Reworking GWAS Data to Understand the Role of Nongenetic Factors in MS Etiopathogenesis

 ,
,  , and
, and
Abstract
:1. Introduction
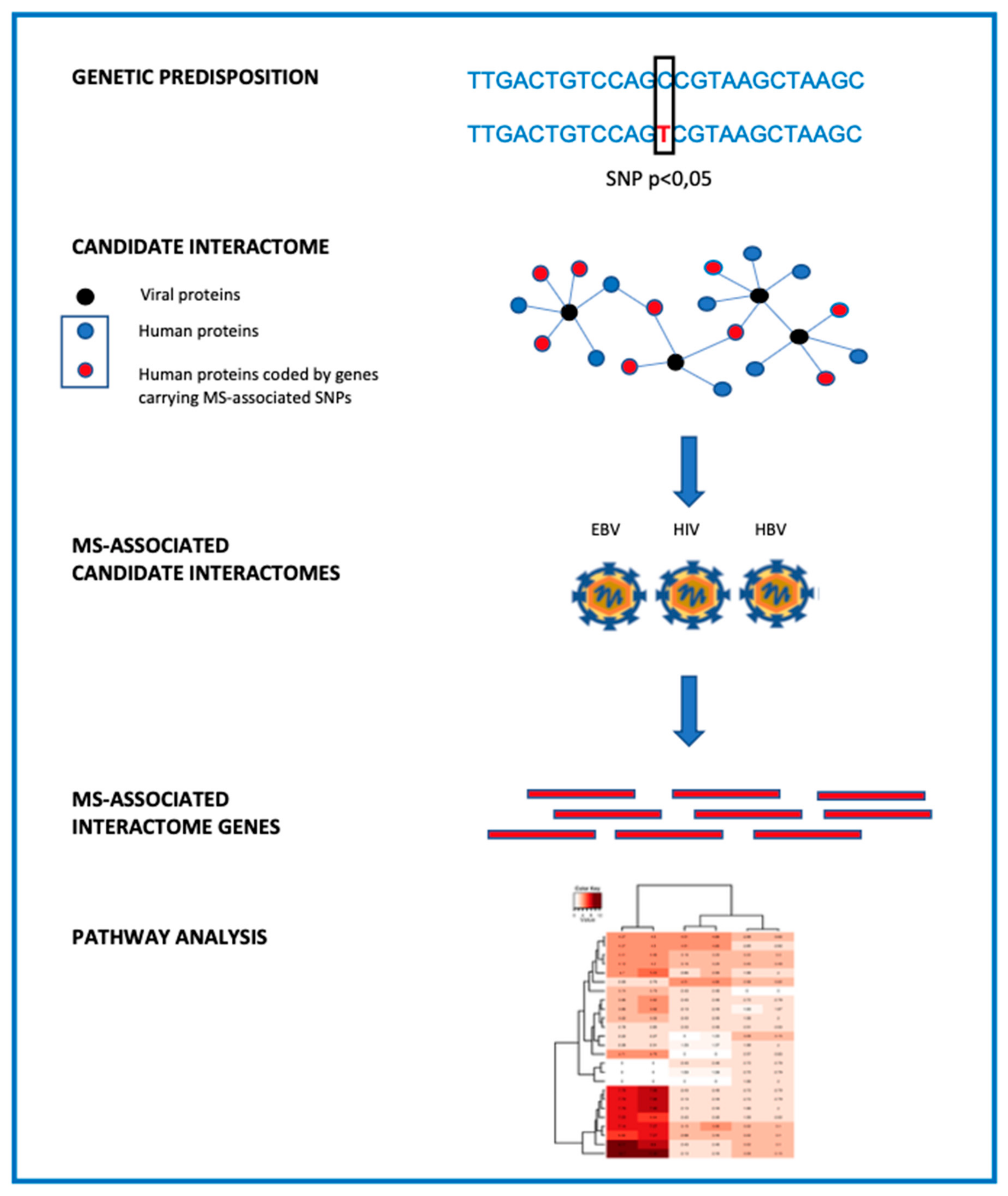
2. Bioinformatic Reworking of GWAS Data
3. Interactome-Based Approach
4. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Olsson, T.; Barcellos, L.F.; Alfredsson, L. Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat. Rev. Neurol. 2017, 13, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Kakalacheva, K.; Lunemann, J.D. Environmental triggers of multiple sclerosis. FEBS Lett. 2011, 585, 3724–3729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ontaneda, D.; Thompson, A.J.; Fox, R.J.; Cohen, J.A. Progressive multiple sclerosis: Prospects for disease therapy, repair, and restoration of function. Lancet 2017, 389, 1357–1366. [Google Scholar] [CrossRef]
- Ramagopalan, S.V.; Dobson, R.; Meier, U.C.; Giovannoni, G. Multiple sclerosis: Risk factors, prodromes, and potential causal pathways. Lancet Neurol. 2010, 9, 727–739. [Google Scholar] [CrossRef]
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bordi, I.; Umeton, R.; Ricigliano, V.A.; Annibali, V.; Mechelli, R.; Ristori, G.; Grassi, F.; Salvetti, M.; Sutera, A. A mechanistic, stochastic model helps understand multiple sclerosis course and pathogenesis. Int. J. Genom. 2013, 2013, 910321. [Google Scholar] [CrossRef] [PubMed]
- Bordi, I.; Ricigliano, V.A.; Umeton, R.; Ristori, G.; Grassi, F.; Crisanti, A.; Sutera, A.; Salvetti, M. Noise in multiple sclerosis: Unwanted and necessary. Ann. Clin. Transl. Neurol. 2014, 1, 502–511. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 2011, 476, 214–219. [Google Scholar] [CrossRef]
- IMSGC. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef]
- Cotsapas, C.; Mitrovic, M. Genome-wide association studies of multiple sclerosis. Clin. Trans. Immunol. 2018, 7, e1018. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, 6460. [Google Scholar]
- International Multiple Sclerosis Genetics Consortium. Low-Frequency and Rare-Coding Variation Contributes to Multiple Sclerosis Risk. Cell 2018, 175, 1679–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilariño-Güell, C.; Zimprich, A.; Martinelli-Boneschi, F.; Herculano, B.; Wang, Z.; Matesanz, F.; Urcelay, E.; Vandenbroeck, K.; Leyva, L.; Gris, D.; et al. Exome sequencing in multiple sclerosis families identifies 12 candidate genes and nominates biological pathways for the genesis of disease. PLoS Genet. 2019, 15, e1008180. [Google Scholar] [CrossRef] [PubMed]
- Vidmar, L.; Maver, A.; Drulović, J.; Sepčić, J.; Novaković, I.; Ristič, S.; Šega, S.; Peterlin, B. Multiple Sclerosis patients carry an increased burden of exceedingly rare genetic variants in the inflammasome regulatory genes. Sci. Rep. 2019, 9, 9171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ristori, G.; Cannoni, S.; Stazi, M.A.; Vanacore, N.; Cotichini, R.; Alfò, M.; Pugliatti, M.; Sotgiu, S.; Solaro, C.; Bomprezzi, R.; et al. Multiple sclerosis in twins from continental Italy and Sardinia: A nationwide study. Ann. Neurol. 2006, 59, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Fagnani, C.; Neale, M.C.; Nisticò, L.; Stazi, M.A.; Ricigliano, V.A.; Buscarinu, M.C.; Salvetti, M.; Ristori, G. Twin studies in multiple sclerosis: A meta-estimation of heritability and environmentality. Mult. Scler. 2015, 21, 1404–1413. [Google Scholar] [CrossRef]
- Maier, L.M.; Lowe, C.E.; Cooper, J.; Downes, K.; Anderson, D.E.; Severson, C.; Clark, P.M.; Healy, B.; Walker, N.; Aubin, C.; et al. IL2RA genetic heterogeneity in multiple sclerosis and type 1 diabetes susceptibility and soluble interleukin-2 receptor production. PLoS Genet. 2009, 5, e1000322. [Google Scholar] [CrossRef]
- Wallace, C.; Cutler, A.J.; Pontikos, N.; Pekalski, M.L.; Burren, O.S.; Cooper, J.D.; García, A.R.; Ferreira, R.C.; Guo, H.; Walker, N.M.; et al. Dissection of a Complex Disease Susceptibility Region Using a Bayesian Stochastic Search Approach to Fine Mapping. PLoS Genet. 2015, 11, e1005272. [Google Scholar] [CrossRef] [Green Version]
- Afanasyeva, M.A.; Putlyaeva, L.V.; Demin, D.E.; Kulakovskiy, I.V.; Vorontsov, I.E.; Fridman, M.V.; Makeev, V.J.; Kuprash, D.V.; Schwartz, A.M. The single nucleotide variant rs12722489 determines differential estrogen receptor binding and enhancer properties of an IL2RA intronic region. PLoS ONE 2017, 12, e0172681. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Cortes, A.; Shipman, L.; Evans, H.G.; Attfield, K.E.; Jostins, L.; Barber, T.; Kaur, G.; Kuttikkatte, S.B.; Leach, O.A.; et al. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci. Transl. Med. 2016, 8, 363ra149. [Google Scholar] [CrossRef] [Green Version]
- Galarza-Muñoz, G.; Briggs, F.B.S.; Evsyukova, I.; Schott-Lerner, G.; Kennedy, E.M.; Nyanhete, T.; Wang, L.; Bergamaschi, L.; Widen, S.G.; Tomaras, G.D.; et al. Human Epistatic Interaction Controls IL7R Splicing and Increases Multiple Sclerosis Risk. Cell 2017, 169, 72–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, A.P.; Dendrou, C.A.; Attfield, K.E.; Haghikia, A.; Xifara, D.K.; Butter, F.; Poschmann, G.; Kaur, G.; Lambert, L.; Leach, O.A.; et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature 2012, 488, 508–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steri, M.; Orrù, V.; Idda, M.L.; Pitzalis, M.; Pala, M.; Zara, I.; Sidore, C.; Faà, V.; Floris, M.; Deiana, M.; et al. Overexpression of the Cytokine BAFF and Autoimmunity Risk. N. Engl. J. Med. 2017, 376, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Milo, R. Therapies for multiple sclerosis targeting B cells. Croat. Med. J. 2019, 60, 87–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kappos, L.; Hartung, H.P.; Freedman, M.S.; Boyko, A.; Radü, E.W.; Mikol, D.D.; Lamarine, M.; Hyvert, Y.; Freudensprung, U.; Plitz, T.; et al. Atacicept in multiple sclerosis (ATAMS): A randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol. 2014, 13, 353–363. [Google Scholar] [CrossRef]
- Damotte, V.; Guillot-Noel, L.; Patsopoulos, N.A.; Madireddy, L.; El Behi, M.; International Multiple Sclerosis Genetics Consortium; Wellcome Trust Case Control Consortium 2; De Jager, P.L.; Baranzini, S.E.; Cournu-Rebeix, I.; et al. A gene pathway analysis highlights the role of cellular adhesion molecules in multiple sclerosis susceptibility. Genes Immun. 2014, 15, 126–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Starza, S.; Ferraldeschi, M.; Buscarinu, M.C.; Romano, S.; Fornasiero, A.; Mechelli, R.; Umeton, R.; Ristori, G.; Salvetti, M. Genome-Wide Multiple Sclerosis Association Data and Coagulation. Front. Neurol. 2019, 10, 95. [Google Scholar] [CrossRef]
- Fang, H.; ULTRA-DD Consortium; De Wolf, H.; Knezevic, B.; Burnham, K.L.; Osgood, J.; Sanniti, A.; Lledó Lara, A.; Kasela, S.; De Cesco, S.; et al. A genetics-led approach defines the drug target landscape of 30 immune-related traits. Nat. Genet. 2019, 51, 1082–1091. [Google Scholar] [CrossRef]
- Baranzini, S.E.; Galwey, N.W.; Wang, J.; Khankhanian, P.; Lindberg, R.; Pelletier, D.; Wu, W.; Uitdehaag, B.M.; Kappos, L.; Pol-Man, C.H.; et al. Pathway and network-based analysis of genome-wide association studies in multiple sclerosis. Hum. Mol. Genet. 2009, 18, 2078–2090. [Google Scholar] [CrossRef]
- IMSGC; Hafler, D.A.; Compston, A.; Sawcer, S.J.; Lander, E.S.; Daly, M.J.; De Jager, P.L.; de Bakker, P.I.; Gabriel, S.B.; Mirel, D.B.; et al. Risk alleles for multiple sclerosis identified by a Genome Wide Study. N. Engl. J. Med. 2007, 357, 851–862. [Google Scholar]
- Baranzini, S.E.; Wang, J.; Gibson, R.A.; Galwey, N.; Naegelin, Y.; Barkhof, F.; Radue, E.W.; Lindberg, R.L.; Uitdehaag, B.M.; Johnson, M.R.; et al. Genome-wide association analysis of susceptibility and clinical phenotype in multiple sclerosis. Hum. Mol. Genet. 2009, 18, 767–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Multiple Sclerosis Genetics Consortium. Network-based multiple sclerosis pathway analysis with GWAS data from 15,000 cases and 30,000 controls. Am. J. Hum. Genet. 2013, 92, 854–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patsopoulos, N.A.; Bayer Pharma MS Genetics Working Group; Steering Committees of Studies Evaluating IFNb-1b and a CCR1- Antagonist; ANZgene Consortium; GeneMSA; International Multiple Sclerosis Genetics Consortium; Reischl, J.; Lehr, S.; Bauer, D.; Heubach, J.; et al. Genome- wide meta-analysis identifies novel multiple sclerosis susceptibility loci. Ann. Neurol. 2011, 70, 897–912. [Google Scholar] [CrossRef] [PubMed]
- Holmans, P.; Green, E.K.; Pahwa, J.S.; Ferreira, M.A.; Purcell, S.M.; Sklar, P.; Owen, M.J.; O’Donovan, M.C.; Craddock, N.; Consortium, W.T.C.C. Gene ontology analysis of GWA study data sets provides insights into the biology of bipolar disorder. Am. J. Hum. Genet. 2009, 85, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricigliano, V.A.; Handel, A.E.; Sandve, G.K.; Annibali, V.; Ristori, G.; Mechelli, R.; Cader, M.Z.; Salvetti, M. EBNA2 binds to genomic intervals associated with multiple sclerosis and overlaps with vitamin D receptor occupancy. PLoS ONE 2015, 10, e0119605. [Google Scholar] [CrossRef] [PubMed]
- Cree, B.A.; Rioux, J.D.; McCauley, J.L.; Gourraud, P.F.D.; Goyette, P.; McElroy, J.; De Jager, P.; Santaniello, A.; Vyse, T.J.; Gregersen, P.K.; et al. A major histocompatibility Class I locus contributes to multiple sclerosis susceptibility independently from HLA-DRB1*15:01. PLoS ONE 2010, 5, e11296. [Google Scholar] [CrossRef] [PubMed]
- Harley, J.B.; Chen, X.; Pujato, M.; Miller, D.; Maddox, A.; Forney, C.; Magnussen, A.F.; Lynch, A.; Chetal, K.; Yukawa, M.; et al. Transcription factors operate across disease loci with EBNA2 implicated in autoimmunity. Nat. Genet. 2018, 50, 699–707. [Google Scholar] [CrossRef]
- International Multiple Sclerosis Genetics Consortium. A systems biology approach uncovers cell-specific gene regulatory effects of genetic associations in multiple sclerosis. Nat. Commun. 2019, 10, 2236. [Google Scholar] [CrossRef] [Green Version]
- Cirillo, E.; Parnell, L.D.; Evelo, C.T. A Review of Pathway-Based Analysis Tools That Visualize Genetic Variants. Front. Genet. 2017, 8, 174. [Google Scholar] [CrossRef] [Green Version]
- Giacalone, G.; Clarelli, F.; Osiceanu, A.M.; Guaschino, C.; Brambilla, P.; Sorosina, M.; Liberatore, G.; Zauli, A.; Esposito, F.; Rodegher, M.; et al. Analysis of genes, pathways and networks involved in disease severity and age at onset in primary-progressive multiple sclerosis. Mult. Scler. 2015, 21, 1431–1442. [Google Scholar] [CrossRef]
- Wang, L.; Mousavi, P.; Baranzini, S.E. iPINBPA: An integrative network-based functional module discovery tool for genome-wide association studies. Pac. Symp. Biocomput. 2015, 255–266. [Google Scholar]
- Afrasiabi, A.; Parnell, G.P.; Fewings, N.; Schibeci, S.D.; Basuki, M.A.; Chandramohan, R.; Zhou, Y.; Taylor, B.; Brown, D.A.; Swaminathan, S.; et al. Evidence from genome wide association studies implicates reduced control of Epstein-Barr virus infection in multiple sclerosis susceptibility. Genome Med. 2019, 11, 26. [Google Scholar] [CrossRef] [Green Version]
- Salvetti, M.; Giovannoni, G.; Aloisi, F. Epstein-Barr virus and multiple sclerosis. Curr. Opin. Neurol. 2009, 22, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Levin, L.I.; Munger, K.L.; O’Reilly, E.J.; Falk, K.I.; Ascherio, A. Primary infection with the Epstein-Barr virus and risk of multiple sclerosis. Ann. Neurol. 2010, 67, 824–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ENCODE Project Consortium. The ENCODE (ENCyclopedia of DNA Elements) Project. Science 2004, 306, 636–640. [Google Scholar] [CrossRef] [Green Version]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [Green Version]
- Jirtle, R.L.; Skinner, M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007, 8, 253–262. [Google Scholar] [CrossRef]
- Mechelli, R.; Annibali, V.; Ristori, G.; Vittori, D.; Coarelli, G.; Salvetti, M. Multiple sclerosis etiology: Beyond genes and environment. Expert Rev. Clin. Immunol. 2010, 6, 481–490. [Google Scholar] [CrossRef]
- Pedre, X.; Mastronardi, F.; Bruck, W.; López-Rodas, G.; Kuhlmann, T.; Casaccia, P. Changed histone acetylation patterns in normal-appearing white matter and early multiple sclerosis lesions. J. Neurosci. 2011, 31, 3435. [Google Scholar] [CrossRef] [Green Version]
- Farh, K.K.H.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.H.; Shishkin, A.A.; et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef]
- Castro, K.; Casaccia, P. Epigenetic modifications in brain and immune cells of multiple sclerosis patients. Mult. Scler. 2018, 24, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, T.; Lindén, M.; Morikawa, H.; Fernandes, S.J.; Ruhrmann, S.; Huss, M.; Brandi, M.; Piehl, F.; Jagodic, M.; Tegnér, J.; et al. Impact of genetic risk loci for multiple sclerosis on expression of proximal genes in patients. Hum. Mol. Genet. 2018, 27, 912–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Jager, P.L.; Baecher-Allan, C.; Maier, L.M.; Arthur, A.T.; Ottoboni, L.; Barcellos, L.; McCauley, J.L.; Sawcer, S.; Goris, A.; Saarela, J.; et al. The role of the CD58 locus in multiple sclerosis. Proc. Natl. Acad. Sci. USA 2009, 106, 5264–5269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hecker, M.; Fitzner, B.; Blaschke, J.; Blaschke, P.; Zettl, U.K. Susceptibility variants in the CD58 gene locus point to a role of microRNA-548ac in the pathogenesis of multiple sclerosis. Mutat. Res. Rev. Mutat. Res. 2015, 763, 161–167. [Google Scholar] [CrossRef]
- Hecker, M.; Boxberger, N.; Illner, N.; Fitzner, B.; Schroder, I.; Winkelmann, A.; Dudesek, A.; Meister, S.; Koczan, D.; Lorenz, P.; et al. A genetic variant associated with multiple sclerosis inversely affects the expression of CD58 and microRNA-548ac from the same gene. PLoS Genet. 2019, 15, e1007961. [Google Scholar] [CrossRef]
- Mechelli, R.; Umeton, R.; Policano, C.; Annibali, V.; Coarelli, G.; Ricigliano, V.A.; Vittori, D.; Fornasiero, A.; Buscarinu, M.C.; International Multiple Sclerosis Genetics Consortium; et al. A “candidate-interactome” aggregate analysis of genome-wide association data in multiple sclerosis. PLoS ONE 2013, 8, e63300. [Google Scholar] [CrossRef] [Green Version]
- Menche, J.; Sharma, A.; Kitsak, M.; Ghiassian, S.D.; Videl, M.; Loscalzo, J.; Barabasi, A.L. Uncovering disease-disease relationships through the incomplete interactome. Science 2015, 347, 841–849. [Google Scholar] [CrossRef] [Green Version]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef]
- Li, J.; Holm, J.; Bergh, J.; Eriksson, M.; Darabi, H.; Lindström, L.S.; Törnberg, S.; Hall, P.; Czene, K. Breast cancer genetic risk profile is differentially associated with interval and screen-detected breast cancers. Ann. Oncol. 2015, 26, 517–522. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef]
- Karlson, E.W.; Chibnik, L.B.; Kraft, P.; Cui, J.; Keenan, B.T.; Ding, B.; Raychaudhuri, S.; Klareskog, L.; Alfredsson, L.; Plenge, R.M. Cumulative association of 22 genetic variants with sero positive rheumatoid arthritis risk. Ann. Rheum. Dis. 2010, 69, 1077–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, S.M.; MacDonald, K.; Fathalli, F.; Yim, A.; Lepage, M.; Iyer, S.; Malla, A.; Joober, R. Polygenic risk score associated with specific symptom dimension sin first-episode psychosis. Schizophr. Res. 2017, 184, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Vassos, E.; Di Forti, M.; Coleman, J.; Iyegbe, C.; Prata, D.; Euesden, J.; O’Reilly, P.; Curtis, C.; Kolliakou, A.; Patel, H.; et al. An examination of polygenic score risk prediction in individuals with first-episode psychosis. Biol. Psychiatry 2017, 81, 470–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Natarajan, P.; Young, R.; Stitziel, N.O.; Padmanabhan, S.; Baber, U.; Mehran, R.; Sartori, S.; Fuster, V.; Reilly, D.F.; Butterworth, A.; et al. Polygenic risk score identifies subgroup with higher burden of atherosclerosis and greater relative benefit from statin therapy in the primary prevention setting. Circulation 2017, 135, 2091–2101. [Google Scholar] [CrossRef] [Green Version]
- Raynor, L.A.; Pankow, J.S.; Rasmussen-Torvik, L.J.; Tang, W.; Prizment, A.; Couper, D.J. Pleiotropy and pathway analyses of genetic variants associated with both type 2 diabetes and prostate cancer. Int. J. Mol. Epidemiol. Genet. 2013, 4, 49–60. [Google Scholar]
- Layton, J.; Li, X.; Shen, C.; De Groot, M.; Lange, L.; Correa, A.; Wessel, J. Type 2 diabetes genetic risk scores are associated with increased type 2 diabetes risk among African Americans by cardiometabolic status. Clin. Med. Insights Endocrinol. Diabetes 2018, 11, 1179551417748942. [Google Scholar] [CrossRef]
- Belsky, D.W.; Sears, M.R.; Hancox, R.J.; Harrington, H.; Houts, R.; Moffitt, T.E.; Sugden, K.; Williams, B.; Poulton, R.; Caspi, A. Polygenic risk and the development and course of asthma: An analysis of data from a four-decade longitudinal study. Lancet Respir. Med. 2013, 1, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Pihlstrom, L.; Morset, K.R.; Grimstad, E.; Vitelli, V.; Toft, M. A cumulative genetic risk score predicts progression in Parkinson’s disease. Mov. Disord. 2016, 31, 487–490. [Google Scholar] [CrossRef]
- Nalls, M.A.; Escott-Price, V.; Williams, N.M.; Lubbe, S.; Keller, M.F.; Morris, H.R.; Singleton, A.B.; International Parkinson’s Disease Genomics Consortium (IPDGC). Genetic risk and age in Parkinson’s disease: Continuum not stratum. Mov. Disord. 2015, 30, 850–854. [Google Scholar] [CrossRef] [Green Version]
- Khera, A.V.; Emdin, C.A.; Drake, I.; Natarajan, P.; Bick, A.G.; Cook, N.R.; Chasman, D.I.; Baber, U.; Mehran, R.; Rader, D.J.; et al. Genetic risk: Adherence to a healthy life style, and coronary disease. N. Engl. J. Med. 2016, 375, 2349–2358. [Google Scholar] [CrossRef] [Green Version]
- Ripatti, S.; Tikkanen, E.; Orho-Melander, M.; Havulinna, A.S.; Silander, K.; Sharma, A.; Guiducci, C.; Perola, M.; Jula, A.; Sinisalo, J.; et al. A multi locus genetic risks core for coronary heart disease: Case-control and prospective cohort analyses. Lancet 2010, 376, 1393–1400. [Google Scholar] [CrossRef] [Green Version]
- Okuda, D.T.; Mowry, E.M.; Beheshtian, A.; Waubant, E.; Baranzini, S.E.; Goodin, D.S.; Hauser, S.L.; Pelletier, D. Incidental MRI anomalies suggestive of multiple sclerosis: The radiologically isolated syndrome. Neurology 2009, 72, 800–805. [Google Scholar] [CrossRef] [PubMed]
- Yea, C.; Tellier, R.; Chong, P.; Westmacott, G.; Marrie, R.A.; Bar-Or, A.; Banwell, B.; Canadian Pediatric Demyelinating Disease Network. Epstein-Barr virus in oral shedding of children with multiple sclerosis. Neurology 2013, 81, 1392–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mechelli, R.; Manzari, C.; Policano, C.; Annese, A.; Picardi, E.; Umeton, R.; Fornasiero, A.; D’Erchia, A.M.; Buscarinu, M.C.; Agliardi, C.; et al. Epstein-Barr virus genetic variants are associated with multiple sclerosis. Neurology 2015, 84, 1362–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wray, N.R.; Wijmenga, C.; Sullivan, P.F.; Yang, J.; Visscher, P.M. Common disease is more complex than implied by the core omnigenic model. Cell 2018, 173, 1573–1580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, M.D.; Chen-Plotkin, A.S. The post-GWAS era: From association to function. Am. J. Hum. Genet. 2018, 102, 717–730. [Google Scholar] [CrossRef]


{kind=link}
| Source | GWAS Datasets | p-Value Cut-Off of MS-Associated SNPs | Analysis | Results |
|---|---|---|---|---|
| Baranzini et al 2009 [29] | IMSGC, 2007 [30]; Baranzini et al. 2009 [31] | p < 0.05 | Pathway analysis and protein–protein interaction (PPI)-network | Identification of immunological and neural pathways enriched in MS |
| IMSGC et al. 2013 [32] | IMSGC, 2011 [8]; Patsopoulos et al. 2011 [33] | p < 0.05 | protein-interaction-network-based pathway analysis (PINBPA) | Identification of new MS susceptibility loci association blocks (groups of contiguous genes with a p-value < 0.05) including BCL10, CD48, REL, TRAF3, and TEC. |
| Mechelli et al. 2013 [34] | IMSGC, 2011 [8] | p < 0.05 | Candidate-interactome approach | EBV is the most MS-associated environmental risk factor interacting with MS-associated SNPs. |
| Ricigliano et al. 2015 [35] | IMSGC 2013 [9]; Cree et al. 2010 [36] | p < 5 × 108 | Transcription factor binding | EBNA2 and VDR binding sites are enriched in MS-associated loci. |
| Harley et al 2018 [37] | NHGRI GWAS catalog | p < 5 × 108 | Transcription factor binding | EBNA2 binding sites are enriched in autoimmune-associated loci, including MS. |
| IMSGC et al. 2019 [38] | IMSGC 2019 [11] | p < 5 × 108 | Prioritization of cell specific gene/protein networks | Explanation of the potential role of GAWS signals in a tissue/cell-specific manner: identification of cell-specific susceptibility pathways. |
| IMSGC 2019 [11] | IMSGC 2019 [11] | p < 5 × 108 | Multiple approaches: cell-specific eQTL, pathway analyses; PPI | Prioritization of genes putatively associated with the disease, and identification of possible major implications for resident microglia and the B cell in MS. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mechelli, R.; Umeton, R.; Manfrè, G.; Romano, S.; Buscarinu, M.C.; Rinaldi, V.; Bellucci, G.; Bigi, R.; Ferraldeschi, M.; Salvetti, M.; et al. Reworking GWAS Data to Understand the Role of Nongenetic Factors in MS Etiopathogenesis. Genes 2020, 11, 97. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010097
Mechelli R, Umeton R, Manfrè G, Romano S, Buscarinu MC, Rinaldi V, Bellucci G, Bigi R, Ferraldeschi M, Salvetti M, et al. Reworking GWAS Data to Understand the Role of Nongenetic Factors in MS Etiopathogenesis. Genes. 2020; 11(1):97. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010097
Chicago/Turabian StyleMechelli, Rosella, Renato Umeton, Grazia Manfrè, Silvia Romano, Maria Chiara Buscarinu, Virginia Rinaldi, Gianmarco Bellucci, Rachele Bigi, Michela Ferraldeschi, Marco Salvetti, and et al. 2020. "Reworking GWAS Data to Understand the Role of Nongenetic Factors in MS Etiopathogenesis" Genes 11, no. 1: 97. https://0-doi-org.brum.beds.ac.uk/10.3390/genes11010097