Identification of a Novel Imprinted Transcript in the Porcine GNAS Complex Locus Using Methylome and Transcriptome of Parthenogenetic Fetuses

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Porcine Oocyte Collection and In Vitro Maturation
2.3. Production of Parthenogenetic Embryos
2.4. Collection of Fertilized and Parthenogenetic Fetuses
2.5. Whole Genome Bisulfite Sequencing (WGBS)
2.6. Analysis of WGBS
2.7. RNA Extraction from the Fetuses
2.8. cDNA Library Construction and RNA-Sequencing
2.9. De novo Assembly of Sequencing Reads
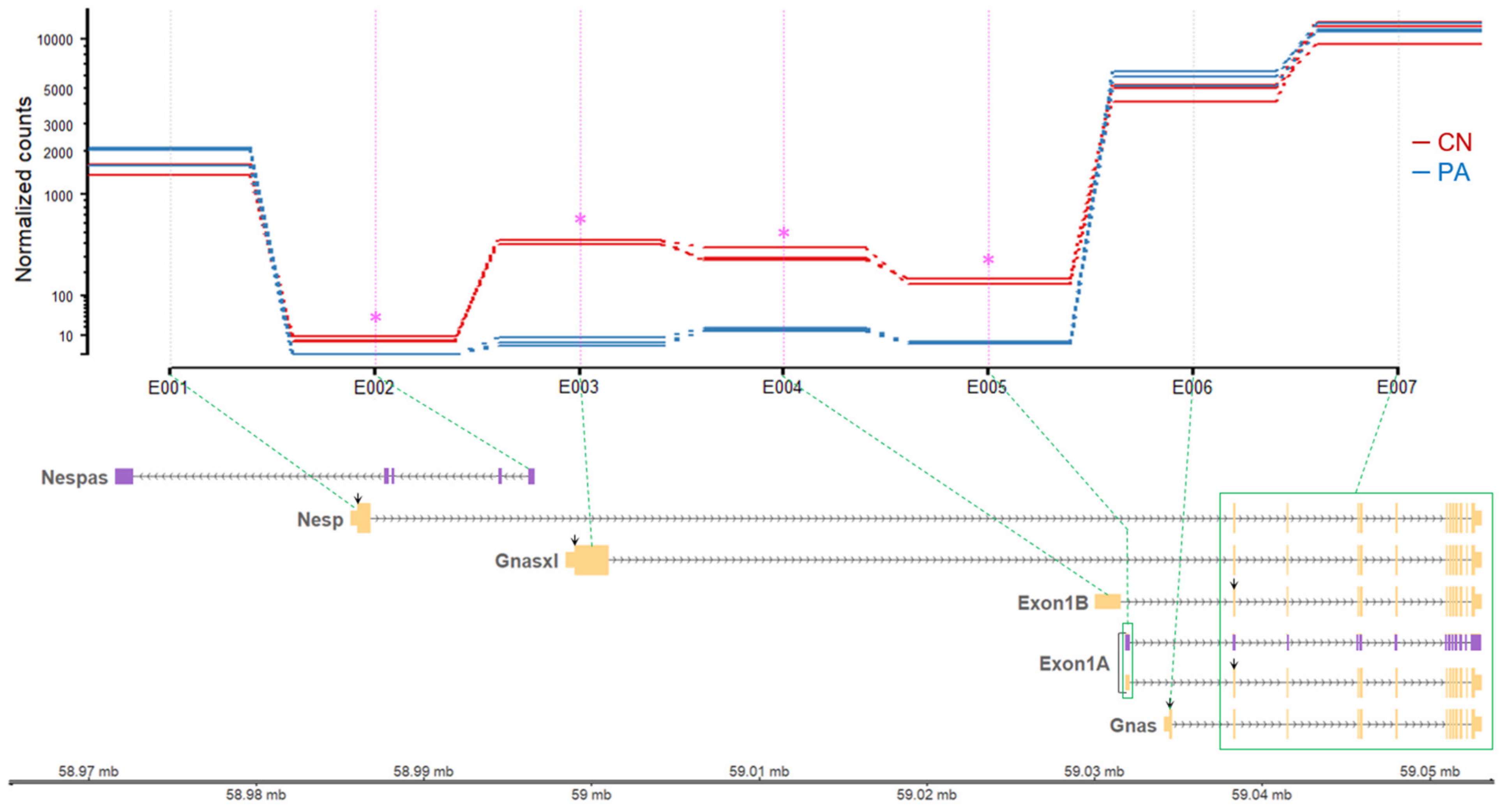
2.10. Analysis of Differential Expression of Exonic Regions
3. Results
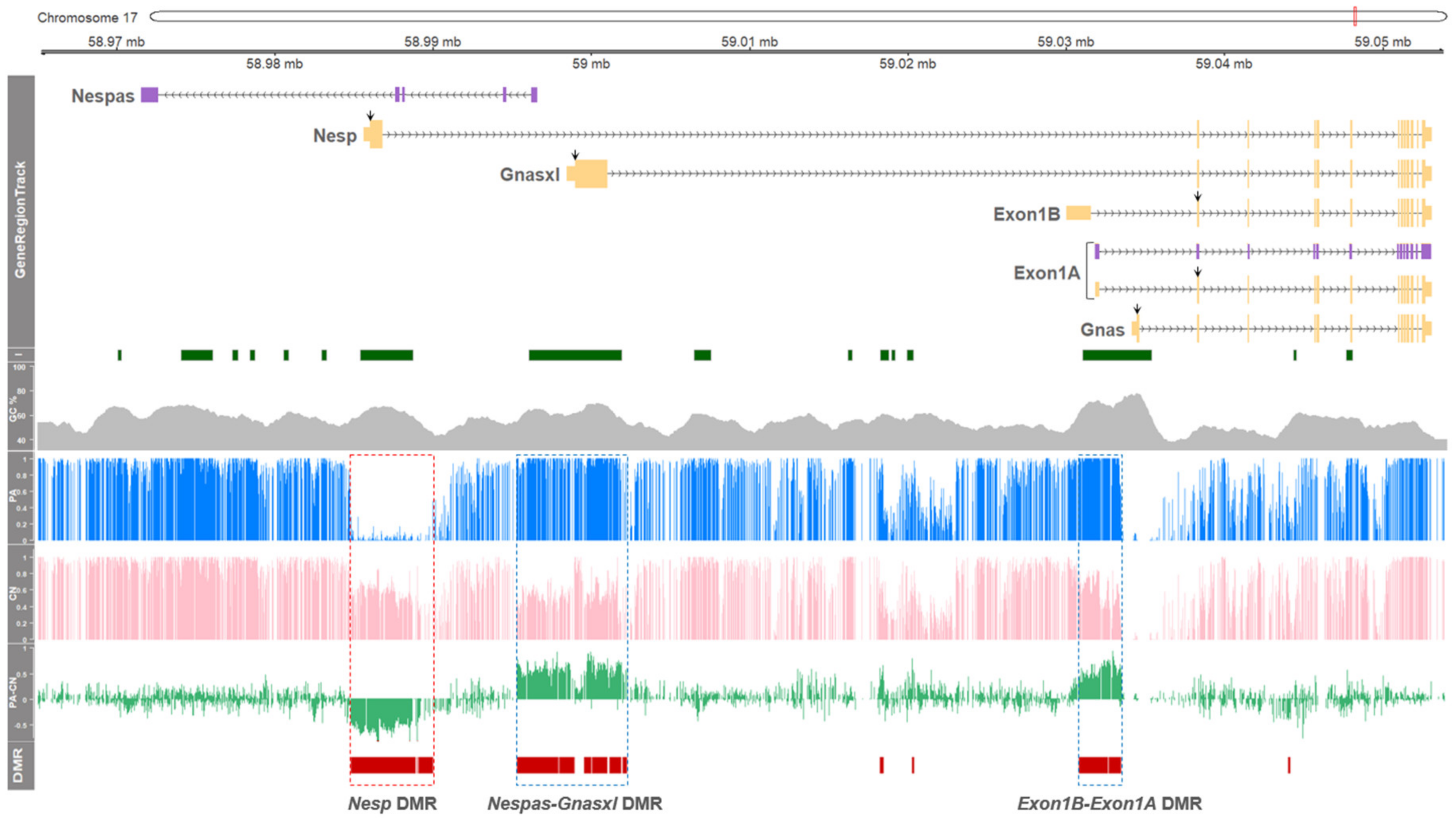
3.1. DNA Methylation in the GNAS Locus of Porcine Parthenogenetic Fetuses is Regulated in an Exon- Specific Manner
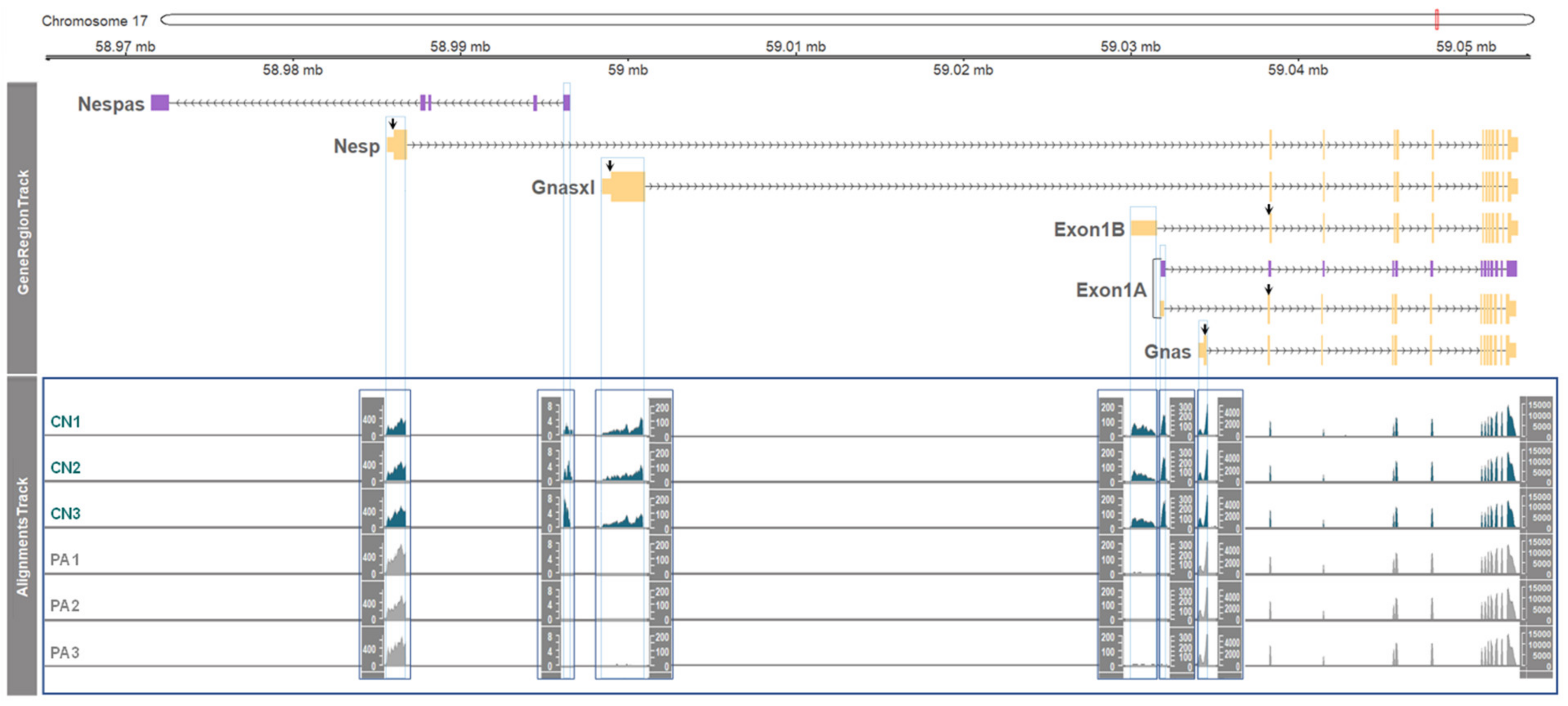
3.2. Paternal or Maternal Gene Expression in the GNAS Locus
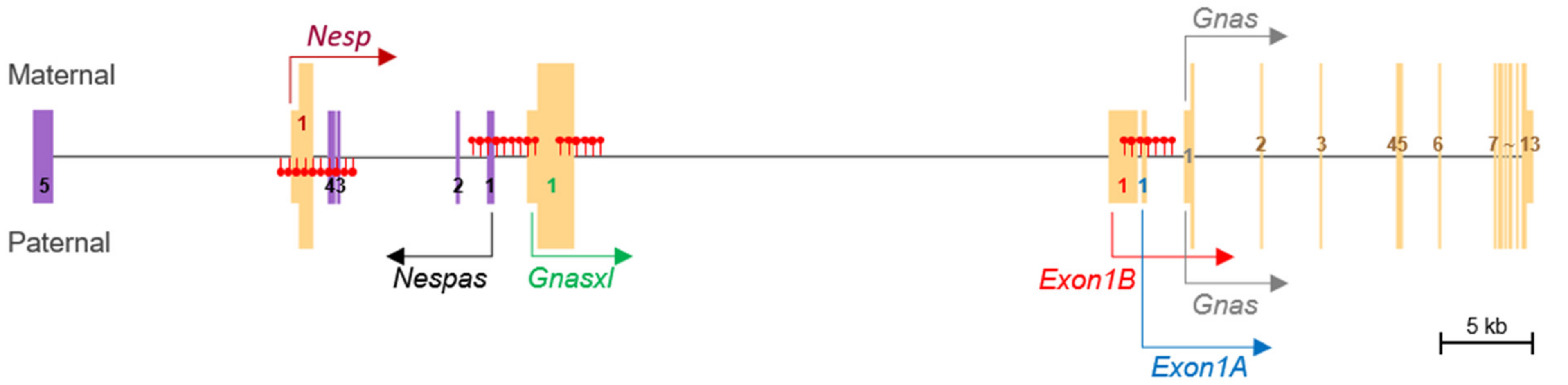
3.3. DNA Methylation and Allele-Specific Expression in the Porcine GNAS Locus Showed a Conserved Pattern
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Reik, W.; Walter, J. Genomic imprinting: Parental influence on the genome. Nat. Rev. Genet. 2001, 2, 21–32. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Babak, T.; DeVeale, B.; Tsang, E.K.; Zhou, Y.; Li, X.; Smith, K.S.; Kukurba, K.R.; Zhang, R.; Li, J.B.; van der Kooy, D.; et al. Genetic conflict reflected in tissue-specific maps of genomic imprinting in human and mouse. Nat. Genet. 2015, 47, 544–549. [Google Scholar] [CrossRef] [Green Version]
- Babak, T.; Deveale, B.; Armour, C.; Raymond, C.; Cleary, M.A.; van der Kooy, D.; Johnson, J.M.; Lim, L.P. Global survey of genomic imprinting by transcriptome sequencing. Curr. Biol. 2008, 18, 1735–1741. [Google Scholar] [CrossRef] [Green Version]
- Plagge, A.; Gordon, E.; Dean, W.; Boiani, R.; Cinti, S.; Peters, J.; Kelsey, G. The imprinted signaling protein XL alpha s is required for postnatal adaptation to feeding. Nat. Genet. 2004, 36, 818–826. [Google Scholar] [CrossRef]
- Chen, M.; Gavrilova, O.; Liu, J.; Xie, T.; Deng, C.; Nguyen, A.T.; Nackers, L.M.; Lorenzo, J.; Shen, L.; Weinstein, L.S. Alternative Gnas gene products have opposite effects on glucose and lipid metabolism. Proc. Natl. Acad. Sci. USA 2005, 102, 7386–7391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, T.; Plagge, A.; Gavrilova, O.; Pack, S.; Jou, W.; Lai, E.W.; Frontera, M.; Kelsey, G.; Weinstein, L.S. The alternative stimulatory G protein alpha-subunit XLalphas is a critical regulator of energy and glucose metabolism and sympathetic nerve activity in adult mice. J. Biol. Chem. 2006, 281, 18989–18999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neugebauer, N.; Luther, H.; Reinsch, N. Parent-of-origin effects cause genetic variation in pig performance traits. Animal 2010, 4, 672–681. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.C. Genomic imprinting in farm animals. Annu. Rev. Anim. Biosci. 2014, 2, 23–40. [Google Scholar] [CrossRef]
- Hayward, B.E.; Moran, V.; Strain, L.; Bonthron, D.T. Bidirectional imprinting of a single gene: GNAS1 encodes maternally, paternally, and biallelically derived proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 15475–15480. [Google Scholar] [CrossRef] [Green Version]
- Oczkowicz, M.; Piestrzyńska-Kajtoch, A.; Ropka-Molik, K.; Rejduch, B.; Eckert, R. Expression and imprinting analysis of the NESP55 gene in pigs. Gene Expr. Patterns 2012, 12, 18–23. [Google Scholar] [CrossRef]
- Kwon, D.J.; Kim, D.H.; Hwang, I.S.; Kim, D.E.; Kim, H.J.; Kim, J.S.; Lee, K.; Im, G.S.; Lee, J.W.; Hwang, S. Generation of alpha-1,3-galactosyltransferase knocked-out transgenic cloned pigs with knocked-in five human genes. Transgenic Res. 2017, 26, 153–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thurston, A.; Taylor, J.; Gardner, J.; Sinclair, K.D.; Young, L.E. Monoallelic expression of nine imprinted genes in the sheep embryo occurs after the blastocyst stage. Reproduction 2008, 135, 29–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischoff, S.; Tsai, S.; Hardison, N.; Motsinger-Reif, A.; Freking, B.; Piedrahita, J. Functional genomic approaches for the study of fetal/placental development in swine with special emphasis on imprinted genes. Control Pig Reprod. VIII 2010, 66, 245. [Google Scholar]
- Juhling, F.; Kretzmer, H.; Bernhart, S.H.; Otto, C.; Stadler, P.F.; Hoffmann, S. metilene: Fast and sensitive calling of differentially methylated regions from bisulfite sequencing data. Genome Res. 2016, 26, 256–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahne, F.; Ivanek, R. Visualizing Genomic Data Using Gviz and Bioconductor. Methods Mol. Biol. 2016, 1418, 335–351. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Reyes, A.; Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012, 22, 2008–2017. [Google Scholar] [CrossRef]
- Linglart, A.; Gensure, R.C.; Olney, R.C.; Juppner, H.; Bastepe, M. A novel STX16 deletion in autosomal dominant pseudohypoparathyroidism type Ib redefines the boundaries of a cis-acting imprinting control element of GNAS. Am. J. Hum. Genet. 2005, 76, 804–814. [Google Scholar] [CrossRef] [Green Version]
- Elli, F.M.; de Sanctis, L.; Peverelli, E.; Bordogna, P.; Pivetta, B.; Miolo, G.; Beck-Peccoz, P.; Spada, A.; Mantovani, G. Autosomal dominant pseudohypoparathyroidism type Ib: A novel inherited deletion ablating STX16 causes loss of imprinting at the A/B DMR. J. Clin. Endocrinol. Metab. 2014, 99, E724–E728. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; TuanMu, L.C.; Wei, H.; Gao, F.; Li, L.; Zhang, S. Development and imprinted gene expression in uniparental preimplantation mouse embryos in vitro. Mol. Biol. Rep. 2015, 42, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Arena, R.; Zacchini, F.; Toschi, P.; Palazzese, L.; Czernik, M.; Ptak, G.E. Developmental peculiarities in placentae of ovine uniparental conceptuses. PLoS ONE 2017, 12, e0188278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozasa, T.; Itoh, H.; Tsukamoto, T.; Kaziro, Y. Isolation and characterization of the human Gs alpha gene. Proc. Natl. Acad. Sci. USA 1988, 85, 2081–2085. [Google Scholar] [CrossRef] [Green Version]
- Williamson, C.M.; Ball, S.T.; Dawson, C.; Mehta, S.; Beechey, C.V.; Fray, M.; Teboul, L.; Dear, T.N.; Kelsey, G.; Peters, J. Uncoupling antisense-mediated silencing and DNA methylation in the imprinted Gnas cluster. PLoS Genet. 2011, 7, e1001347. [Google Scholar] [CrossRef] [Green Version]
- Tafaj, O.; Juppner, H. Pseudohypoparathyroidism: One gene, several syndromes. J. Endocrinol. Invest. 2017, 40, 347–356. [Google Scholar] [CrossRef]
- Liu, J.; Yu, S.; Litman, D.; Chen, W.; Weinstein, L.S. Identification of a methylation imprint mark within the mouse Gnas locus. Mol. Cell. Biol. 2000, 20, 5808–5817. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, L.S.; Xie, T.; Zhang, Q.H.; Chen, M. Studies of the regulation and function of the Gs alpha gene Gnas using gene targeting technology. Pharmacol. Ther. 2007, 115, 271–291. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Yu, D.; Lee, E.; Eckhaus, M.; Lee, R.; Corria, Z.; Accili, D.; Westphal, H.; Weinstein, L.S. Variable and tissue-specific hormone resistance in heterotrimeric Gs protein alpha-subunit (Gsalpha) knockout mice is due to tissue-specific imprinting of the gsalpha gene. Proc. Natl. Acad. Sci. USA 1998, 95, 8715–8720. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Wang, J.; Dickerson, K.E.; Kelleher, J.; Xie, T.; Gupta, D.; Lai, E.W.; Pacak, K.; Gavrilova, O.; Weinstein, L.S. Central nervous system imprinting of the G protein G(s)alpha and its role in metabolic regulation. Cell Metab. 2009, 9, 548–555. [Google Scholar] [CrossRef] [Green Version]
- Williamson, C.M.; Ball, S.T.; Nottingham, W.T.; Skinner, J.A.; Plagge, A.; Turner, M.D.; Powles, N.; Hough, T.; Papworth, D.; Fraser, W.D.; et al. A cis-acting control region is required exclusively for the tissue-specific imprinting of Gnas. Nat. Genet. 2004, 36, 894–899. [Google Scholar] [CrossRef]
- Williamson, C.M.; Turner, M.D.; Ball, S.T.; Nottingham, W.T.; Glenister, P.; Fray, M.; Tymowska-Lalanne, Z.; Plagge, A.; Powles-Glover, N.; Kelsey, G.; et al. Identification of an imprinting control region affecting the expression of all transcripts in the Gnas cluster. Nat. Genet. 2006, 38, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Jaritz, M.; Guenzl, P.; Vlatkovic, I.; Sommer, A.; Tamir, I.M.; Marks, H.; Klampfl, T.; Kralovics, R.; Stunnenberg, H.G.; et al. An RNA-Seq strategy to detect the complete coding and non-coding transcriptome including full-length imprinted macro ncRNAs. PLoS ONE 2011, 6, e27288. [Google Scholar] [CrossRef] [PubMed]
- Ogata, T.; Kagami, M. Kagami-Ogata syndrome: A clinically recognizable upd(14)pat and related disorder affecting the chromosome 14q32.2 imprinted region. J. Hum. Genet 2016, 61, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Eggermann, T.; Perez de Nanclares, G.; Maher, E.R.; Temple, I.K.; Tumer, Z.; Monk, D.; Mackay, D.J.; Gronskov, K.; Riccio, A.; Linglart, A.; et al. Imprinting disorders: A group of congenital disorders with overlapping patterns of molecular changes affecting imprinted loci. Clin. Epigenetics 2015, 7, 123. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ECB 1 | Width 2 | Exon | M.N.count 3 (CN) | M.N.count (PA) | log2fold 4 (PA/CN) | p-Value 5 | Padjust 6 |
|---|---|---|---|---|---|---|---|
| E001 | 1220 | Nesp 1st exon | 1473.00 ± 115.11 b | 2017.00 ± 176.12 b | 0.45 | 0.18 | 0.92 |
| E002 | 350 | Nespas l 1st exon | 6.00 ± 1.53 a | 0.0033 ± 0.0033 a | −10.81 | 1.59E − 06 | 7.32E − 04 |
| E003 | 1972 | Gnasxl 1st exon | 402.00 ± 4.04 ab | 5.00 ± 2.08 a | −6.33 | 0.00E + 00 7 | 0.00E + 00 |
| E004 | 1506 | Exon1B 1st exon | 298.00 ± 21.96 ab | 17.33 ± 1.86 a | −4.10 | 3.74E − 82 | 2.07E − 77 |
| E005 | 205 | Exon1A 1st exon | 155.67 ± 11.89 ab | 3.67 ± 0.67 a | −5.41 | 6.63E − 84 | 4.88E − 79 |
| E006 | 481 | Gnas 1st exon | 4621.00 ± 423.42 c | 6147.67 ± 536.13 c | 0.41 | 0.13 | 0.88 |
| E007 | 14,754 | Shared exons (2nd–13th) 8 | 10,757.33 ± 610.78 d | 12,251.00 ± 568.18 d | 0.19 | 0.81 | 1.00 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ahn, J.; Wu, H.; Lee, J.; Hwang, I.-S.; Yu, D.; Ahn, J.-S.; Lee, J.-W.; Hwang, S.; Lee, K. Identification of a Novel Imprinted Transcript in the Porcine GNAS Complex Locus Using Methylome and Transcriptome of Parthenogenetic Fetuses. Genes 2020, 11, 96. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010096
Ahn J, Wu H, Lee J, Hwang I-S, Yu D, Ahn J-S, Lee J-W, Hwang S, Lee K. Identification of a Novel Imprinted Transcript in the Porcine GNAS Complex Locus Using Methylome and Transcriptome of Parthenogenetic Fetuses. Genes. 2020; 11(1):96. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010096
Chicago/Turabian StyleAhn, Jinsoo, Huiguang Wu, Joonbum Lee, In-Sul Hwang, Debing Yu, Jin-Seop Ahn, Jeong-Woong Lee, Seongsoo Hwang, and Kichoon Lee. 2020. "Identification of a Novel Imprinted Transcript in the Porcine GNAS Complex Locus Using Methylome and Transcriptome of Parthenogenetic Fetuses" Genes 11, no. 1: 96. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010096