax6ΔPD—which lacks the complete PD and it is predicted to result in a truncated protein [34,35]. However, Kim and Lauderdale showed that Pax6ΔPD has a distinct function in mammalian eye development than Pax6 and Pax6(5a), since the overexpression of Pax6ΔPD, while using BAC and YAC transgene systems, lead to severe microphthalmia in both wildtype and Pax6-deficient mice [36,37]. Dual reporter systems during mouse embryonic development showed that Pax6ΔPD isoform, although at lower levels than PD-containing isoforms, was mostly detected in the peripheral neural retina and developing ciliary body, but it was absent from developing lens and cornea [36,38].
The regulation and target genes of each isoform in the eye are not entirely understood. It is hypothesized that canonical PAX6 is more prominent during embryonic development of ocular tissues and is related to differentiation and cell fate determination, while PAX6(5a) seems to be more relevant in the later development or postnatally and for cell proliferation [39,40,41]. Accordingly, expression studies in the mouse lens showed that, during embryonic development, canonical Pax6 expression is much higher (8:1) when compared to Pax6(5a). However, in adult eye tissues (lens, cornea and retina), the ratio changes to 1:1 [42,43]. The same tendency was observed in the retina of chick embryos, where canonical Pax6 is highly expressed during early stages in the eye primordium and lens placode, whereas Pax6(5a) expression gradually increases at later stages, particularly in the cornea and lens [39]. The expression of Pax6(5a) actually exceeds canonical Pax6 expression in the posterior retina of chicks, as birds possess a high density of photoreceptors, comparable to the fovea in primates [39]. These expression studies seem to corroborate human phenotypes, since mutations affecting the CTS, which is the main DNA-binding subdomain of PAX6(5a) isoform, have been associated with isolated foveal hypoplasia (discussed in Section 6.3.1.) [33,44,45]. In the cornea, PAX6 and PAX6(5a) are both expressed in the epithelial layer with a correlation between the expression levels of epithelial-specific keratins and PAX6 isoforms. Overexpression studies showed that canonical PAX6 seems to induce KRT3, while PAX6(5a) induced KRT12 expression via each of their respective PD subdomains [46].
Although the PAX6 isoforms seem to have independent roles and downstream targets in eye development, the existence of a positive feedback system between Pax6 and Pax6(5a) in mice was also reported [47,48]. Furthermore, the expression ratio between both of the isoforms seems to be essential for normal eye development, and altered ratios has been associated with eye and brain abnormalities [49,50,51].
3. PAX6 Regulation
Due to the high similarity of mice, Drosophila, or quail Pax6 to human PAX6, these organisms have been extensively used as model systems to understand its function and complex regulatory network. Promoters P0 and P1 mainly regulate the transcription of PAX6 and, to a lesser extent, by internal promoter Pα, giving rise to the many transcripts that encode the different isoforms (Figure 1) [52,53,54,55]. In situ hybridization experiments in the developing mouse eye showed that P0 promoter initiates gene expression in the cornea and conjunctival epithelia, lens placode, and retina, whereas the P1 promoter mainly initiates transcription in the lens placode, optic vesicle, and CNS, but only weakly in the cornea and conjunctiva. Pα directs the expression in retinal amacrine cells, ciliary body, and iris [41,52,53,55].
However, tissue- and time-specific regulation of PAX6 is still largely unknown. Furthermore, there is no direct relationship between the promoters and expression of specific transcripts. In mice, P0 and P1 both initiate the expression of Pax6 and Pax6(5a) transcripts, while Pα-derived transcripts seems to be more directly linked to the Pax6ΔPD isoform [34,54]. These results point to additional regulation elements that act together with promoters in the complex regulation and expression patterns of PAX6. Conserved cis regulatory regions have been identified both upstream and downstream of PAX6 and they are summarized in Table 1.
The ectodermal enhancer (EE) is located ~3.5 Kb upstream of the P0 promoter and regulates the expression of PAX6 during the development of surface ectodermal-derived tissues (lens, cornea, conjunctiva) [48,54,60,64,65]. In mice, Pax6 was found to directly interact with the EE for autoregulation mechanisms, but it can also interact with Sox2 and Sox3, transcription factors that are also involved in early eye and lens development [48,66]. The Pax6-Sox2 complex can also act on the Sox2 enhancer N-3, showing a synergistic mechanism between these two transcription factors in the regulation of crystallin gene expression [67].
Approximately 150 Kb downstream of PAX6, within introns 7 to 9 of neighbour gene ELP4, resides an essential region for PAX6 regulation—Downstream Regulatory Region (DRR). DRR contains several conserved elements that, if absent, affect PAX6 expression and normal eye development [25,37,57,58]. Although the role of all the elements has not been uncovered, important retinal and iris enhancers are located within this region, since the deletion of DRR in mice completely abolished Pax6 expression in both tissues, as well as in the ciliary body [37]. The deletion of DRR did not dramatically alter Pax6 expression in the lens; the same was observed when the deletion of EE reduced, but did not abolish, Pax6 expression in the lens [64]. These results show that multiple enhancers in multiple regions regulate tissue-specific transcription [37].
Within the DRR, an 800 bp specific enhancer element—SIMO—was found to have a PAX6 PD consensus binding sequence, guiding expression in early and later stages of eye formation, i.e., early surface ectoderm, lens, and neural retinal differentiation, as well as in adulthood, in the lens epithelium, retina, and iris [26]. SIMO was shown to be critical for Pax6 expression, as the disruption of this element alone was sufficient for abolishing expression in the developing lens in transgenic mice and zebrafish [26]. Importantly, deletion or point mutations in SIMO were described to cause aniridia phenotype in humans (discussed in Section 4.4.) [26,68]. However, the disruption of PAX6 expression in the lens when SIMO is affected is most likely due to a self-regulation mechanism of PAX6 itself, which, as in EE, can bind to SIMO via PD and regulate its own expression during eye development in a positive feedback loop [26]. In the mouse lens, two transcription factors from the TALE homeoproteins family, Meis1 and Meis2, have been identified as upstream regulators of Pax6 by regulating the activity of both EE and SIMO enhancers [69,70]. However, the exact role and regulation mechanisms of these regions on PAX6 expression in other eye tissues are still largely unknown.
Comparative genomic hybridisation (CGH) analysis of aniridia patients that lacked mutations in PAX6 coding sequence, pointing to additional regulatory elements located in this region, have identified deletions in PAX6 3’ regulatory region (Figure 2) [71,72,73,74,75]. As a result, Ansari et al. proposed a critical region for PAX6 transcriptional activation that spanned approximately 245 Kb and encompassed neighbour genes DNAJC24, IMMP1L, and ELP4 [72]. More recently, Plaisancie et al. refined it to a 18 Kb region within ELP4 that does not include SIMO, but another highly conserved enhancer in the DRR, E180B (Figure 2) [56]. These findings highlight a possible role of E180B in aniridia [75].
4. PAX6 Mutation Spectrum
The “PAX6 Mutation Database” (http://lsdb.hgu.mrc.ac.uk/home.php?select_db=PAX6) catalogs all reported mutations in PAX6 and had, until its last update in 2018, nearly 500 unique variants registered. Intragenic mutations cover ~96% of variants in the database, while the remaining 4% represent the whole gene deletions or variants described in 5’ and 3’ regulatory regions. 80% of intragenic mutations are spread through the complete coding sequence of the gene, but the majority seem to be located within exon 5, 6 and 9, which represent the paired (5 and 6) and homeo- (9) domains of PAX6 protein. While considering the mutation type, the most common intragenic mutations that were observed in PAX6 are nonsense (39%), followed by frameshifts (27%), missense (12%), splice site (15%), small indels (2%), and C-terminal extension (or run-ons) (2%) (Figure 3) [15,78].
4.1. Premature Termination Codon (PTC) Mutations—Nonsense, Frameshift, and Splice Site Variants
Nucleotide changes that introduce a premature termination codon (PTC) are the most common mutations that are found in PAX6. Nonsense, frameshift (out of frame insertions or deletions), and most splice site mutations result in the insertion of a PTC and the consequent termination of translation, accounting for 65–70% of all PAX6 mutations in the database (Figure 3A). Patients with PTC variants tend to present classical aniridia phenotype [15].
Although PTC mutations were first thought to cause truncated proteins with a dominant negative effect, it is now accepted that mRNA transcripts containing PTCs located until 50 bp upstream of the last exon junction may be subjected to nonsense-mediated decay (NMD) [79]. Hence, PTCs in PAX6 mostly result in the degradation of the mutated transcript and the consequent loss of 50% of PAX6 protein levels [15,80]. Four specific nonsense variants are the most common PTC mutations and they account for more than 20% of all entries in the database: c.607C>T, p.Arg203* (exon 8); c.718C>T, p.Arg240* (exon 9); c.781C>T, p.Arg261* (exon 10); and, c.949C>T, p.Arg317* (exon 11) [15]. These mutations are located in known methylated CpG islands in exons 8–13, which constitute mutational “hotspots” for aniridia [78].
Further evidence that NMD is acting on PTC-containing PAX6 transcripts is the absence of these variants at the 3’ end of the gene [78]. PTCs in this region are expected to escape NMD and they would result in truncated proteins with a possible dominant negative effect and more severe phenotypes [81,82]. However, within the region that was predicted to escape NMD, i.e., the last 50 bp from exon 12 and exon 13, there are no nonsense mutations reported aside from c.1183G>T (p.Gly395*) in the last nucleotide of exon 12, but with predicted mRNA missplicing [83], and all frameshift changes are predicted to cause C-terminal extensions [78].
4.2. C-Terminal Exptension (CTE) Variants
Frameshift or point mutations in PAX6 that alter the stop codon location and allow for translation to continue into the 3’UTR region are less frequent than the variants described earlier, with only 13 entries being reported. Large aniridia cohort studies show that CTEs are associated with aniridia-like phenotypes, with a severity comparable to PTC-causing variants [84,85,86]. These observations seem to suggest that CTE mutations also generate PAX6 haploinsufficiency; however, NMD does not degrade these transcripts, since there is no PTC introduced, which means that these changes should indeed produce a longer protein that would most likely be unstable with the affected PST domain transactivation activity. The mechanisms of how CTE mutations cause haploinsufficiency are still unexplained.
4.3. Missense Variants
Missense mutations, which cause one amino acid to be replaced by a different one during translation, are reported in ~12% of all entries in the PAX6 Mutation Database. They are more concentrated in the PD and functional studies predict these variants cause differences in DNA binding and the transactivation activities of PAX6 [87,88,89,90]. These mutations are usually associated with milder, but atypical ocular phenotypes, in some cases without the presence of iris defects [86]. In fact, Tzoulaki et al. reported that missense mutations in PAX6 are responsible for nearly 70% of non-aniridia eye disorders that were registered in the database (Figure 3C) [78], like microphthalmia, optic nerve anomalies, coloboma, isolated foveal hypoplasia, and anterior segment dysgenesis (discussed in Section 6.3.) [44,90,91,92,93].
4.4. Non-Coding Variants
An increasing number of reports point to the implication of mutations in non-coding regions in neurodevelopmental and eye disorders [94,95,96]. In the PAX6 Mutations Database, approximately 15% of all variants are located in the intronic regions and they are generally associated with classical aniridia phenotypes. Although the great majority are located in donor and acceptor sites at the intron-exon borders, deep intronic variants have recently been found in large aniridia patient cohorts [75,83]. The deletions or point mutations in non-translated 5’ and 3’UTR of PAX6 have also been reported [75,97,98]. Minigene assays or in silico analysis revealed that most of these variants are likely to affect the normal splicing patterns, which results in the formation of PTCs and the consequent PAX6 haploinsufficiency [99].
Changes in 3’ regulatory PAX6 regions were also identified in patients presenting with classical aniridia. A single nucleotide change (chr11: 31,685,945G>T) in the SIMO enhancer, ~150Kb downstream of PAX6, was described by Bhatia et al., who showed that the phenotype is due to the change affecting a PAX6 recognition site, disrupting its autoregulation loop, and ultimately resulting in decreased PAX6 expression and haploinsufficiency [26]. Several deletions that encompass other 3’ regulatory regions have also been reported to cause aniridia (Figure 2) [71,72,73,74,75,76,77].
4.5. Chromosomal Rearrangements and Large Deletions
Chromosomal rearrangements (deletions, duplications, translocations, or inversions) involving part or whole PAX6 gene or regulatory elements account for up to 10% of all aniridia cases. Some reports suggest that the PAX6 deletions are more frequent in sporadic as compared to familial aniridia patients, while others found no significant difference [84,100,101,102]. Large deletions that encompass PAX6 and other neighbour genes, such as WT1, result in systemic disease, such as WAGR, caracterised by the presence of Wilms tumour, aniridia, genitourinary anomalies, and retardation characterize (described in Section 6.2).
4.6. Biallelic Mutations
Mutations that affect both PAX6 alleles have rarely been described and cause very severe ocular and neurodevelopmental abnormalities, in most cases leading to embryonic death [103,104]. A surviving patient with compound heterozygous mutations in PAX6 was described as having microphthalmia, neonatal diabetes mellitus, hypopituitarism, and microcephaly, as well as trisomy 21. The patient inherited a missense mutation affecting the PD from the father (c.112C>T, p.Arg38Trp), who had microcornea and severe cataracts, and a nonsense mutation in HD from the mother (c.718C>T, p.Arg240*), who presented with classical familial aniridia [105]. It is plausible to assume that, although affected, the missense variant still contributed for some amount of PAX6 function, since PAX6 is a very dosage-sensitive gene. Another patient with compound heterozygous nonsense mutations (c.607C>T, p.Arg203* and c.1058C>G, p.Ser353*), presenting with anophthalmia, severe CNS defects, including microcephaly and the complete absence of the corpus callosum and olfactory bulbs, sadly died a few days after birth [103].
5. Inheritance of PAX6 Mutations
Most PAX6 mutations causing aniridia are heterozygous, sporadic, or familial in an autosomal dominant manner, with significant phenotypic variability. It is estimated that nearly two-thirds of aniridia cases are familial, while the remaining third are considered to be sporadic [86,106]. Accordingly, sporadic occurrence was described in nearly 40% of entries in the PAX6 Mutation Database. Up to one-third of sporadic cases of aniridia are associated with PAX6 and WT1 deletions, while the remaining two-thirds are considered to be most likely caused by de novo point mutations [89].
Recent studies reported that the rate of mosaicism could be as high as 17.5% among apparent de novo cases for different dominant disorders [83,107]. Indeed, several reports have suggested mosaicism as the cause for the variable phenotypes seen in some sporadic PAX6-affected patients [83,93,108,109]. A recent study from Tarilonte et al. proved the existence of post-zygotic parental mosaicism in three unrelated Spanish families with variable aniridia or microphthalmia phenotypes caused by heterozygous nonsense (c.771G>A, p.Trp257* and c.120C>A, p.Cys40*) or missense (c.178T>C, p.Tyr60His) PAX6 mutations, respectively [110]. Quantitative analysis of parental PAX6 gene showed that all of the proband’s fathers have mutant allele fractions that range from 13 to 29%, depending on the tissue analysed, and have mild or no ocular features as compared to their affected offspring. Similarly, Bai et al. reported the presence of male gonadal mosaicism in a Chinese family with aniridia caused by PTC-inducing mutation c.879_880delCA, p.Ser294Cysfs*46 [109].
The presence of parental mosaicism in PAX6 contributes to partially explaining intra-familiar variabilities very often seen in patients. Furthermore, mosaicism is particularly relevant in sporadic aniridia cases, as it might be the underlying transmittance mechanism and not, in fact, de novo mutations, which has deep implications for genetic testing and counselling.
6. Genotype-Phenotype Correlations
6.1. Aniridia (MIM 106210)
Aniridia is a pan-ocular disorder that bilaterally affects the formation of the iris, cornea, lens, fovea, and optic nerve. Its prevalence is 1:40,000–100,000 with complete penetrance and variable expressivity [15,100,111].
The most obvious ocular feature is complete or partial iris hypoplasia, being accompanied by nystagmus and foveal hypoplasia, with the latter being the main cause of reduced visual acuity from birth. Further vision loss can occur from later onset cataracts, aniridia-related corneal keratopathy (ARK), and glaucoma (Figure 4A–C). Cataracts tend to develop during the late teens to early adulthood in 50–85% of aniridia patients, while up to two-thirds of patients can develop glaucoma between late childhood and early adulthood [15,112]. ARK is the most relevant feature contributing to visual loss in aniridia patients and it affects ~20% patients, but up to 90% can present with corneal irregularities [113]. Foveal hypoplasia and nystagmus were estimated to range between 85–95% of aniridia patients [86]. Optic nerve hypoplasia, although less common, is associated with ~10% of aniridia cases [90,114].
However, aniridia patients can present with a wide phenotypic spectrum without clear correlation between genotype and phenotype, even in patients within the same family [15]. Accordingly, a recent report from Pedersen et al. showed that family members with the same PAX6 mutation, a 2 bp deletion in intron 2, presented with variable iris involvement, which ranged from almost normal to no iris, as well as different degrees of foveal hypoplasia [115,116]. In contrast, Lagali et al. recently found a correlation between PAX6 variants and the severity and progression of ARK [117,118]. In a cohort of 46 aniridia patients, the authors found a minimal level of keratopathy and an increase in cornea thickness in all aniridia patients from early age. Patients with whole gene deletions presented the most severe and early onset ARK, followed by those with PTC or CTE mutations. Patients with missense mutations showed milder non-progressive ARK and, lastly, non-PAX6 mutations had the mildest forms of disease and generally the best visual acuity [118]. However, the clinical phenotype of ARK is heterogenous and patients with the same mutation can often display different degrees of ARK.
PAX6 haploinsufficiency, which is caused by intragenic PTC-causing mutations, whole gene deletions, or inactivation of regulatory regions, is the cause of up to 85% of aniridia cases (Figure 3B). Independent of the type or location of the PTC-containing transcripts, NMD is thought to be activated, resulting in null alleles. Iris hypoplasia is the most common feature in patients with mutations that cause PAX6 haploinsufficiency, whereas patients with missense mutations tend to have less affected iris [86]. CTE variants usually translate to severe iris hypoplasia similar to PTCs, although Hingorani et al. examination of 10 patients with CTE showed that the iris phenotypes tend to be milder in these patients compared to patients with PTCs in the same study [86,119].
Aniridia-like phenotypes have been found in patients with rare variants in FOXC1 and PITX2 [72,120,121]. These genes are usually associated with anterior segment dysgenesis (ASD), which are characterized by iris hypoplasia (or atrophy) and corectopia, and, frequently, childhood-onset glaucoma. However, patients with ASD usually have better visual acuity than aniridia patients, without nystagmus or foveal abnormalities [15,122]. In mice, Foxc1 was recently found to be a downstream target of Pax6 in the iris, and the deletion of this gene also caused cornea neo-vascularisation, which indicated that both genes belong to a common network in the formation of the anterior segment [123,124]. Recently, missense mutations in TRIM44 gene, located 4 Mb away from PAX6, were identified in a Chinese family with aniridia, cataracts and glaucoma [125]. TRIM44 was shown to negatively regulate PAX6 expression, but only one pedigree was reported to date.
It is estimated that 5% of aniridia patients remain without molecular diagnosis, pointing to more PAX6 regulatory regions, modifiers, or even novel genes to be discovered [102].
6.2. WAGR (MIM 194072)
WAGR (Wilms tumour, Aniridia, Genitourinary abnormalities and mental Retardation) is an autosomal dominant disorder with a prevalence of 1:500,000 [15]. Chromosomal rearrangements or small deletions in 11p region encompassing PAX6 and WT1 loci cause it, but can be variable in size [71,126]. Approximately 30% of patients with sporadic aniridia suffer from this syndrome and diagnosed children have 50 to 70% risk of developing Wilms tumour involving the kidney, so regular screening is necessary for increasing the detection and obtaining better prognosis [127].
While the absence of one copy of PAX6 and WT1 is established as the cause for aniridia and Wilms tumour (and genitourinary anomalies), respectively, the genetic causes that are behind the neurodevelopmental defects are less clear. Within the critical region in 11p, several genes have been associated with neurodevelopmental problems, like autism and ADHD. BDNF, a gene encoding the brain-derived neurotrophic factor, is located ~4 Mb from PAX6 in 11p14.1. Patients with BDNF haploinsufficiency have variable degrees of developmental delay, as well as behavioural problems [128,129]. SLC1A2, encoding a glutamate transporter, and PRRG4, encoding a vitamin K-dependent membrane protein, are also located in 11p13-p12, a region that is identified by linkage analysis as an autism candidate region [130]. However, PAX6 itself should also be considered, since some patients with PAX6 intragenic mutations present with cerebral abnormalities as well as development delays and autism [16,17,131].
WAGRO (MIM 612469) is a variant syndrome of WAGR that includes obesity [132,133]. Patients that were diagnosed with WAGRO have deletions that also encompass BDNF. BDNF is also involved in energy homeostasis in humans and haploinsufficiency is correlated with higher BMI (Body Mass Index), increased appetite, and childhood-onset obesity compared to WAGR patients without BDNF deletion [126,128].
6.3. Non-Aniridia Phenotypes
6.3.1. Isolated Foveal Hypoplasia
Isolated foveal hypoplasia, which is usually accompanied by nystagmus, has been described in few families with PAX6 missense mutations in the paired domain: c.227C>G, p.Pro76Arg and c.382C>T, p.Arg128Cys [44,45,134]. These mutations are located in the CTS subdomain of PD, which modulates the DNA-binding activity of PAX6(5a) isoform [32]. It was previously reported that the PAX6(5a) isoform was highly expressed in the fovea, and that mutations in exon 5a, which affect CTS binding activity, also caused foveal hypoplasia (not isolated) [39]. These results point to foveal hypoplasia being linked to variants that affect the PAX6(5a) isoform in particular, but further experimental evidence is required to confirm this. However, it should also be noted that a missense (c.214G>A, p.Gly72Ser) in the end of NTS and two PTC mutations in the PSTD (c.1035_1048del14, p.Pro346Aspfs*20 and c.1061_1070del10, p.Tyr354Cysfs*8) were also identified by Hingorani et al. in patients with foveal hypoplasia and mild structural abnormalities in the iris (Figure 5) [86].
6.3.2. Microphthalmia, Anophthalmia and Coloboma (MAC)
MAC is a group of developmental eye disorders characterised by reduced size orabsence of the ocular globe and it is caused by mutations in more than 90 genes, including PAX6 (Figure 4) [92]. Of the 13 different mutations that are associated with microphthalmia in the PAX6 Mutation Database, the vast majority (8) are missense, although PTC-inducing mutations have been reported (two nonsense and two frameshifts) (Figure 5) [108,110,135,136]. Bilateral microphthalmia in a patient with WAGR syndrome was also identified [137].
MAC phenotypes are significantly associated with mutations that are likely to disrupt the PAX6-DNA interaction, with mutations being more common in the PD, which is the domain that is known to interact with SOX2 [67,138,139,140]. Mutations in SOX2 are the most common cause of bilateral anophthalmia and severe microphthalmia [89,90]. Accordingly, a recent report has linked novel missense mutations in PAX6 (c.160A>C, p.Ser54Arg and c.372C>A, p.Asn124Lys) to severe bilateral microphthalmia, with phenotype resembling SOX2-associated MAC [140]. The authors showed that these mutations significantly reduce the binding affinity of PAX6 to a specific regulatory sequence in the EE, LE9, which also synergistically responds to SOX2 [48]. This could lead to an inability of both PAX6 and SOX2 to cooperatively bind to LE9 or other target DNA sequences, which suggests that, aside from being required during lens development, the PAX6-SOX2 partnership can have an additional role earlier in eye field development.
Anophthalmia is associated with homozygous PAX6 variants, where there is biallelic loss of function. The affected individuals are usually still births or die soon after birth with severe brain abnormalities [27,104].
The most common form of ocular coloboma resulting from PAX6 defects is that affecting the iris, which have been identified in patients with both nonsense, missense, and frameshift changes in PAX6, as well as the deletion of 3’regulatory region [141,142,143,144]. Optic nerve coloboma are generally associated with microphthalmia, with missense mutations affecting all of the functional domains of PAX6 (Figure 5) [91,92]. It is thought that the inability of PAX6 to bind PAX2 promoter and repress its expression in the optic cup during early eye development cause PAX6-related optic nerve anomalies. PAX2 is another PAX-family transcription factor that is mainly expressed in the optic stalk and represses PAX6 expression in this region. This mutual repression is essential for defining the boundaries between the optic stalk, which will later form the optic nerve, and the optic cup [90,145].
6.3.3. Gillespie Syndrome (MIM 206700)
Another syndrome with aniridia-like features is Gillespie syndrome (GS), which is a rare disorder that is characterized by nonprogressive cerebellar ataxia, intellectual disability, hypotonia, and iris hypoplasia with the presence of scalloped edges, fixed dilated pupils, and remnants of pupillary membrane [102]. Mutations in the ITPR1 gene were identified as causative in GS [146,147]. However, a T>A substitution in intron 2 of PAX6 was identified in two individuals that were described with Gillespie syndrome, but with atypical features, like corectopia and ptosis [148]. Later, a chromosomal deletion encompassing the 3’ regulatory region of PAX6 was also identified in a patient that was diagnosed with Gillespie syndrome [72].
6.3.4. Anterior Segment Dysgenesis–Peters Anomaly (MIM 604229)
Peters anomaly is part of a larger spectrum of anterior segment dysgeneses characterized by abnormalities in the cornea, iris and lens, including central cornea opacity, defects in corneal stroma and Descemet’s membrane, and iridocorneal and corneolenticular adhesions [122]. Mutations in PAX6, but also PITX2 and CYP1B1, have been identified in patients with Peters anomaly [122,149]. In the PAX6 Mutation Database there are 11 different mutations associated with this syndrome: 10 are missense mutations, localized primarily in the PD, and one is a nonsense variant in the PSTD (Figure 5) [33,45,149,150].
7. Conclusions
PAX6 plays a number of important roles both during eye development and maintenance of adult eye tissues. Defects in PAX6 lead to the perturbations in the dosage, time, and tissue specific expression, as well as disruption of its regulatory network. Due to this complex picture, individuals with mutations affecting PAX6 present highly variable pan-ocular features, which makes genotype-phenotype correlations difficult to establish.
Developments in transcriptomics and epigenomics have allowed for increasing the understanding of PAX6 transcriptional targets, regulators, and interactors in the eye [12,151,152,153]. Accordingly, growing evidence supports the importance of PAX6 regulation through microRNAs, as well as long non-coding RNAs [154,155,156,157,158,159]. These key advances mainly arose from mouse and Drosophila studies. However, it is still far from clear how PAX6 operates during eye development and how mutations translate into the variability of phenotypes that are described in this review. More representative models are needed for this purpose, and to overcome innate differences in eye development between human and other model organisms.
The generation of induced pluripotent stem cells (iPSCs)s allowed for the development of more representative in vitro models of human eye diseases, as well as of human eye development. This strategy would be particularly suitable for complex diseases, like aniridia, since iPSCs have the ability to differentiate into different cell types. Several ocular cell types where PAX6 activity is important have been successfully derived from iPSCs, like corneal and lens epithelial cells [157,160,161,162]. Additionally, iPSC-derived retinal organoids are now standardly used to study disease mechanisms and develop novel therapeutic approaches for eye disorders, like inherited retinal dystrophies [163,164,165,166,167]. However, this promising approach has not yet been applied to study PAX6- related eye diseases.
The majority of PAX6 mutations result in null alleles and consequent PAX6 haploinsufficiency and they are known to cause aniridia. Hence, understanding the PAX6 dosage requirements in each affected tissue is of particular importance, not only to understand phenotypic outcomes, but also to predict therapeutic targets. Nonsense suppression therapy targets PTCs that are caused by nonsense mutations and bypasses them to produce full length protein [168]. A clinical trial on the oral administration of ataluren (PTC124) in aniridia patients is currently ongoing (NCT02647359), after promising results were seen in Sey+/− mice that were treated with this compound [169,170]. Postnatal administration of ataluren was shown to increase the levels of Pax6 protein up to 80% of control levels, improving lens and corneal epithelium morphology, as well as retina function, as shown by increased ERG responses. However, the exact mechanism of action of ataluren is still unknown, and there is criticism over the mode of drug administration, as topical/local application might permit lower dose, higher penetration, and less off-target effects. The results from the clinical trial are eagerly awaited.
Author Contributions
Writing: original draft preparation, D.L.C.; writing: review and editing, D.L.C., G.A., M.C., M.M.; funding acquisition, M.C., M.M.
Funding
This research was funded by Wellcome Trust (205174/Z/16/Z), National Institute for Health Research (NIHR) Biomedical Research Centre at Moorfields Eye Hospital NHS Foundation Trust and UCL Institute of Ophthalmology, Fight for Sight UK and Moorfields Eye Charity to M.M. GA is supported by a Fight for Sight UK Early Career Investigator Award, National Institute for Health Research (NIHR) Biomedical Research Centre at Great Ormond Street Hospital Institute of Child Health and Moorfields Eye Charity. MC is supported by the Spanish Institute of Health Carlos III (ISCIII)/European Regional Development Fund (ERDF) (PI17_01164 and CPII17_00006), the Regional Government of Madrid (CAM, B2017/BMD3721) and Spanish Federation of Rare Diseases (FEDER).
Acknowledgments
The authors would like to acknowledge Alex Yeong for helpful discussions and assistance with clinical data.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Callaerts, P.; Halder, G.; Gehring, W.J. PAX-6 in development and evolution. Annu. Rev. Neurosci. 1997, 20, 483–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ton, C.C.; Hirvonen, H.; Miwa, H.; Weil, M.M.; Monaghan, P.; Jordan, T.; van Heyningen, V.; Hastie, N.D.; Meijers-Heijboer, H.; Drechsler, M.; et al. Positional cloning and characterization of a paired box- and homeobox-containing gene from the aniridia region. Cell 1991, 67, 1059–1074. [Google Scholar] [CrossRef] [Green Version]
- Walther, C.; Guenet, J.L.; Simon, D.; Deutsch, U.; Jostes, B.; Goulding, M.D.; Plachov, D.; Balling, R.; Gruss, P. Pax: A murine multigene family of paired box-containing genes. Genomics 1991, 11, 424–434. [Google Scholar] [CrossRef]
- Hill, R.E.; Favor, J.; Hogan, B.L.; Ton, C.C.; Saunders, G.F.; Hanson, I.M.; Prosser, J.; Jordan, T.; Hastie, N.D.; van Heyningen, V. Mouse small eye results from mutations in a paired-like homeobox-containing gene. Nature 1991, 354, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Jordan, T.; Hanson, I.; Zaletayev, D.; Hodgson, S.; Prosser, J.; Seawright, A.; Hastie, N.; van Heyningen, V. The human PAX6 gene is mutated in two patients with aniridia. Nat. Genet. 1992, 1, 328–332. [Google Scholar] [CrossRef]
- Krauss, S.; Johansen, T.; Korzh, V.; Fjose, A. Expression pattern of zebrafish pax genes suggests a role in early brain regionalization. Nature 1991, 353, 267–270. [Google Scholar] [CrossRef]
- Martin, P.; Carriere, C.; Dozier, C.; Quatannens, B.; Mirabel, M.A.; Vandenbunder, B.; Stehelin, D.; Saule, S. Characterization of a paired box- and homeobox-containing quail gene (Pax-QNR) expressed in the neuroretina. Oncogene 1992, 7, 1721–1728. [Google Scholar]
- Quiring, R.; Walldorf, U.; Kloter, U.; Gehring, W.J. Homology of the eyeless gene of Drosophila to the Small eye gene in mice and Aniridia in humans. Science 1994, 265, 785–789. [Google Scholar] [CrossRef]
- Halder, G.; Callaerts, P.; Gehring, W.J. Induction of ectopic eyes by targeted expression of the eyeless gene in Drosophila. Science 1995, 267, 1788–1792. [Google Scholar] [CrossRef] [Green Version]
- Chow, R.L.; Altmann, C.R.; Lang, R.A.; Hemmati-Brivanlou, A. Pax6 induces ectopic eyes in a vertebrate. Development 1999, 126, 4213–4222. [Google Scholar]
- Terzic, J.; Saraga-Babic, M. Expression pattern of PAX3 and PAX6 genes during human embryogenesis. Int. J. Dev. Biol. 1999, 43, 501–508. [Google Scholar] [PubMed]
- Shaham, O.; Menuchin, Y.; Farhy, C.; Ashery-Padan, R. Pax6: A multi-level regulator of ocular development. Prog. Retin. Eye Res. 2012, 31, 351–376. [Google Scholar] [CrossRef] [PubMed]
- Stanescu, D.; Iseli, H.P.; Schwerdtfeger, K.; Ittner, L.M.; Reme, C.E.; Hafezi, F. Continuous expression of the homeobox gene Pax6 in the ageing human retina. Eye 2007, 21, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Richardson, R.; Hingorani, M.; Van Heyningen, V.; Gregory-Evans, C.; Moosajee, M. Clinical utility gene card for: Aniridia. Eur. J. Hum. Genet. 2016, 24, 1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moosajee, M.; Hingorani, M.; Moore, A.T. PAX6-Related Aniridia. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 2018. [Google Scholar]
- Maekawa, M.; Iwayama, Y.; Nakamura, K.; Sato, M.; Toyota, T.; Ohnishi, T.; Yamada, K.; Miyachi, T.; Tsujii, M.; Hattori, E.; et al. A novel missense mutation (Leu46Val) of PAX6 found in an autistic patient. Neurosci. Lett. 2009, 462, 267–271. [Google Scholar] [CrossRef]
- Kikkawa, T.; Casingal, C.R.; Chun, S.H.; Shinohara, H.; Hiraoka, K.; Osumi, N. The role of Pax6 in brain development and its impact on pathogenesis of autism spectrum disorder. Brain Res. 2019, 1705, 95–103. [Google Scholar] [CrossRef]
- Davis, L.K.; Meyer, K.J.; Rudd, D.S.; Librant, A.L.; Epping, E.A.; Sheffield, V.C.; Wassink, T.H. Pax6 3′ deletion results in aniridia, autism and mental retardation. Hum. Genet. 2008, 123, 371–378. [Google Scholar] [CrossRef] [Green Version]
- Hergott-Faure, L.; Borot, S.; Kleinclauss, C.; Abitbol, M.; Penfornis, A. Pituitary function and glucose tolerance in a family with a PAX6 mutation. Ann. D’Endocrinol. 2012, 73, 510–514. [Google Scholar] [CrossRef]
- Xatzipsalti, M.; Voutetakis, A.; Stamoyannou, L.; Chrousos, G.P.; Kanaka-Gantenbein, C. Congenital Hypopituitarism: Various Genes, Various Phenotypes. Horm. Metab. Res. 2019, 51, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Hanish, A.E.; Butman, J.A.; Thomas, F.; Yao, J.; Han, J.C. Pineal hypoplasia, reduced melatonin and sleep disturbance in patients with PAX6 haploinsufficiency. J. Sleep Res. 2016, 25, 16–22. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, T.N.; Free, S.L.; Williamson, K.A.; Stevens, J.M.; Churchill, A.J.; Hanson, I.M.; Shorvon, S.D.; Moore, A.T.; van Heyningen, V.; Sisodiya, S.M. Polymicrogyria and absence of pineal gland due to PAX6 mutation. Ann. Neurol. 2003, 53, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Boese, E.A.; Tollefson, M.R.; Schnieders, M.J.; Darbro, B.W.; Alward, W.L.M.; Fingert, J.H. Novel Intragenic PAX6 Deletion in a Pedigree with Aniridia, Morbid Obesity, and Diabetes. Curr. Eye Res. 2020, 45, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Panneerselvam, A.; Kannan, A.; Mariajoseph-Antony, L.F.; Prahalathan, C. PAX proteins and their role in pancreas. Diabetes Res. Clin. Pract. 2019, 155, 107792. [Google Scholar] [CrossRef] [PubMed]
- Kleinjan, D.A.; Seawright, A.; Schedl, A.; Quinlan, R.A.; Danes, S.; van Heyningen, V. Aniridia-associated translocations, DNase hypersensitivity, sequence comparison and transgenic analysis redefine the functional domain of PAX6. Hum. Mol. Genet. 2001, 10, 2049–2059. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Bengani, H.; Fish, M.; Brown, A.; Divizia, M.T.; de Marco, R.; Damante, G.; Grainger, R.; van Heyningen, V.; Kleinjan, D.A. Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia. Am. J. Hum. Genet. 2013, 93, 1126–1134. [Google Scholar] [CrossRef] [Green Version]
- Glaser, T.; Walton, D.S.; Maas, R.L. Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat. Genet. 1992, 2, 232–239. [Google Scholar] [CrossRef]
- Bruun, J.A.; Thomassen, E.I.; Kristiansen, K.; Tylden, G.; Holm, T.; Mikkola, I.; Bjorkoy, G.; Johansen, T. The third helix of the homeodomain of paired class homeodomain proteins acts as a recognition helix both for DNA and protein interactions. Nucleic Acids Res. 2005, 33, 2661–2675. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Chao, L.Y.; Mishra, R.; Davies, J.; Saunders, G.F. Missense mutation at the C-terminus of PAX6 negatively modulates homeodomain function. Hum. Mol. Genet. 2001, 10, 911–918. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.K.; Singh, S.; Saunders, G.F. Dissection of the transactivation function of the transcription factor encoded by the eye developmental gene PAX6. J. Biol. Chem. 1998, 273, 7210–7221. [Google Scholar] [CrossRef] [Green Version]
- Epstein, J.A.; Glaser, T.; Cai, J.; Jepeal, L.; Walton, D.S.; Maas, R.L. Two independent and interactive DNA-binding subdomains of the Pax6 paired domain are regulated by alternative splicing. Genes Dev. 1994, 8, 2022–2034. [Google Scholar] [CrossRef] [Green Version]
- Epstein, J.; Cai, J.; Glaser, T.; Jepeal, L.; Maas, R. Identification of a Pax paired domain recognition sequence and evidence for DNA-dependent conformational changes. J. Biol. Chem. 1994, 269, 8355–8361. [Google Scholar]
- Azuma, N.; Yamaguchi, Y.; Handa, H.; Hayakawa, M.; Kanai, A.; Yamada, M. Missense mutation in the alternative splice region of the PAX6 gene in eye anomalies. Am. J. Hum. Genet. 1999, 65, 656–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Lauderdale, J.D. Analysis of Pax6 expression using a BAC transgene reveals the presence of a paired-less isoform of Pax6 in the eye and olfactory bulb. Dev. Biol. 2006, 292, 486–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carriere, C.; Plaza, S.; Martin, P.; Quatannens, B.; Bailly, M.; Stehelin, D.; Saule, S. Characterization of quail Pax-6 (Pax-QNR) proteins expressed in the neuroretina. Mol. Cell. Biol. 1993, 13, 7257–7266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Lauderdale, J.D. Overexpression of pairedless Pax6 in the retina disrupts corneal development and affects lens cell survival. Dev. Biol. 2008, 313, 434–454. [Google Scholar] [CrossRef]
- Kleinjan, D.A.; Seawright, A.; Mella, S.; Carr, C.B.; Tyas, D.A.; Simpson, T.I.; Mason, J.O.; Price, D.J.; van Heyningen, V. Long-range downstream enhancers are essential for Pax6 expression. Dev. Biol. 2006, 299, 563–581. [Google Scholar] [CrossRef] [Green Version]
- Lakowski, J.; Majumder, A.; Lauderdale, J.D. Mechanisms controlling Pax6 isoform expression in the retina have been conserved between teleosts and mammals. Dev. Biol. 2007, 307, 498–520. [Google Scholar] [CrossRef] [Green Version]
- Azuma, N.; Tadokoro, K.; Asaka, A.; Yamada, M.; Yamaguchi, Y.; Handa, H.; Matsushima, S.; Watanabe, T.; Kohsaka, S.; Kida, Y.; et al. The Pax6 isoform bearing an alternative spliced exon promotes the development of the neural retinal structure. Hum. Mol. Genet. 2005, 14, 735–745. [Google Scholar] [CrossRef]
- Dominguez, M.; Ferres-Marco, D.; Gutierrez-Avino, F.J.; Speicher, S.A.; Beneyto, M. Growth and specification of the eye are controlled independently by Eyegone and Eyeless in Drosophila melanogaster. Nat. Genet. 2004, 36, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Xu, P.X.; Zhang, X.; Heaney, S.; Yoon, A.; Michelson, A.M.; Maas, R.L. Regulation of Pax6 expression is conserved between mice and flies. Development 1999, 126, 383–395. [Google Scholar]
- Zhang, W.; Cveklova, K.; Oppermann, B.; Kantorow, M.; Cvekl, A. Quantitation of PAX6 and PAX6 (5a) transcript levels in adult human lens, cornea, and monkey retina. Mol. Vis. 2001, 7, 1–5. [Google Scholar] [PubMed]
- Cvekl, A.; Callaerts, P. PAX6: 25th anniversary and more to learn. Exp. Eye Res. 2017, 156, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Azuma, N.; Nishina, S.; Yanagisawa, H.; Okuyama, T.; Yamada, M. PAX6 missense mutation in isolated foveal hypoplasia. Nat. Genet. 1996, 13, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Van Heyningen, V.; Williamson, K.A. PAX6 in sensory development. Hum. Mol. Genet. 2002, 11, 1161–1167. [Google Scholar] [CrossRef]
- Sasamoto, Y.; Hayashi, R.; Park, S.J.; Saito-Adachi, M.; Suzuki, Y.; Kawasaki, S.; Quantock, A.J.; Nakai, K.; Tsujikawa, M.; Nishida, K. PAX6 Isoforms, along with Reprogramming Factors, Differentially Regulate the Induction of Cornea-specific Genes. Sci. Rep. 2016, 6, 20807. [Google Scholar] [CrossRef] [Green Version]
- Pinson, J.; Simpson, T.I.; Mason, J.O.; Price, D.J. Positive autoregulation of the transcription factor Pax6 in response to increased levels of either of its major isoforms, Pax6 or Pax6 (5a), in cultured cells. BMC Dev. Biol. 2006, 6, 25. [Google Scholar] [CrossRef] [Green Version]
- Aota, S.; Nakajima, N.; Sakamoto, R.; Watanabe, S.; Ibaraki, N.; Okazaki, K. Pax6 autoregulation mediated by direct interaction of Pax6 protein with the head surface ectoderm-specific enhancer of the mouse Pax6 gene. Dev. Biol. 2003, 257, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Dora, N.; Ou, J.; Kucerova, R.; Parisi, I.; West, J.D.; Collinson, J.M. PAX6 dosage effects on corneal development, growth, and wound healing. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2008, 237, 1295–1306. [Google Scholar] [CrossRef] [Green Version]
- Pinson, J.; Mason, J.O.; Simpson, T.I.; Price, D.J. Regulation of the Pax6: Pax6 (5a) mRNA ratio in the developing mammalian brain. BMC Dev. Biol. 2005, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, B.K.; Yang, Y.; Cveklova, K.; Cvekl, A. Functional interactions between alternatively spliced forms of Pax6 in crystallin gene regulation and in haploinsufficiency. Nucleic Acids Res. 2004, 32, 1696–1709. [Google Scholar] [CrossRef] [Green Version]
- Plaza, S.; Dozier, C.; Langlois, M.C.; Saule, S. Identification and characterization of a neuroretina-specific enhancer element in the quail Pax-6 (Pax-QNR) gene. Mol. Cell. Biol. 1995, 15, 892–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaza, S.; Dozier, C.; Turque, N.; Saule, S. Quail Pax-6 (Pax-QNR) mRNAs are expressed from two promoters used differentially during retina development and neuronal differentiation. Mol. Cell. Biol. 1995, 15, 3344–3353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kammandel, B.; Chowdhury, K.; Stoykova, A.; Aparicio, S.; Brenner, S.; Gruss, P. Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity. Dev. Biol. 1999, 205, 79–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.P.; Saunders, G.F. Transcriptional regulation of the human PAX6 gene promoter. J. Biol. Chem. 1997, 272, 3430–3436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, S.; Monahan, J.; Ravi, V.; Gautier, P.; Murdoch, E.; Brenner, S.; van Heyningen, V.; Venkatesh, B.; Kleinjan, D.A. A survey of ancient conserved non-coding elements in the PAX6 locus reveals a landscape of interdigitated cis-regulatory archipelagos. Dev. Biol. 2014, 387, 214–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McBride, D.J.; Buckle, A.; van Heyningen, V.; Kleinjan, D.A. DNaseI hypersensitivity and ultraconservation reveal novel, interdependent long-range enhancers at the complex Pax6 cis-regulatory region. PLoS ONE 2011, 6, e28616. [Google Scholar] [CrossRef] [Green Version]
- Griffin, C.; Kleinjan, D.A.; Doe, B.; van Heyningen, V. New 3′ elements control Pax6 expression in the developing pretectum, neural retina and olfactory region. Mech. Dev. 2002, 112, 89–100. [Google Scholar] [CrossRef] [Green Version]
- Kleinjan, D.A.; Seawright, A.; Childs, A.J.; van Heyningen, V. Conserved elements in Pax6 intron 7 involved in (auto)regulation and alternative transcription. Dev. Biol. 2004, 265, 462–477. [Google Scholar] [CrossRef] [Green Version]
- Williams, S.C.; Altmann, C.R.; Chow, R.L.; Hemmati-Brivanlou, A.; Lang, R.A. A highly conserved lens transcriptional control element from the Pax-6 gene. Mech. Dev. 1998, 73, 225–229. [Google Scholar] [CrossRef]
- Vance, K.W.; Sansom, S.N.; Lee, S.; Chalei, V.; Kong, L.; Cooper, S.E.; Oliver, P.L.; Ponting, C.P. The long non-coding RNA Paupar regulates the expression of both local and distal genes. EMBO J. 2014, 33, 296–311. [Google Scholar] [CrossRef]
- Buckle, A.; Nozawa, R.S.; Kleinjan, D.A.; Gilbert, N. Functional characteristics of novel pancreatic Pax6 regulatory elements. Hum. Mol. Genet. 2018, 27, 3434–3448. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.; Bhatia, S.; Gautier, P.; Loosli, F.; Tay, B.H.; Tay, A.; Murdoch, E.; Coutinho, P.; van Heyningen, V.; Brenner, S.; et al. Sequencing of Pax6 loci from the elephant shark reveals a family of Pax6 genes in vertebrate genomes, forged by ancient duplications and divergences. PLoS Genet. 2013, 9, e1003177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimanlig, P.V.; Faber, S.C.; Auerbach, W.; Makarenkova, H.P.; Lang, R.A. The upstream ectoderm enhancer in Pax6 has an important role in lens induction. Development 2001, 128, 4415–4424. [Google Scholar] [PubMed]
- Rowan, S.; Siggers, T.; Lachke, S.A.; Yue, Y.; Bulyk, M.L.; Maas, R.L. Precise temporal control of the eye regulatory gene Pax6 via enhancer-binding site affinity. Genes Dev. 2010, 24, 980–985. [Google Scholar] [CrossRef] [Green Version]
- Goudreau, G.; Petrou, P.; Reneker, L.W.; Graw, J.; Loster, J.; Gruss, P. Mutually regulated expression of Pax6 and Six3 and its implications for the Pax6 haploinsufficient lens phenotype. Proc. Natl. Acad. Sci. USA 2002, 99, 8719–8724. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Kamachi, Y.; Matsunami, H.; Imada, K.; Uchikawa, M.; Kondoh, H. PAX6 and SOX2-dependent regulation of the Sox2 enhancer N-3 involved in embryonic visual system development. Genes Cells Devoted Mol. Cell. Mech. 2007, 12, 1049–1061. [Google Scholar] [CrossRef]
- Lauderdale, J.D.; Wilensky, J.S.; Oliver, E.R.; Walton, D.S.; Glaser, T. 3′ deletions cause aniridia by preventing PAX6 gene expression. Proc. Natl. Acad. Sci. USA 2000, 97, 13755–13759. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Friedman, A.; Heaney, S.; Purcell, P.; Maas, R.L. Meis homeoproteins directly regulate Pax6 during vertebrate lens morphogenesis. Genes Dev. 2002, 16, 2097–2107. [Google Scholar] [CrossRef] [Green Version]
- Antosova, B.; Smolikova, J.; Klimova, L.; Lachova, J.; Bendova, M.; Kozmikova, I.; Machon, O.; Kozmik, Z. The Gene Regulatory Network of Lens Induction Is Wired through Meis-Dependent Shadow Enhancers of Pax6. PLoS Genet. 2016, 12, e1006441. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Kelly, F.; Palomares, M.; Vallespin, E.; Villaverde, C.; Martin-Arenas, R.; Velez-Monsalve, C.; Lorda-Sanchez, I.; Nevado, J.; Trujillo-Tiebas, M.J.; Lapunzina, P.; et al. Improving molecular diagnosis of aniridia and WAGR syndrome using customized targeted array-based CGH. PLoS ONE 2017, 12, e0172363. [Google Scholar] [CrossRef]
- Ansari, M.; Rainger, J.; Hanson, I.M.; Williamson, K.A.; Sharkey, F.; Harewood, L.; Sandilands, A.; Clayton-Smith, J.; Dollfus, H.; Bitoun, P.; et al. Genetic Analysis of ‘PAX6-Negative’ Individuals with Aniridia or Gillespie Syndrome. PLoS ONE 2016, 11, e0153757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syrimis, A.; Nicolaou, N.; Alexandrou, A.; Papaevripidou, I.; Nicolaou, M.; Loukianou, E.; Christophidou-Anastasiadou, V.; Malas, S.; Sismani, C.; Tanteles, G.A. Aniridia due to a novel microdeletion affecting PAX6 regulatory enhancers: Case report and review of the literature. J. Genet. 2018, 97, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, G.C.; Hesselson, S.E.; Chan, J.Y.; Jenkins, A.B.; Laybutt, D.R.; Hesselson, D.; Campbell, L.V. Deletion distal to the PAX6 coding region reveals a novel basis for familial cosegregation of aniridia and diabetes mellitus. Diabetes Res. Clin. Pract. 2019, 148, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Plaisancie, J.; Tarilonte, M.; Ramos, P.; Jeanton-Scaramouche, C.; Gaston, V.; Dollfus, H.; Aguilera, D.; Kaplan, J.; Fares-Taie, L.; Blanco-Kelly, F.; et al. Implication of non-coding PAX6 mutations in aniridia. Hum. Genet. 2018, 137, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Bayrakli, F.; Guney, I.; Bayri, Y.; Ercan-Sencicek, A.G.; Ceyhan, D.; Cankaya, T.; Mason, C.; Bilguvar, K.; Bayrakli, S.; Mane, S.M.; et al. A novel heterozygous deletion within the 3’ region of the PAX6 gene causing isolated aniridia in a large family group. J. Clin. Neurosci. 2009, 16, 1610–1614. [Google Scholar] [CrossRef] [PubMed]
- Addis, L.; Ahn, J.W.; Dobson, R.; Dixit, A.; Ogilvie, C.M.; Pinto, D.; Vaags, A.K.; Coon, H.; Chaste, P.; Wilson, S.; et al. Microdeletions of ELP4 Are Associated with Language Impairment, Autism Spectrum Disorder, and Mental Retardation. Hum. Mutat. 2015, 36, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Tzoulaki, I.; White, I.M.; Hanson, I.M. PAX6 mutations: Genotype-phenotype correlations. BMC Genet. 2005, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Celik, A.; Kervestin, S.; Jacobson, A. NMD: At the crossroads between translation termination and ribosome recycling. Biochimie 2015, 114, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Vincent, M.C.; Pujo, A.L.; Olivier, D.; Calvas, P. Screening for PAX6 gene mutations is consistent with haploinsufficiency as the main mechanism leading to various ocular defects. Eur. J. Hum. Genet. 2003, 11, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.; Tang, H.K.; Lee, J.Y.; Saunders, G.F. Truncation mutations in the transactivation region of PAX6 result in dominant-negative mutants. J. Biol. Chem. 1998, 273, 21531–21541. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Khajavi, M.; Ohyama, T.; Hirabayashi, S.; Wilson, J.; Reggin, J.D.; Mancias, P.; Butler, I.J.; Wilkinson, M.F.; Wegner, M.; et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 2004, 36, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Vasilyeva, T.A.; Voskresenskaya, A.A.; Kasmann-Kellner, B.; Khlebnikova, O.V.; Pozdeyeva, N.A.; Bayazutdinova, G.M.; Kutsev, S.I.; Ginter, E.K.; Semina, E.V.; Marakhonov, A.V.; et al. Molecular analysis of patients with aniridia in Russian Federation broadens the spectrum of PAX6 mutations. Clin. Genet. 2017, 92, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.O.; Howarth, R.J.; Williamson, K.A.; van Heyningen, V.; Beal, S.J.; Crolla, J.A. Genetic analysis of chromosome 11p13 and the PAX6 gene in a series of 125 cases referred with aniridia. Am. J. Med. Genet. Part A 2008, 146, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Bobilev, A.M.; McDougal, M.E.; Taylor, W.L.; Geisert, E.E.; Netland, P.A.; Lauderdale, J.D. Assessment of PAX6 alleles in 66 families with aniridia. Clin. Genet. 2016, 89, 669–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, M.; Williamson, K.A.; Moore, A.T.; van Heyningen, V. Detailed ophthalmologic evaluation of 43 individuals with PAX6 mutations. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2581–2590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chao, L.Y.; Mishra, R.; Strong, L.C.; Saunders, G.F. Missense mutations in the DNA-binding region and termination codon in PAX6. Hum. Mutat. 2003, 21, 138–145. [Google Scholar] [CrossRef]
- Tang, H.K.; Chao, L.Y.; Saunders, G.F. Functional analysis of paired box missense mutations in the PAX6 gene. Hum. Mol. Genet. 1997, 6, 381–386. [Google Scholar] [CrossRef] [Green Version]
- Hanson, I.M. PAX6 and congenital eye malformations. Pediatr. Res. 2003, 54, 791–796. [Google Scholar] [CrossRef] [Green Version]
- Azuma, N.; Yamaguchi, Y.; Handa, H.; Tadokoro, K.; Asaka, A.; Kawase, E.; Yamada, M. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am. J. Hum. Genet. 2003, 72, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Williamson, K.A.; FitzPatrick, D.R. The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur. J. Med. Genet. 2014, 57, 369–380. [Google Scholar] [CrossRef]
- Harding, P.; Moosajee, M. The Molecular Basis of Human Anophthalmia and Microphthalmia. J. Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deml, B.; Reis, L.M.; Lemyre, E.; Clark, R.D.; Kariminejad, A.; Semina, E.V. Novel mutations in PAX6, OTX2 and NDP in anophthalmia, microphthalmia and coloboma. Eur. J. Hum. Genet. 2016, 24, 535–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perenthaler, E.; Yousefi, S.; Niggl, E.; Barakat, T.S. Beyond the Exome: The Non-coding Genome and Enhancers in Neurodevelopmental Disorders and Malformations of Cortical Development. Front. Cell. Neurosci. 2019, 13, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protas, M.E.; Weh, E.; Footz, T.; Kasberger, J.; Baraban, S.C.; Levin, A.V.; Katz, L.J.; Ritch, R.; Walter, M.A.; Semina, E.V.; et al. Mutations of conserved non-coding elements of PITX2 in patients with ocular dysgenesis and developmental glaucoma. Hum. Mol. Genet. 2017, 26, 3630–3638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisenberger, T.; Neuhaus, C.; Khan, A.O.; Decker, C.; Preising, M.N.; Friedburg, C.; Bieg, A.; Gliem, M.; Charbel Issa, P.; Holz, F.G.; et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: The example of retinal dystrophies. PLoS ONE 2013, 8, e78496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronskov, K.; Rosenberg, T.; Sand, A.; Brondum-Nielsen, K. Mutational analysis of PAX6: 16 novel mutations including 5 missense mutations with a mild aniridia phenotype. Eur. J. Hum. Genet. 1999, 7, 274–286. [Google Scholar] [CrossRef] [Green Version]
- Axton, R.A.; Hanson, I.M.; Love, J.; Seawright, A.; Prosser, J.; van Heyningen, V. Combined SSCP/heteroduplex analysis in the screening for PAX6 mutations. Mol. Cell. Probes 1997, 11, 287–292. [Google Scholar] [CrossRef]
- Filatova, A.Y.; Vasilyeva, T.A.; Marakhonov, A.V.; Voskresenskaya, A.A.; Zinchenko, R.A.; Skoblov, M.Y. Functional reassessment of PAX6 single nucleotide variants by in vitro splicing assay. Eur. J. Hum. Genet. 2019, 27, 488–493. [Google Scholar] [CrossRef] [Green Version]
- Yokoi, T.; Nishina, S.; Fukami, M.; Ogata, T.; Hosono, K.; Hotta, Y.; Azuma, N. Genotype-phenotype correlation of PAX6 gene mutations in aniridia. Hum. Genome Var. 2016, 3, 15052. [Google Scholar] [CrossRef] [Green Version]
- Crolla, J.A.; van Heyningen, V. Frequent chromosome aberrations revealed by molecular cytogenetic studies in patients with aniridia. Am. J. Hum. Genet. 2002, 71, 1138–1149. [Google Scholar] [CrossRef] [Green Version]
- Hall, H.N.; Williamson, K.A.; FitzPatrick, D.R. The genetic architecture of aniridia and Gillespie syndrome. Hum. Genet. 2019, 138, 881–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glaser, T.; Jepeal, L.; Edwards, J.G.; Young, S.R.; Favor, J.; Maas, R.L. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat. Genet. 1994, 7, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Sidor, B.; Szymanska, K.; Williamson, K.; van Heyningen, V.; Roszkowski, T.; Wierzba-Bobrowicz, T.; Zaremba, J. Malformations of the brain in two fetuses with a compound heterozygosity for two PAX6 mutations. Folia Neuropathol. 2009, 47, 372–382. [Google Scholar] [PubMed]
- Solomon, B.D.; Pineda-Alvarez, D.E.; Balog, J.Z.; Hadley, D.; Gropman, A.L.; Nandagopal, R.; Han, J.C.; Hahn, J.S.; Blain, D.; Brooks, B.; et al. Compound heterozygosity for mutations in PAX6 in a patient with complex brain anomaly, neonatal diabetes mellitus, and microophthalmia. Am. J. Med. Genet. Part A 2009, 149, 2543–2546. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Colby, K.A. A review of the clinical and genetic aspects of aniridia. Semin. Ophthalmol. 2013, 28, 306–312. [Google Scholar] [CrossRef]
- Qin, L.; Wang, J.; Tian, X.; Yu, H.; Truong, C.; Mitchell, J.J.; Wierenga, K.J.; Craigen, W.J.; Zhang, V.W.; Wong, L.C. Detection and Quantification of Mosaic Mutations in Disease Genes by Next-Generation Sequencing. J. Mol. Diagn. 2016, 18, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Riera, M.; Wert, A.; Nieto, I.; Pomares, E. Panel-based whole exome sequencing identifies novel mutations in microphthalmia and anophthalmia patients showing complex Mendelian inheritance patterns. Mol. Genet. Genom. Med. 2017, 5, 709–719. [Google Scholar] [CrossRef]
- Bai, Z.; Kong, X. Extension of the mutation spectrum of PAX6 from three Chinese congenital aniridia families and identification of male gonadal mosaicism. Mol. Genet. Genom. Med. 2018, 6, 1053–1067. [Google Scholar] [CrossRef]
- Tarilonte, M.; Morin, M.; Ramos, P.; Galdos, M.; Blanco-Kelly, F.; Villaverde, C.; Rey-Zamora, D.; Rebolleda, G.; Munoz-Negrete, F.J.; Tahsin-Swafiri, S.; et al. Parental Mosaicism in PAX6 Causes Intra-Familial Variability: Implications for Genetic Counseling of Congenital Aniridia and Microphthalmia. Front. Genet. 2018, 9, 479. [Google Scholar] [CrossRef] [Green Version]
- Dubey, S.K.; Mahalaxmi, N.; Vijayalakshmi, P.; Sundaresan, P. Mutational analysis and genotype-phenotype correlations in southern Indian patients with sporadic and familial aniridia. Mol. Vis. 2015, 21, 88–97. [Google Scholar]
- Gramer, E.; Reiter, C.; Gramer, G. Glaucoma and frequency of ocular and general diseases in 30 patients with aniridia: A clinical study. Eur. J. Ophthalmol. 2012, 22, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.T.; Kim, D.H.; Kim, H. PAX6 aniridia syndrome: Clinics, genetics, and therapeutics. Curr. Opin. Ophthalmol. 2017, 28, 436–447. [Google Scholar] [CrossRef] [PubMed]
- McCulley, T.J.; Mayer, K.; Dahr, S.S.; Simpson, J.; Holland, E.J. Aniridia and optic nerve hypoplasia. Eye 2005, 19, 762–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pedersen, H.R.; Hagen, L.A.; Landsend, E.C.S.; Gilson, S.J.; Utheim, O.A.; Utheim, T.P.; Neitz, M.; Baraas, R.C. Color Vision in Aniridia. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2142–2152. [Google Scholar] [CrossRef]
- Pedersen, H.R.; Neitz, M.; Gilson, S.J.; Landsend, E.C.S.; Utheim, O.A.; Utheim, T.P.; Baraas, R.C. The Cone Photoreceptor Mosaic in Aniridia: Within-Family Phenotype-Genotype Discordance. Ophthalmol. Retin. 2019, 3, 523–534. [Google Scholar] [CrossRef] [Green Version]
- Lagali, N.; Wowra, B.; Fries, F.N.; Latta, L.; Moslemani, K.; Utheim, T.P.; Wylegala, E.; Seitz, B.; Kasmann-Kellner, B. Early phenotypic features of aniridia-associated keratopathy and association with PAX6 coding mutations. Ocul. Surf. 2019. [Google Scholar] [CrossRef]
- Lagali, N.; Wowra, B.; Fries, F.N.; Latta, L.; Moslemani, K.; Utheim, T.P.; Wylegala, E.; Seitz, B.; Kasmann-Kellner, B. PAX6 Mutational Status Determines Aniridia-Associated Keratopathy Phenotype. Ophthalmology 2019. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, S.; Jinda, W.; Limwongse, C.; Atchaneeyasakul, L.O.; Phadke, S.R. Run-on mutation in the PAX6 gene and chorioretinal degeneration in autosomal dominant aniridia. Mol. Vis. 2011, 17, 1305–1309. [Google Scholar]
- Ito, Y.A.; Footz, T.K.; Berry, F.B.; Mirzayans, F.; Yu, M.; Khan, A.O.; Walter, M.A. Severe molecular defects of a novel FOXC1 W152G mutation result in aniridia. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3573–3579. [Google Scholar] [CrossRef] [Green Version]
- Law, S.K.; Sami, M.; Piri, N.; Coleman, A.L.; Caprioli, J. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol. Vis. 2011, 17, 1231–1238. [Google Scholar]
- Ito, Y.A.; Walter, M.A. Genomics and anterior segment dysgenesis: A review. Clin. Exp. Ophthalmol. 2014, 42, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shan, X.; Gregory-Evans, C.Y. A mouse model of aniridia reveals the in vivo downstream targets of Pax6 driving iris and ciliary body development in the eye. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Singh, H.P.; Lacal, P.M.; Sasman, A.; Fatima, A.; Liu, T.; Schultz, K.M.; Losordo, D.W.; Lehmann, O.J.; Kume, T. Forkhead box transcription factor FoxC1 preserves corneal transparency by regulating vascular growth. Proc. Natl. Acad. Sci. USA 2012, 109, 2015–2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Qin, G.; Chen, G.; Li, T.; Gao, L.; Huang, L.; Zhang, Y.; Ouyang, K.; Wang, Y.; Pang, Y.; et al. Variants in TRIM44 Cause Aniridia by Impairing PAX6 Expression. Hum. Mutat. 2015, 36, 1164–1167. [Google Scholar] [CrossRef] [PubMed]
- Han, J.C.; Liu, Q.R.; Jones, M.; Levinn, R.L.; Menzie, C.M.; Jefferson-George, K.S.; Adler-Wailes, D.C.; Sanford, E.L.; Lacbawan, F.L.; Uhl, G.R.; et al. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N. Engl. J. Med. 2008, 359, 918–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischbach, B.V.; Trout, K.L.; Lewis, J.; Luis, C.A.; Sika, M. WAGR syndrome: A clinical review of 54 cases. Pediatrics 2005, 116, 984–988. [Google Scholar] [CrossRef]
- Shinawi, M.; Sahoo, T.; Maranda, B.; Skinner, S.A.; Skinner, C.; Chinault, C.; Zascavage, R.; Peters, S.U.; Patel, A.; Stevenson, R.E.; et al. 11p14.1 microdeletions associated with ADHD, autism, developmental delay, and obesity. Am. J. Med. Genet. Part A 2011, 155, 1272–1280. [Google Scholar] [CrossRef]
- Harcourt, B.E.; Bullen, D.V.R.; Kao, K.T.; Tassoni, D.; Alexander, E.J.; Burgess, T.; White, S.M.; Sabin, M.A. Maternal inheritance of BDNF deletion, with phenotype of obesity and developmental delay in mother and child. Am. J. Med. Genet. Part A 2018, 176, 194–200. [Google Scholar] [CrossRef]
- Szatmari, P.; Paterson, A.D.; Zwaigenbaum, L.; Roberts, W.; Brian, J.; Liu, X.Q.; Vincent, J.B.; Skaug, J.L.; Thompson, A.P.; Senman, L.; et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007, 39, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Sisodiya, S.M.; Free, S.L.; Williamson, K.A.; Mitchell, T.N.; Willis, C.; Stevens, J.M.; Kendall, B.E.; Shorvon, S.D.; Hanson, I.M.; Moore, A.T.; et al. PAX6 haploinsufficiency causes cerebral malformation and olfactory dysfunction in humans. Nat. Genet. 2001, 28, 214–216. [Google Scholar] [CrossRef]
- Marlin, S.; Couet, D.; Lacombe, D.; Cessans, C.; Bonneau, D. Obesity: A new feature of WAGR (del 11p) syndrome. Clin. Dysmorphol. 1994, 3, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Gul, D.; Ogur, G.; Tunca, Y.; Ozcan, O. Third case of WAGR syndrome with severe obesity and constitutional deletion of chromosome (11)(p12p14). Am. J. Med. Genet. 2002, 107, 70–71. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Thomas, M.G.; Andrews, C.; Chan, W.M.; Proudlock, F.A.; McLean, R.J.; Pradeep, A.; Engle, E.C.; Gottlob, I. Autosomal-dominant nystagmus, foveal hypoplasia and presenile cataract associated with a novel PAX6 mutation. Eur. J. Hum. Genet. 2014, 22, 344–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Li, S.; Zhang, Q. Microphthalmia, late onset keratitis, and iris coloboma/aniridia in a family with a novel PAX6 mutation. Ophthalmic Genet. 2012, 33, 119–121. [Google Scholar] [CrossRef] [Green Version]
- Dansault, A.; David, G.; Schwartz, C.; Jaliffa, C.; Vieira, V.; de la Houssaye, G.; Bigot, K.; Catin, F.; Tattu, L.; Chopin, C.; et al. Three new PAX6 mutations including one causing an unusual ophthalmic phenotype associated with neurodevelopmental abnormalities. Mol. Vis. 2007, 13, 511–523. [Google Scholar]
- Kawase, E.; Tanaka, K.; Honna, T.; Azuma, N. A case of atypical WAGR syndrome with anterior segment anomaly and microphthalmos. Arch. Ophthalmol. 2001, 119, 1855–1856. [Google Scholar]
- Kamachi, Y.; Uchikawa, M.; Tanouchi, A.; Sekido, R.; Kondoh, H. Pax6 and SOX2 form a co-DNA-binding partner complex that regulates initiation of lens development. Genes Dev. 2001, 15, 1272–1286. [Google Scholar] [CrossRef] [Green Version]
- Aberdam, E.; Petit, I.; Sangari, L.; Aberdam, D. Induced pluripotent stem cell-derived limbal epithelial cells (LiPSC) as a cellular alternative for in vitro ocular toxicity testing. PLoS ONE 2017, 12, e0179913. [Google Scholar] [CrossRef] [Green Version]
- Williamson, K.A.; Hall, H.N.; Owen, L.J.; Livesey, B.J.; Hanson, I.M.; Adams, G.G.W.; Bodek, S.; Calvas, P.; Castle, B.; Clarke, M.; et al. Recurrent heterozygous PAX6 missense variants cause severe bilateral microphthalmia via predictable effects on DNA-protein interaction. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Dai, L.; Huang, Y.; Liao, Q.; Bai, Y. A large novel deletion downstream of PAX6 gene in a Chinese family with ocular coloboma. PLoS ONE 2013, 8, e83073. [Google Scholar] [CrossRef] [Green Version]
- Goolam, S.; Carstens, N.; Ross, M.; Bentley, D.; Lopes, M.; Peden, J.; Kingsbury, Z.; Tsogka, E.; Barlow, R.; Carmichael, T.R.; et al. Familial congenital cataract, coloboma, and nystagmus phenotype with variable expression caused by mutation in PAX6 in a South African family. Mol. Vis. 2018, 24, 407–413. [Google Scholar] [PubMed]
- Gregory-Evans, C.Y.; Williams, M.J.; Halford, S.; Gregory-Evans, K. Ocular coloboma: A reassessment in the age of molecular neuroscience. J. Med. Genet. 2004, 41, 881–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.; Choi, D.G.; Chun, B.Y.; Oh, E.H.; Lee, Y.J.; Kim, U.K.; Park, J.S. A family with a mild form of congenital nystagmus and optic disc coloboma caused by a novel PAX6 mutation. Gene 2019, 705, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.; Cecconi, F.; Bernier, G.; Andrejewski, N.; Kammandel, B.; Wagner, M.; Gruss, P. Spatial specification of mammalian eye territories by reciprocal transcriptional repression of Pax2 and Pax6. Development 2000, 127, 4325–4334. [Google Scholar]
- Gerber, S.; Alzayady, K.J.; Burglen, L.; Bremond-Gignac, D.; Marchesin, V.; Roche, O.; Rio, M.; Funalot, B.; Calmon, R.; Durr, A.; et al. Recessive and Dominant De Novo ITPR1 Mutations Cause Gillespie Syndrome. Am. J. Hum. Genet. 2016, 98, 971–980. [Google Scholar] [CrossRef] [Green Version]
- McEntagart, M.; Williamson, K.A.; Rainger, J.K.; Wheeler, A.; Seawright, A.; De Baere, E.; Verdin, H.; Bergendahl, L.T.; Quigley, A.; Rainger, J.; et al. A Restricted Repertoire of De Novo Mutations in ITPR1 Cause Gillespie Syndrome with Evidence for Dominant-Negative Effect. Am. J. Hum. Genet. 2016, 98, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Ticho, B.H.; Hilchie-Schmidt, C.; Egel, R.T.; Traboulsi, E.I.; Howarth, R.J.; Robinson, D. Ocular findings in Gillespie-like syndrome: Association with a new PAX6 mutation. Ophthalmic Genet. 2006, 27, 145–149. [Google Scholar] [CrossRef]
- Hanson, I.M.; Fletcher, J.M.; Jordan, T.; Brown, A.; Taylor, D.; Adams, R.J.; Punnett, H.H.; van Heyningen, V. Mutations at the PAX6 locus are found in heterogeneous anterior segment malformations including Peters’ anomaly. Nat. Genet. 1994, 6, 168–173. [Google Scholar] [CrossRef]
- Wolf, M.T.; Lorenz, B.; Winterpacht, A.; Drechsler, M.; Schumacher, V.; Royer-Pokora, B.; Blankenagel, A.; Zabel, B.; Wildhardt, G. Ten novel mutations found in Aniridia. Hum. Mutat. 1998, 12, 304–313. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, Y.; McGreal, R.; Cohen-Tayar, Y.; Rockowitz, S.; Wilczek, C.; Ashery-Padan, R.; Shechter, D.; Zheng, D.; Cvekl, A. Pax6 associates with H3K4-specific histone methyltransferases Mll1, Mll2, and Set1a and regulates H3K4 methylation at promoters and enhancers. Epigenet. Chromatin 2016, 9, 37. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Rockowitz, S.; Xie, Q.; Ashery-Padan, R.; Zheng, D.; Cvekl, A. Identification of in vivo DNA-binding mechanisms of Pax6 and reconstruction of Pax6-dependent gene regulatory networks during forebrain and lens development. Nucleic Acids Res. 2015, 43, 6827–6846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsui, S.; Wang, J.; Wang, L.; Dai, W.; Lu, L. CTCF-Mediated and Pax6-Associated Gene Expression in Corneal Epithelial Cell-Specific Differentiation. PLoS ONE 2016, 11, e0162071. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.C.; Lowe, K.; Hanson, L.; Gil, T.; Braun, L.; Howard, P.L.; Chow, R.L. Mapping the Pax6 3′ untranslated region microRNA regulatory landscape. BMC Genom. 2018, 19, 820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhinge, A.; Poschmann, J.; Namboori, S.C.; Tian, X.; Jia Hui Loh, S.; Traczyk, A.; Prabhakar, S.; Stanton, L.W. MiR-135b is a direct PAX6 target and specifies human neuroectoderm by inhibiting TGF-beta/BMP signaling. EMBO J. 2014, 33, 1271–1283. [Google Scholar] [CrossRef] [Green Version]
- Shaham, O.; Gueta, K.; Mor, E.; Oren-Giladi, P.; Grinberg, D.; Xie, Q.; Cvekl, A.; Shomron, N.; Davis, N.; Keydar-Prizant, M.; et al. Pax6 regulates gene expression in the vertebrate lens through miR-204. PLoS Genet. 2013, 9, e1003357. [Google Scholar] [CrossRef] [Green Version]
- Shalom-Feuerstein, R.; Serror, L.; De La Forest Divonne, S.; Petit, I.; Aberdam, E.; Camargo, L.; Damour, O.; Vigouroux, C.; Solomon, A.; Gaggioli, C.; et al. Pluripotent stem cell model reveals essential roles for miR-450b-5p and miR-184 in embryonic corneal lineage specification. Stem Cells 2012, 30, 898–909. [Google Scholar] [CrossRef]
- Berdasco, M.; Gomez, A.; Rubio, M.J.; Catala-Mora, J.; Zanon-Moreno, V.; Lopez, M.; Hernandez, C.; Yoshida, S.; Nakama, T.; Ishikawa, K.; et al. DNA Methylomes Reveal Biological Networks Involved in Human Eye Development, Functions and Associated Disorders. Sci. Rep. 2017, 7, 11762. [Google Scholar] [CrossRef] [Green Version]
- Singer, R.A.; Arnes, L.; Cui, Y.; Wang, J.; Gao, Y.; Guney, M.A.; Burnum-Johnson, K.E.; Rabadan, R.; Ansong, C.; Orr, G.; et al. The Long Noncoding RNA Paupar Modulates PAX6 Regulatory Activities to Promote Alpha Cell Development and Function. Cell Metab. 2019, 30, 1091–1106.e8. [Google Scholar] [CrossRef]
- Hayashi, R.; Ishikawa, Y.; Sasamoto, Y.; Katori, R.; Nomura, N.; Ichikawa, T.; Araki, S.; Soma, T.; Kawasaki, S.; Sekiguchi, K.; et al. Co-ordinated ocular development from human iPS cells and recovery of corneal function. Nature 2016, 531, 376–380. [Google Scholar] [CrossRef]
- Qiu, X.; Yang, J.; Liu, T.; Jiang, Y.; Le, Q.; Lu, Y. Efficient generation of lens progenitor cells from cataract patient-specific induced pluripotent stem cells. PLoS ONE 2012, 7, e32612. [Google Scholar] [CrossRef] [Green Version]
- Foster, J.W.; Wahlin, K.; Adams, S.M.; Birk, D.E.; Zack, D.J.; Chakravarti, S. Cornea organoids from human induced pluripotent stem cells. Sci. Rep. 2017, 7, 41286. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamm, D.M.; Clark, E.; Capowski, E.E.; Singh, R. The Role of FGF9 In The Production of Neural Retina And RPE In A Pluripotent Stem Cell Model of Early Human Retinal Development. Am. J. Ophthalmol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, D.A.; Lane, A.; Ramsden, C.M.; Carr, A.J.; Munro, P.M.; Jovanovic, K.; Schwarz, N.; Kanuga, N.; Muthiah, M.N.; Hull, S.; et al. Identification and Correction of Mechanisms Underlying Inherited Blindness in Human iPSC-Derived Optic Cups. Cell Stem Cell 2016, 18, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Blau, H.M.; Daley, G.Q. Stem Cells in the Treatment of Disease. N. Engl. J. Med. 2019, 380, 1748–1760. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.L.; Gao, M.L.; Lei, X.L.; Lv, J.N.; Zhao, H.; He, K.W.; Xia, X.X.; Li, L.Y.; Chen, Y.C.; Li, Y.P.; et al. Gene Correction Reverses Ciliopathy and Photoreceptor Loss in iPSC-Derived Retinal Organoids from Retinitis Pigmentosa Patients. Stem Cell Rep. 2018, 10, 1267–1281. [Google Scholar] [CrossRef] [Green Version]
- Richardson, R.; Smart, M.; Tracey-White, D.; Webster, A.R.; Moosajee, M. Mechanism and evidence of nonsense suppression therapy for genetic eye disorders. Exp. Eye Res. 2017, 155, 24–37. [Google Scholar] [CrossRef]
- Wang, X.; Gregory-Evans, K.; Wasan, K.M.; Sivak, O.; Shan, X.; Gregory-Evans, C.Y. Efficacy of Postnatal In Vivo Nonsense Suppression Therapy in a Pax6 Mouse Model of Aniridia. Mol. Ther. Nucleic Acids 2017, 7, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Gregory-Evans, C.Y.; Wang, X.; Wasan, K.M.; Zhao, J.; Metcalfe, A.L.; Gregory-Evans, K. Postnatal manipulation of Pax6 dosage reverses congenital tissue malformation defects. J. Clin. Investig. 2014, 124, 111–116. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Human PAX6 locus. (A) PAX6 gene structure in 11p13, with 14 exons (boxes, colours represent respective protein domains), promoters P0, P1 and Pα (boxes with diagonal lines) and regulatory elements (oval shapes with horizontal lines) including ectodermal enhancer (EE), located upstream PAX6, and Downstream Regulatory Region (DRR) and SIMO, within the introns of neighbour gene ELP4 (last four exons represented by orange boxes). EE: ectodermal enhancer; DRR: downstream regulatory region. (B) Main PAX6 isoforms, canonical PAX6 and PAX6(5a) and their respective structure with functional domains. PD: paired domain; NTS: N-terminal subdomain; CTS: C-terminal subdomain; LNK: linker region; HD: homeodomain; PST: proline-serine-threonine domain; aa: amino acids.
Figure 1.
Human PAX6 locus. (A) PAX6 gene structure in 11p13, with 14 exons (boxes, colours represent respective protein domains), promoters P0, P1 and Pα (boxes with diagonal lines) and regulatory elements (oval shapes with horizontal lines) including ectodermal enhancer (EE), located upstream PAX6, and Downstream Regulatory Region (DRR) and SIMO, within the introns of neighbour gene ELP4 (last four exons represented by orange boxes). EE: ectodermal enhancer; DRR: downstream regulatory region. (B) Main PAX6 isoforms, canonical PAX6 and PAX6(5a) and their respective structure with functional domains. PD: paired domain; NTS: N-terminal subdomain; CTS: C-terminal subdomain; LNK: linker region; HD: homeodomain; PST: proline-serine-threonine domain; aa: amino acids.

Figure 2.
Schematic representation of previously reported deletions in 11p13 encompassing regulatory regions 3’ of PAX6. Genes are represented by grey boxes and arrows indicate direction of transcription. PAX6 is highlighted in black. Known PAX6 enhancers are indicated by oval shapes with horizontal lines with focus on SIMO and E180B. Coloured bars represent deletions identified in different cohorts of aniridia patients without PAX6 mutations [68,72,73,75,76,77]. The vertical dashed lines indicate a common region deleted in all mentioned aniridia patients, which was defined first as 245 Kb long by Ansari et al. (blue dashed lines) and later reduced to 18 Kb by Plaisancie et al. (red dashed lines) [72,75]. Genomic coordinates are based on human genome assembly hg19.
Figure 2.
Schematic representation of previously reported deletions in 11p13 encompassing regulatory regions 3’ of PAX6. Genes are represented by grey boxes and arrows indicate direction of transcription. PAX6 is highlighted in black. Known PAX6 enhancers are indicated by oval shapes with horizontal lines with focus on SIMO and E180B. Coloured bars represent deletions identified in different cohorts of aniridia patients without PAX6 mutations [68,72,73,75,76,77]. The vertical dashed lines indicate a common region deleted in all mentioned aniridia patients, which was defined first as 245 Kb long by Ansari et al. (blue dashed lines) and later reduced to 18 Kb by Plaisancie et al. (red dashed lines) [72,75]. Genomic coordinates are based on human genome assembly hg19.

Figure 3.
Distribution of different mutation types in the PAX6 Mutation Database. (A) All of the mutations reported in the database. (B) Mutation frequencies associated to aniridia. (C) PAX6 mutation distribution in non-aniridia phenotypes. PTC (premature termination codon) include all mutations predicted to cause a premature stop codon and include nonsense, frameshift and splice site variants. CTE refer to variants that cause C-terminal extension of PAX6.
Figure 3.
Distribution of different mutation types in the PAX6 Mutation Database. (A) All of the mutations reported in the database. (B) Mutation frequencies associated to aniridia. (C) PAX6 mutation distribution in non-aniridia phenotypes. PTC (premature termination codon) include all mutations predicted to cause a premature stop codon and include nonsense, frameshift and splice site variants. CTE refer to variants that cause C-terminal extension of PAX6.

Figure 4.
Phenotypic spectrum of patients with PAX6 mutations. (A) Classical aniridia phenotype showing complete iris hypoplasia with central corneal opacity and vascularisation, (B) fundus imaging indicating absence of foveal reflex and (C) optical coherence tomography (OCT) of the macula showing foveal hypoplasia. (D) Left eye of a patient diagnosed with bilateral microphthalmia with microcornea. (E) Patient diagnosed with dominant nystagmus with no iris abnormalities and (F) foveal hypoplasia on OCT.
Figure 4.
Phenotypic spectrum of patients with PAX6 mutations. (A) Classical aniridia phenotype showing complete iris hypoplasia with central corneal opacity and vascularisation, (B) fundus imaging indicating absence of foveal reflex and (C) optical coherence tomography (OCT) of the macula showing foveal hypoplasia. (D) Left eye of a patient diagnosed with bilateral microphthalmia with microcornea. (E) Patient diagnosed with dominant nystagmus with no iris abnormalities and (F) foveal hypoplasia on OCT.

Figure 5.
Spectrum of coding PAX6 mutations causing non-aniridia phenotypes. A representation of canonical PAX6 structure is shown with respective domains and amino acid distribution. Mutations and respective phenotypes were collected from the PAX6 Mutation Database and the Human Genome Mutation Database (HGMD). The majority of mutations are included in the paired domain (NTS and CTS) and constitute missense mutations (squares). Mutations in alternative exon 5a are shown in blue. a Isolated foveal hypoplasia and nystagmus without iris abnormalities; variants shown in grey show cases presenting full iris but with possible mild structural defects [86]; b Optic nerve malformations include optic nerve coloboma, aplasia, and morning glory disc; NTS: N-terminal subdomain; CTS: C-terminal subdomain; LNK: linker region; HD: homeodomain; PSTD: proline-serine-threonine domain; aa: amino acids; ASD: anterior segment dysgenesis.
Figure 5.
Spectrum of coding PAX6 mutations causing non-aniridia phenotypes. A representation of canonical PAX6 structure is shown with respective domains and amino acid distribution. Mutations and respective phenotypes were collected from the PAX6 Mutation Database and the Human Genome Mutation Database (HGMD). The majority of mutations are included in the paired domain (NTS and CTS) and constitute missense mutations (squares). Mutations in alternative exon 5a are shown in blue. a Isolated foveal hypoplasia and nystagmus without iris abnormalities; variants shown in grey show cases presenting full iris but with possible mild structural defects [86]; b Optic nerve malformations include optic nerve coloboma, aplasia, and morning glory disc; NTS: N-terminal subdomain; CTS: C-terminal subdomain; LNK: linker region; HD: homeodomain; PSTD: proline-serine-threonine domain; aa: amino acids; ASD: anterior segment dysgenesis.


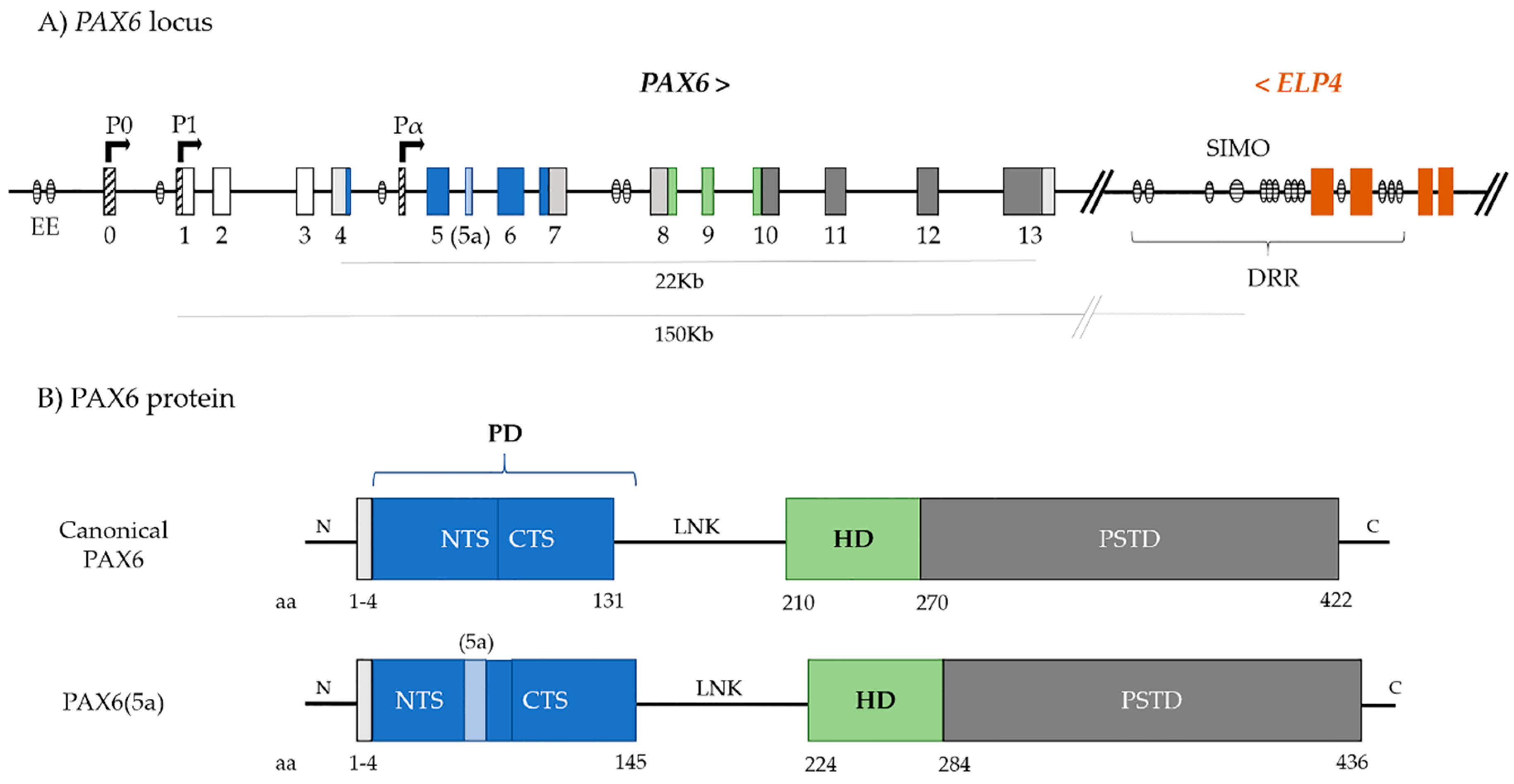{kind=link}
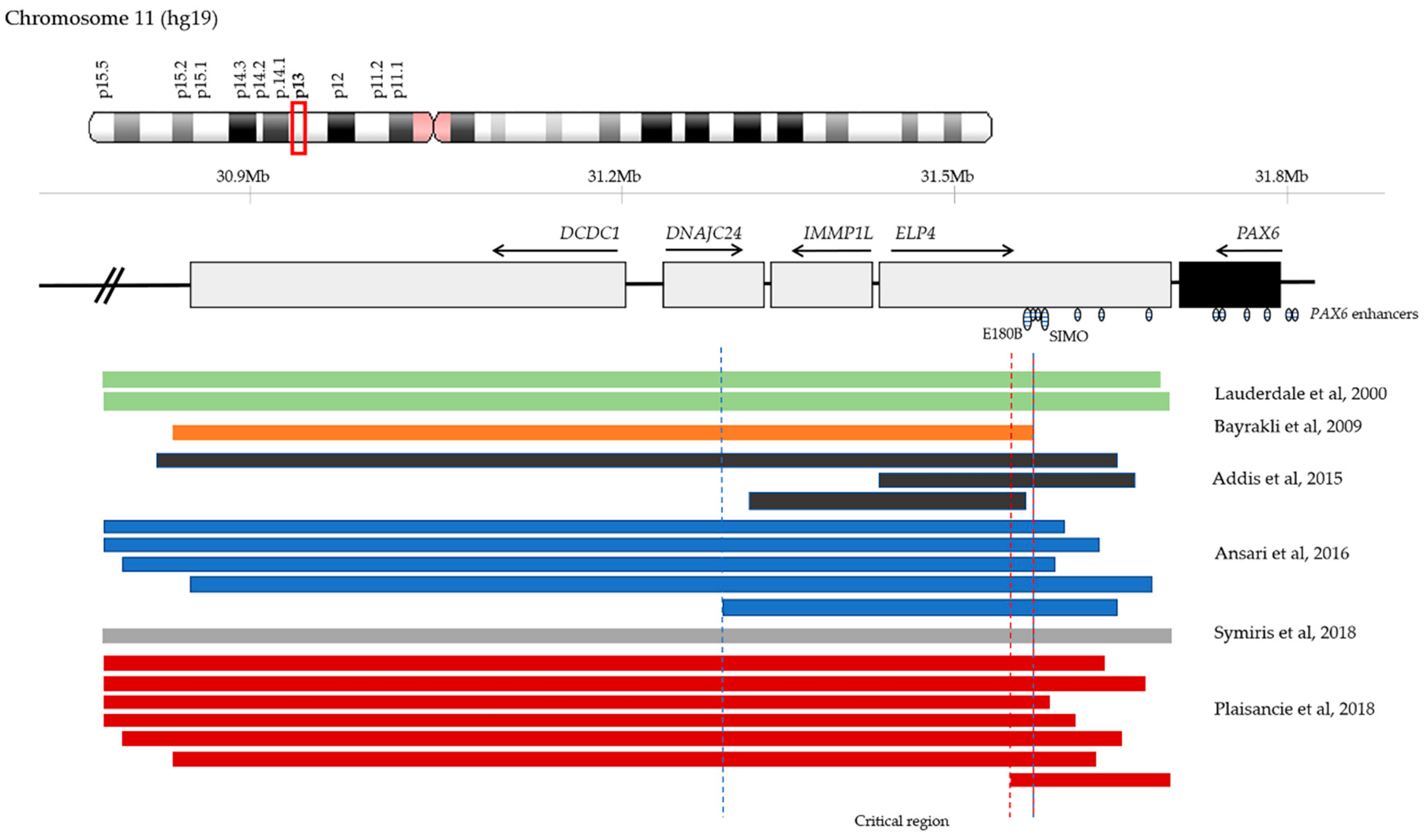{kind=link}
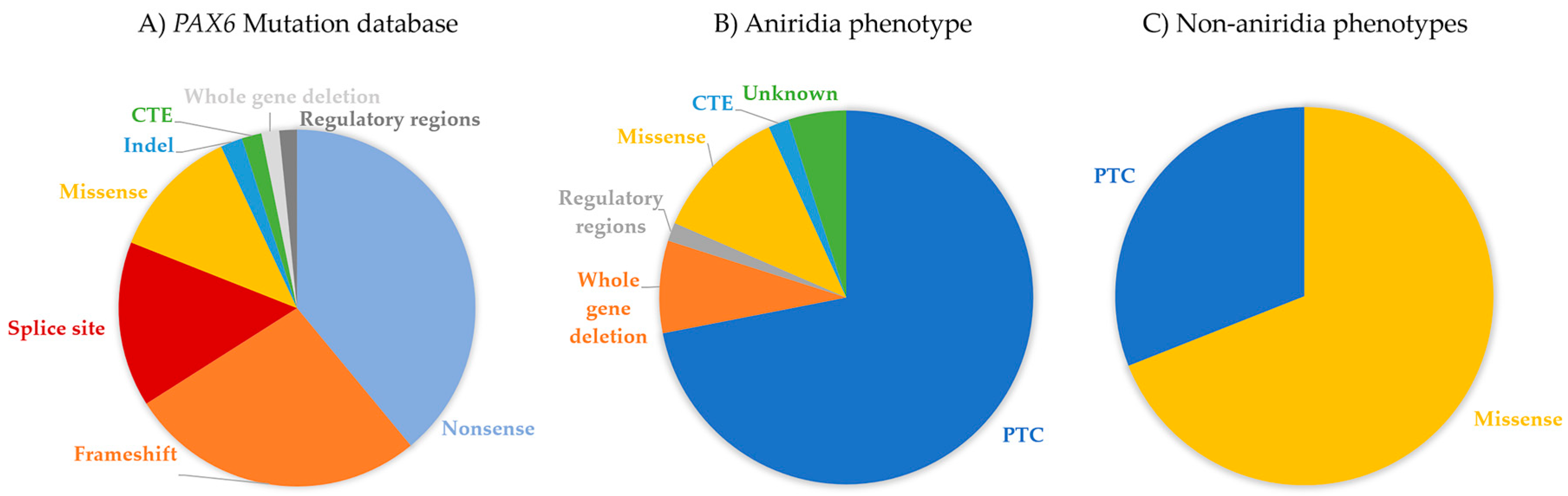{kind=link}
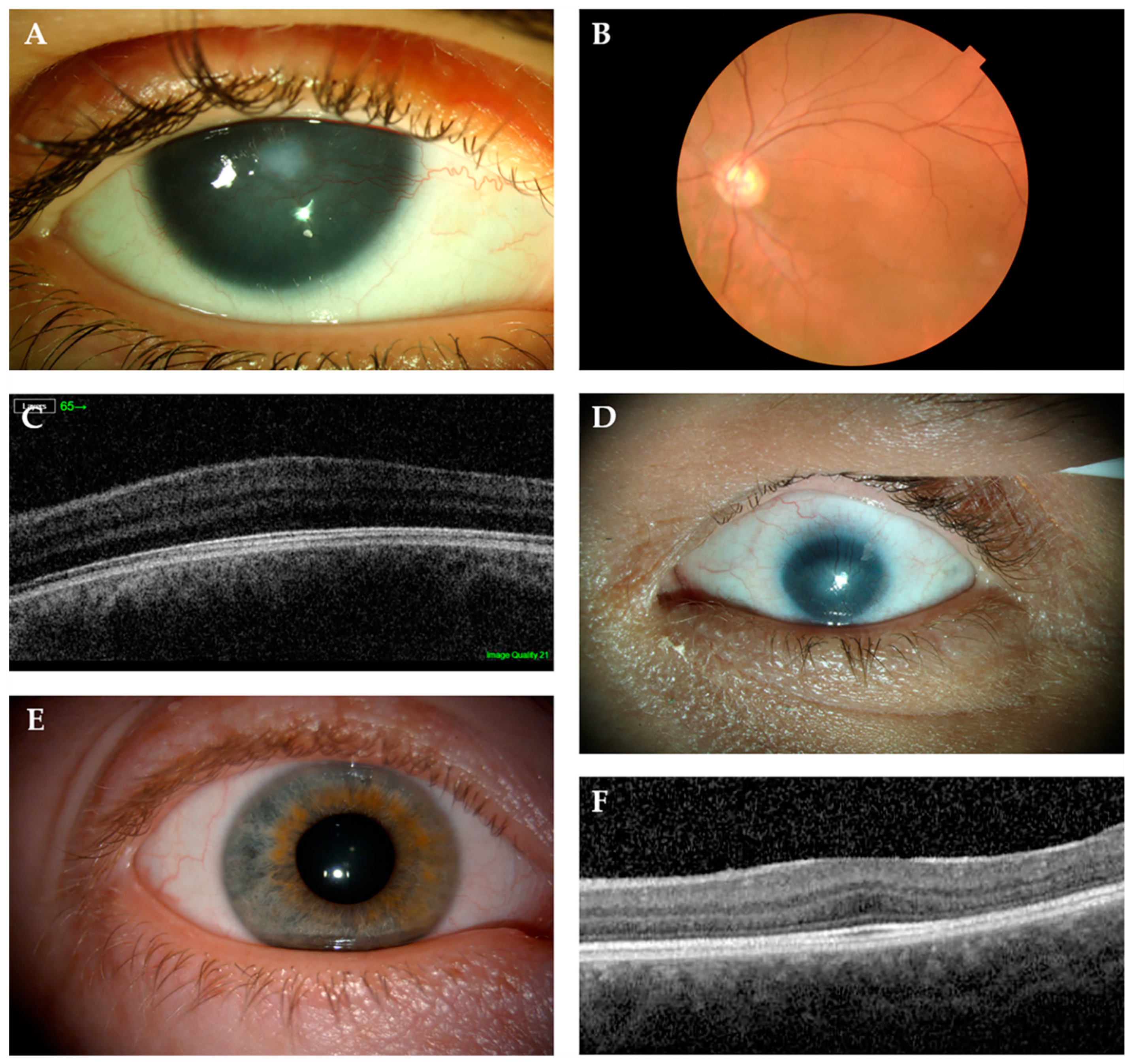{kind=link}
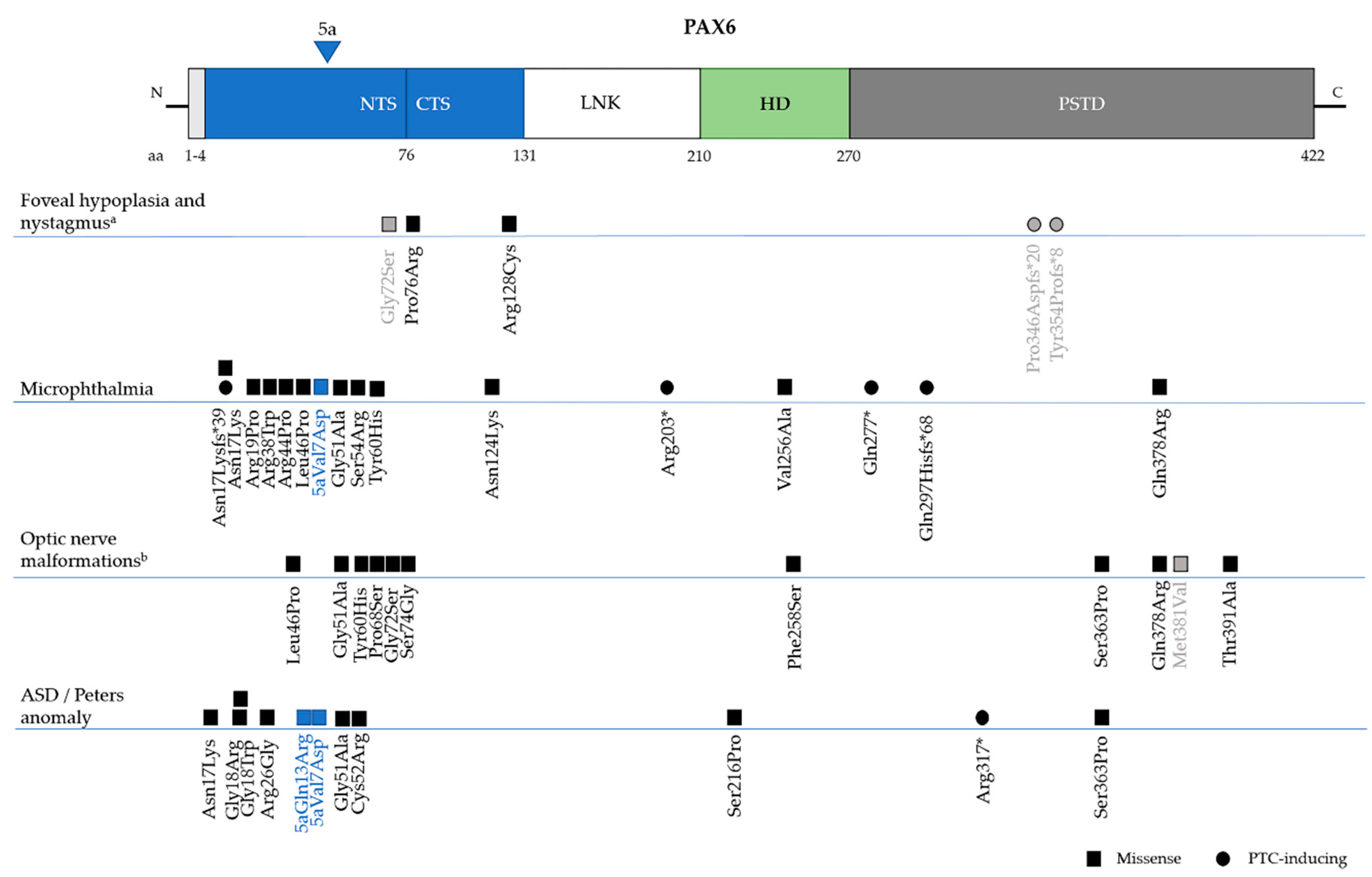{kind=link}
Table 1.
Summary of conserved regulatory elements in the PAX6 locus and their regulation of PAX6 ocular and extra-ocular expression.
Table 1.
Summary of conserved regulatory elements in the PAX6 locus and their regulation of PAX6 ocular and extra-ocular expression.
| Regulatory Element | Distance to PAX6 (Kb) | Location | Eye Expression | Extra-Ocular Expression | Reference |
|---|---|---|---|---|---|
| RB | −215 | Intron ELP4 | Diencephalon, telencephalon, pineal gland | [37] | |
| E180B | −176 | Intron ELP4 | RGCs and optic nerve | Trigeminal ganglia, dorsal spinal cord neurons | [56] |
| HS8B | −177 | Intron ELP4 | [25] | ||
| HS8A | −176 | Intron ELP4 | [25] | ||
| HS6 | −172 | Intron ELP4 | Neural retina | Neural structures | [57] |
| HS5 | −168 | Intron ELP4 | Optic cup and neural retina | Diencephalon | [57] |
| HS3 | −162 | Intron ELP4 | Neural retina | [25] | |
| HS2 | −161 | Intron ELP4 | Neural retina | [25] | |
| SIMO | −153 | Intron ELP4 | Lens and neural retina | Diencephalon, hindbrain, neural tube | [25] |
| E120 | −126 | Intron ELP4 | Olfactory bulbs, brain | [56] | |
| E100 | −104 | Intron ELP4 | Neural retina | Diencephalon, olfactory region | [58] |
| E60A | −54 | Intron ELP4 | Optic cup and neural retina | Neural structures | [57] |
| 7CE1 | −18 | Intron PAX6 | Late eye development | [59] | |
| NRE | −13 | Intron PAX6 | Neural retina, ciliary body, iris | [54] | |
| 0CE1 | −1 | 5’UTR PAX6 | [26] | ||
| agCNE14 (P2) | 4 | Pancreas | [56] | ||
| EE | 5 | Cornea, lens, conjunctiva and lacrimal gland | [54,60] | ||
| agCNE12 (P) | 7 | Pancreatic islets | |||
| agCNE11 | 8 | Pineal gland | [56] | ||
| agCNE10 (Up-9) | 9 | No specific pattern | [56] | ||
| agCNE9 (Up-10) | 12 | Pineal gland | [56] | ||
| Mouse-like Paupar | 54 | [61] | |||
| PE3 (E-52) | 57 | Developing pancreas and brain | [62] | ||
| E-55/C | 59 | No specific pattern | [56] | ||
| E-55/B | 60 | No specific pattern | [56] | ||
| E-55/A | 70 | Hindbrain | [56] | ||
| E-72 | 71 | No specific pattern | [56] | ||
| PE4 (E120) | 110 | Developing pancreas, olfactory tract, cerebellum, hindbrain | [62] | ||
| agCNE5 (Id855) | 178 | Forebrain | [56] | ||
| agCNE4 | 186 | Hindbrain | [56] | ||
| agCNE3 | 214 | Olfactory bulbs | [56] | ||
| E200 | 214 | Olfactory bulbs, cerebellum | [63] | ||
| agCNE1 | 224 | Trigeminal ganglia, dorsal spinal cord neurons | [56] | ||
| E250 | 247 | No specific pattern | [56] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lima Cunha, D.; Arno, G.; Corton, M.; Moosajee, M. The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye. Genes 2019, 10, 1050. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10121050
AMA Style
Lima Cunha D, Arno G, Corton M, Moosajee M. The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye. Genes. 2019; 10(12):1050. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10121050
Chicago/Turabian StyleLima Cunha, Dulce, Gavin Arno, Marta Corton, and Mariya Moosajee. 2019. "The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye" Genes 10, no. 12: 1050. https://0-doi-org.brum.beds.ac.uk/10.3390/genes10121050
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.